一种双核镉配合物及其制备方法和应用与流程
1.本发明涉及合成化合物技术领域,尤其涉及一种双核镉配合物及其制备方法和应用。
背景技术:
2.一氧化氮广泛分布于生物体内各组织中,特别是神经组织中,是一种新型生物信使分子。一氧化氮是一种极不稳定的生物自由基,分子小结构简单,常温下为气体,微溶于水具有脂溶性,可快速透过生物膜扩散。一氧化氮生物半衰期只有3~5s,其生成依赖于一氧化氮合成酶,并在心、脑血管调节、神经、免疫调节等方面有着十分重要的生物学作用,因此受到人们的普遍重视。在空气中一氧化氮会很快转变为二氧化氮产生刺激作用,产生的氮氧化物会造成呼吸道刺激,损害呼吸道;人体处于过量的氮氧化物环境中,会出现胸闷、呼吸窘迫、紫绀等症状,一氧化氮浓度高可致高铁血红蛋白血症。在进行检查时,一氧化氮的检测是一项重要的检测项目,但是由于其半衰期短和快速扩散的特性,检测结果会出现偏差。所以如何完成对一氧化氮的检测,成为了当下研究的热点问题。
技术实现要素:
3.本发明的目的在于提供克服现有技术中的问题,提供一种双核镉配合物及其制备方法和应用。
4.为了实现上述发明目的,本发明提供以下技术方案:
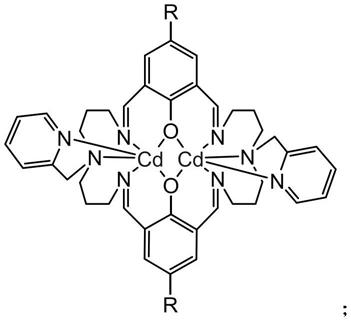
5.本发明提供了一种双核镉配合物,具有如下所示结构:
[0006][0007]
所述r为甲基、甲氧基、f、cl或br。
[0008]
本发明提供了所述双核镉配合物的制备方法,包含下列步骤:
[0009]
(1)将取代苯酚、乙酸铅和无水乙醇混合,得到混合溶液;
[0010]
(2)将混合溶液和n,n
‑
二(3
‑
氨丙基)
‑2‑
吡啶甲胺的无水乙醇溶液混合后进行配位缩合反应,得到铅配合物;
[0011]
(3)将铅配合物和硫酸镉的无水甲醇溶液混合后进行置换反应,即得所述双核镉配合物;
[0012]
所述取代苯酚为2,6
‑
二甲酰基
‑4‑
甲基苯酚、2,6
‑
二甲酰基
‑4‑
甲氧基苯酚、2,6
‑
二甲酰基
‑4‑
氟苯酚、2,6
‑
二甲酰基
‑4‑
氯苯酚或2,6
‑
二甲酰基
‑4‑
溴苯酚。
[0013]
作为优选,所述步骤(1)中取代苯酚和乙酸铅的摩尔比为0.4~0.6:0.4~0.6;
[0014]
所述取代苯酚和无水乙醇的摩尔体积比为0.4~0.6mmol:10~20ml。
[0015]
作为优选,所述步骤(1)中混合的方式为搅拌,所述搅拌的转速为200~400rpm,所述搅拌的时间为1.8~2.2h。
[0016]
作为优选,所述步骤(2)中n,n
‑
二(3
‑
氨丙基)
‑2‑
吡啶甲胺和步骤(1)中取代苯酚的摩尔比为0.4~0.6:0.4~0.6;
[0017]
所述n,n
‑
二(3
‑
氨丙基)
‑2‑
吡啶甲胺和n,n
‑
二(3
‑
氨丙基)
‑2‑
吡啶甲胺的无水乙醇溶液的摩尔体积比为0.4~0.6mmol:8~12ml。
[0018]
作为优选,所述步骤(2)中混合的方式为将n,n
‑
二(3
‑
氨丙基)
‑2‑
吡啶甲胺的无水乙醇溶液滴加进混合溶液中;
[0019]
所述滴加的速率为2~4滴/秒。
[0020]
作为优选,所述步骤(2)中配位缩合反应的时间为2.5~3.5h。
[0021]
作为优选,所述步骤(3)中硫酸镉和步骤(1)中取代苯酚的摩尔比为0.15~0.184:0.4~0.6;
[0022]
所述硫酸镉和硫酸镉的无水甲醇溶液的摩尔体积比为0.15~0.184mmol:10~20ml。
[0023]
作为优选,所述步骤(3)中混合的方式为将硫酸镉的无水甲醇溶液滴加进铅配合物中;
[0024]
所述滴加的速率为2~4滴/秒;
[0025]
所述置换反应的时间为3.5~4.5h。
[0026]
本发明还提供了所述双核镉配合物在制备一氧化氮荧光探针产品中的应用。
[0027]
本发明提供了一种双核镉配合物,本发明以取代苯酚和n,n
‑
二(3
‑
氨丙基)
‑2‑
吡啶甲胺为原料,以大配位数的铅离子为模板,进行配位缩合反应得到了铅配合物;将铅配合物和硫酸镉混合,将配合物中的铅离子置换,得到了双核镉配合物,置换出来的铅离子形成硫酸铅沉淀,便于后续的过滤分离。本发明提供的制备方法简单,核心步骤为配位缩合反应和置换反应,能高效的制备得到目标配合物。经过实验验证,本发明制备的双核镉配合物和一氧化氮能形成稳定的复合物,检测限低,可用于制备一氧化氮荧光探针产品。
附图说明
[0028]
图1为实施例1制备的双核镉配合物的红外光谱图;
[0029]
图2为实施例1制备的双核镉配合物的电喷雾质谱图;
[0030]
图3为实施例1制备的双核镉配合物的分子结构图;
[0031]
图4为实施例1制备的双核镉配合物的十面体结构图;
[0032]
图5为实施例1制备的双核镉配合物的分子间氢键图;
[0033]
图6为实施例1制备的双核镉配合物的沿a轴氢键图;
[0034]
图7为实施例1制备的双核镉配合物通入no的颜色对比图;
[0035]
图8为实施例1制备的双核镉配合物通入no的紫外吸收光谱图;
[0036]
图9为实施例1制备的双核镉配合物通入no的紫外发光图;
[0037]
图10为实施例1制备的双核镉配合物在可见光下通入no的发射光谱图;
[0038]
图11为实施例1制备的双核镉配合物在450nm处的
△
a/b对
△
a的关系图。
具体实施方式
[0039]
本发明提供了一种双核镉配合物,具有如下所示结构:
[0040][0041]
所述r为甲基、甲氧基、f、cl或br。
[0042]
本发明还提供了所述双核镉配合物的制备方法,包含下列步骤:
[0043]
(1)将取代苯酚、乙酸铅和无水乙醇混合,得到混合溶液;
[0044]
(2)将混合溶液和n,n
‑
二(3
‑
氨丙基)
‑2‑
吡啶甲胺的无水乙醇溶液混合后进行配位缩合反应,得到铅配合物;
[0045]
(3)将铅配合物和硫酸镉的无水甲醇溶液混合后进行置换反应,即得所述双核镉配合物;
[0046]
所述取代苯酚为2,6
‑
二甲酰基
‑4‑
甲基苯酚、2,6
‑
二甲酰基
‑4‑
甲氧基苯酚、2,6
‑
二甲酰基
‑4‑
氟苯酚、2,6
‑
二甲酰基
‑4‑
氯苯酚或2,6
‑
二甲酰基
‑4‑
溴苯酚。
[0047]
在本发明中,所述步骤(1)中乙酸铅优选为四水合乙酸铅。
[0048]
在本发明中,所述步骤(1)中取代苯酚和乙酸铅的摩尔比优选为0.4~0.6:0.4~0.6,进一步优选为0.45~0.55:0.45~0.55,更优选为0.48~0.52:0.48~0.52。
[0049]
在本发明中,所述取代苯酚和无水乙醇的摩尔体积比优选为0.4~0.6mmol:10~20ml,进一步优选为0.45~0.55mmol:12~18ml,更优选为0.48~0.52mmol:14~16ml。
[0050]
在本发明中,所述步骤(1)中混合的方式优选为搅拌,所述搅拌的转速优选为200~400rpm,进一步优选为250~350rpm,更优选为280~320rpm;所述搅拌的时间优选为1.8~2.2h,进一步优选为1.9~2.1h,更优选为1.95~2.05h。
[0051]
在本发明中,所述步骤(2)中n,n
‑
二(3
‑
氨丙基)
‑2‑
吡啶甲胺和步骤(1)中取代苯酚的摩尔比优选为0.4~0.6:0.4~0.6,进一步优选为0.45~0.55:0.45~0.55,更优选为0.48~0.52:0.48~0.52。
[0052]
在本发明中,所述n,n
‑
二(3
‑
氨丙基)
‑2‑
吡啶甲胺和n,n
‑
二(3
‑
氨丙基)
‑2‑
吡啶甲胺的无水乙醇溶液的摩尔体积比优选为0.4~0.6mmol:8~12ml,进一步优选为0.45~0.55mmol:9~11ml,更优选为0.48~0.52mmol:9.5~10.5ml。
[0053]
在本发明中,所述步骤(2)中混合的方式优选为将n,n
‑
二(3
‑
氨丙基)
‑2‑
吡啶甲胺
的无水乙醇溶液滴加进混合溶液中。
[0054]
在本发明中,所述滴加的速率优选为2~4滴/秒,更优选为3滴/秒。
[0055]
在本发明中,所述步骤(2)中配位缩合反应的时间优选为2.5~3.5h,进一步优选为2.6~3.4h,更优选为2.8~3.2h;所述配位缩合反应优选在搅拌条件下进行,所述搅拌的速率优选为200~400rpm,进一步优选为250~350rpm,更优选为280~320rpm。
[0056]
在本发明中,以r为甲基的情况为例,所述步骤(2)的配位缩合反应如下所示:
[0057][0058]
在本发明中,所述步骤(3)中硫酸镉优选为硫酸镉的八水合物。
[0059]
在本发明中,所述步骤(3)中硫酸镉和步骤(1)中取代苯酚的摩尔比优选为0.15~0.184:0.4~0.6,进一步优选为0.16~0.174:0.45~0.55,更优选为0.165~0.169:0.48~0.52。
[0060]
在本发明中,所述硫酸镉和硫酸镉的无水甲醇溶液的摩尔体积比优选为0.15~0.184mmol:10~20ml,进一步优选为0.16~0.174mmol:12~18ml,更优选为0.165~0.169mmol:14~16ml。
[0061]
在本发明中,所述步骤(3)中混合的方式优选为将硫酸镉的无水甲醇溶液滴加进铅配合物中。
[0062]
在本发明中,所述滴加的速率优选为2~4滴/秒,更优选为3滴/秒。
[0063]
在本发明中,所述置换反应的时间优选为3.5~4.5h,进一步优选为3.6~4.4h,更优选为3.8~4.2h;所述置换反应优选在搅拌条件下进行,所述搅拌的速率优选为200~400rpm,进一步优选为250~350rpm,更优选为280~320rpm。
[0064]
在本发明中,所述置换反应为硫酸镉中的镉离子置换铅配合物中的铅离子,铅离子和硫酸根离子生成硫酸铅沉淀,置换反应搅拌结束后进行砂芯过滤,将过滤得到的硫酸铅进行洗涤,所述洗涤所用试剂优选为无水甲醇,所述无水甲醇和步骤(1)中取代苯酚的体积摩尔比优选为10~20ml:0.4~0.6mmol,进一步优选为12~18ml:0.45~0.55mmol,更优选为14~16ml:0.48~0.52mmol;洗涤结束后合并滤液和洗液,得到混合体系。
[0065]
在本发明中,将混合体系和高氯酸钠的无水甲醇溶液混合后得到沉淀。
[0066]
在本发明中,所述高氯酸钠优选为一水高氯酸钠,所述高氯酸钠和步骤(1)中取代苯酚的质量摩尔比优选为0.15~0.25g:0.4~0.6mmol,进一步优选为0.16~0.24g:0.45~0.55mmol,更优选为0.18~0.22g:0.48~0.52mmol;所述高氯酸钠和高氯酸钠的无水甲醇溶液的质量体积比优选为0.15~0.25g:8~12ml,进一步优选为0.16~0.24g:9~11ml,更优选为0.18~0.22g:9.5~10.5ml。
[0067]
在本发明中,将高氯酸钠的无水甲醇溶液滴加进行混合体系中,所述滴加的速率优选为2~4滴/秒,更优选为3滴/秒。
[0068]
在本发明中,所述高氯酸钠的无水甲醇溶液和混合体系的混合在搅拌条件下进行,所述搅拌的速率优选为200~400rpm,进一步优选为250~350rpm,更优选为280~320rpm;滴加完毕后将溶液使用砂芯进行抽滤获得沉淀。
[0069]
在本发明中,将沉淀溶于乙醇乙腈溶液中静置,所述乙醇乙腈溶液和步骤(1)中取代苯酚的质量摩尔比优选为1.6~2.0g:0.4~0.6mmol,进一步优选为1.7~1.9g:0.45~0.55mmol,更优选为1.75~1.85g:0.48~0.52mmol;所述乙醇乙腈溶液中乙醇和乙腈的体积比优选为1:0.5~1.5,进一步优选为1:0.6~1.4,更优选为1:0.8~1.2;所述静置的时间优选大于等于14天,进一步优选大于等于16天,更优选大于等于18天。
[0070]
在本发明中,静置结束后得到黄色块状单晶即为双核镉配合物。
[0071]
在本发明中,以r为甲基的情况为例,所述步骤(3)中置换反应如下所示:
[0072][0073]
本发明还提供了所述双核镉配合物在制备一氧化氮荧光探针产品中的应用。
[0074]
在本发明中,所述一氧化氮荧光探针产品用于生物医疗、食品检测、环境监测中一氧化氮的检测和监测;可以精确的测定被测对象的一氧化氮含量。
[0075]
下面结合实施例对本发明提供的技术方案进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
[0076]
实施例1
[0077]
将0.5mmol的2,6
‑
二甲酰基
‑4‑
甲基苯酚、0.5mmol的四水合乙酸铅和15ml的无水乙醇混合在300rpm转速下搅拌2h获得混合溶液;将含0.5mmoln,n
‑
二(3
‑
氨丙基)
‑2‑
吡啶甲胺的10ml无水乙醇溶液以3滴/秒的速度滴加进混合溶液中,滴加完毕后溶液由亮黄色浑浊变为黄色透明溶液,在300rpm的转速下搅拌进行配位缩合反应,3h后获得铅配合物;然后将含0.167mmol的3cdso4·
8h2o的15ml无水乙醇溶液以3滴/秒的速度滴加进行铅配合物中,滴加完毕后溶液变成黄色浑浊,在300rpm的转速下搅拌进行置换反应,搅拌4h后使用砂芯过滤;将获得的硫酸铅沉淀使用15ml的无水甲醇洗涤,合并滤液和洗液得到混合体系;控制转速为300rpm,将混合体系和高氯酸钠的无水甲醇溶液混合,混合的方式为滴加,将含0.2g一水高氯酸钠的10ml无水甲醇溶液以3滴/秒的速度滴加进混合体系中,滴加完毕后有大量的黄色沉淀生成,并使用砂芯抽滤获得沉淀;将沉淀溶于1.8g乙醇乙腈溶液中,乙腈和乙醇的体积比为1:1,静置挥发14天后得到0.2g黄色块状单晶,产率为69%,即为双核镉配合物。
[0078]
将本实施例制备得到的双核镉配合物进行红外光谱分析,结果如图1所示。元素分析(%):实测值:c为45.12,h为4.33,n为10.21;计算值(cd2c
42
h
50
n8o
10
cl2):c为44.93,h为4.49,n为9.98。ir(kbr,cm
‑1):3449(ν
o
‑
h
),2923,2851(ν
ch
),1635(ν
c=n
),1093(ν
clo4
‑
),622(δ
clo4
‑
)。
[0079]
将本实施例制备得到的双核镉配合物溶于无水甲醇中,进行电喷雾质谱分析,结果如图2所示,从图中可以看出m/z 1022.92的质谱峰丰度为100%,归属于[cd2l9(clo4)]
+
,为分子离子峰,l9为配合物中除去金属离子后剩余的后配体。m/z 462.17的质谱峰丰度也为100%,对应于[cd2l9]
2+
,表明配合物正确合成。图中其它的碎片峰的丰度均很小,这表明双核镉配合物在甲醇溶液中cd2l9基团能稳定存在。
[0080]
本实施例制备的双核镉配合物的晶体结构数据和结构优化数据如表1所示,部分键长键角数据如表2所示。
[0081]
表1
[0082]
[0083][0084]
表2
[0085]
[0086][0087]
本实施例制备的双核镉配合物的分子结构图如图3所示,从图中可以看出,整个大环配体由于金属的配位作用而变得扭曲,配体上两个苯环平面的夹角为64.8
°
。配合物的十面体结构如图4所示。从图中可以看出,cd1与大环配体上的两个亚胺氮原子n2和n5、一个叔胺氮原子n3、一个功能悬臂吡啶环上氮原子n4、两个酚氧原子o1和o2、高氯酸根上的氧原子o14结合形成配位的十面体结构;cd1与周围配位原子的距离范围为cd2与大环配体上另一边的两个亚胺氮原子n1和n6、叔胺氮原子n7、悬臂吡啶环上氮原子n8、酚氧原子o1和o2结合形成6配位的八面体结构,cd2与周围的配位原子的距离范围为cd1与cd2配位环境的差别在cd2比cd1少一个高氯酸根配位,其原因是与cd1配位的高氯酸根占据了很大的空间,由于空间阻力,其它的高氯酸根无法与cd2靠近成键。
[0088]
本实施例制备的双核镉配合物的分子间氢键图如图5所示,从图中可以看出配合物分子间的氢键将分子与分子牢固地结合在一起,为配合物分子间形成的三维空间结构起到了重要作用。晶体结构中所有的氢键受体都来自于高氯酸根中的氧原子,而所有的氢键给体都为配合物分子上的碳原子。一个分子中与cd原子配位的高氯酸根上的氧原子o13与另一个分子中的碳原子c12上的氢原子h12b形成氢键从而形成一维结构。氢键c12
–
h12b...o13中h...o的距离为氢键键角为117.0
°
。
[0089]
本实施例制备的双核镉配合物的沿a轴氢键图如图6所示,作为分子中起抗衡电荷作用的游离高氯酸根上的三个氧原子和三个配合物分子上的氢原子形成氢键,为c9
–
h9a...o23,c22
–
h22...o21,c29
–
h29a...o22,其中h9a...o23、h22...o21、h29a...o22的距离分别为和氢键键角分别为176.2
°
、172.4
°
和158.6
°
。
[0090]
本实施例制备的双核镉配合物的氢键数据如表3所示。
[0091]
表3
[0092][0093]
将双核镉配合物配制成浓度为5
×
10
‑5m的甲醇溶液,然后通入no,结果如图7所示,图中左边为加入no前的配合物溶液,右边为加入no后的配合物溶液,从图中可以看出,在通入no前后其颜色在可见光下有显著改变,溶液颜色明显变深,说明no能与双核镉配合物发生作用。将上述甲醇溶液进行紫外吸收研究,得到的紫外吸收光谱图如图8所示,图8中曲线a为加入no前的配合物溶液,曲线b为加入no后的配合物溶液,从图8中可以看出,与通入no前相比较,通入no后,双核镉配合物的由配体到金属的荷移跃迁吸收峰由398nm红移到451nm,说明no参与配位使得配体到金属的共轭体系延长,从结构上看,在双核镉配合物中与镉配位的高氯酸根容易离去(这在质谱结论中也得到证明),而使配位能力更强的no与镉原子配位。
[0094]
将上述制备得到的含双核镉配合物的甲醇溶液,在302nm的紫外光下进行照射,得到的发光图如图9所示,图中左边为加入no前的配合物溶液,右边为加入no后的配合物溶液,从图中可以看出,在302nm的紫外光照射下,未加no的配合物溶液发出蓝色荧光,加入no后的配合物溶液发出绿色荧光,且亮度明显增强,说明配合物与no能有效结合。
[0095]
将上述制备得到的含双核镉配合物的甲醇溶液在400nm的可见光激发波下进行照射,得到的发射光谱如图10所示,图中曲线a为未加入no的配合物溶液,曲线b为加入no的配合物溶液,从图中可以看出,在未加入no时,配合物在460nm出现强的发射光谱;在加入no后配合物的发射光谱红移到506nm,且荧光强度增大了3.2倍。可以看出配合物产生荧光的激发能量低,对细胞的伤害小;有良好的荧光性质,对no有很高的灵敏度。这表明配合物可以作为应用在机体内的潜在的no荧光探针试剂产品。
[0096]
利用紫外分光光度法,通过no与配合物之间的结合平衡方程研究了no与配合物的结合常数,no与配合物(方程中用c表示)反应的方程式可以表示为:配合物与no的结合常数为k:[no],[c],[c
‑
no]分别表示no,配合物,c
‑
no的平衡浓度。由物料守恒定律可得:[c]+[c
‑
no]=a(3);[c
‑
no]+[no]=b(4);a和b分别代表配合物和no的分析浓度,(4)中的[c
‑
no]可以忽略,即因为b远远大于a,因此式[c
‑
no]可以忽略,即[no]≈b(5);将式(3)和式(5)代入式(2),得:令a
0,(c)
=ε
c
al,a
0,[no]
=ε
no
bl,则有(8);将公式(8)代入
(6)中,整理得到:
[0097]
公式(8)中a
0,(c)
和a
0,(no)
可以由测定配合物溶液和饱和no溶液求得。以δa/b对δa作图,通过计算曲线斜率可求得结合常数k。图11为双核镉配合物的
△
a/b对
△
a的关系图,由图中斜率求得no和双核镉配合物的结合常数为7.4
×
103mol
·
l
–1。
[0098]
本实施例制备的双核镉配合物在no加入前后明显的荧光颜色和强度的变化、低的激发能量以及对no高的结合能力,使其具有no分子探针的应用价值。
[0099]
实施例2
[0100]
将0.4mmol的2,6
‑
二甲酰基
‑4‑
氯苯酚、0.6mmol的四水合乙酸铅和10ml的无水乙醇混合在200rpm转速下搅拌1.8h获得混合溶液;将含0.4mmoln,n
‑
二(3
‑
氨丙基)
‑2‑
吡啶甲胺的12ml无水乙醇溶液以3滴/秒的速度滴加进混合溶液中,滴加完毕后溶液由亮黄色浑浊变为黄色透明溶液,在200rpm的转速下搅拌进行配位缩合反应,2.5h后获得铅配合物;然后将含0.15mmol的3cdso4·
8h2o的20ml无水乙醇溶液以3滴/秒的速度滴加进行铅配合物中,滴加完毕后溶液变成黄色浑浊,在200rpm的转速下搅拌进行置换反应,搅拌3.5h后使用砂芯过滤;将获得的硫酸铅沉淀使用10ml的无水甲醇洗涤,合并滤液和洗液得到混合体系;控制转速为200rpm,将混合体系和高氯酸钠的无水甲醇溶液混合,混合的方式为滴加,将含0.15g一水高氯酸钠的12ml无水甲醇溶液以3滴/秒的速度滴加进混合体系中,滴加完毕后有大量的黄色沉淀生成,并使用砂芯抽滤获得沉淀;将沉淀溶于1.6g乙醇乙腈溶液中,乙腈和乙醇的体积比为0.5:1,静置挥发16天后得到0.155g黄色块状单晶,产率为67%,即为双核镉配合物。本实施例制备得到的双核镉配合物进行了与实施例1相同的试验,在no加入前后具有明显的荧光颜色和强度的变化、低的激发能量,具有优异的对no高的结合能力。
[0101]
实施例3
[0102]
将0.6mmol的2,6
‑
二甲酰基
‑4‑
氯苯酚、0.4mmol的四水合乙酸铅和20ml的无水乙醇混合在400rpm转速下搅拌2.2h获得混合溶液;将含0.6mmoln,n
‑
二(3
‑
氨丙基)
‑2‑
吡啶甲胺的8ml无水乙醇溶液以3滴/秒的速度滴加进混合溶液中,滴加完毕后溶液由亮黄色浑浊变为黄色透明溶液,在400rpm的转速下搅拌进行配位缩合反应,3.5h后获得铅配合物;然后将含0.184mmol的3cdso4·
8h2o的10ml无水乙醇溶液以3滴/秒的速度滴加进行铅配合物中,滴加完毕后溶液变成黄色浑浊,在400rpm的转速下搅拌进行置换反应,搅拌4.5h后使用砂芯过滤;将获得的硫酸铅沉淀使用20ml的无水甲醇洗涤,合并滤液和洗液得到混合体系;控制转速为400rpm,将混合体系和高氯酸钠的无水甲醇溶液混合,混合的方式为滴加,将含0.25g一水高氯酸钠的8ml无水甲醇溶液以3滴/秒的速度滴加进混合体系中,滴加完毕后有大量的黄色沉淀生成,并使用砂芯抽滤获得沉淀;将沉淀溶于2.0g乙醇乙腈溶液中,乙腈和乙醇的体积比为1.5:1,静置挥发18天后得到0.236g黄色块状单晶,产率为68%,即为双核镉配合物。本实施例制备得到的双核镉配合物进行了与实施例1相同的试验,在no加入前后具有明显的荧光颜色和强度的变化、低的激发能量,具有优异的对no高的结合能力。
[0103]
由以上实施例可知,本发明提供了一种双核镉配合物,经过配位缩合反应和置换反应后即得。本发明提供的配合物中一个镉原子配位环境为七配位的十面体构型,另一个镉原子为六配位的八面体构型,两个镉原子通过配体上的两个酚氧原子桥联起来。由于分子中含有参与配位和游离的高氯酸根离子,使配合物存在分子自组装行为。高氯酸根中的
氧原子容易和大环配体上的氢原子形成氢键,使得配合物的分子间形成一维链状结构和二维网状结构。本发明提供的配合物与no结合效果好,能形成稳定的复合物,检测限低,配合物发射荧光的激发能量低,在加入no前后其荧光发射光谱的波长和强度的变化都非常明显,用作机体内no分子检测探针产品。
[0104]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1