一种放射性同位素氚标记梓醇及其合成方法与流程
1.本发明涉及放射性化学合成领域,尤其涉及一种放射性同位素氚标记梓醇及其合成方法。
背景技术:
2.玄参科植物地黄(rehmannia glutinosa libosch)是我国很重要的一种大宗常用中药材,它能提高免疫功能,具有降血糖、抗肿瘤、抗过敏、保护心血管系统、抗真菌、止血、抗弥漫性血管内凝血和抗炎免疫等作用。
3.梓醇(cas号:2415
‑
24
‑
9;英文名称:catalpol)是源于该植物的环烯醚萜葡萄糖苷类化合物,具有广泛的药理作用,如具有抗癌、神经保护、抗炎、利尿、降血糖、止血、抗肝炎病毒等功能(王朴.生地黄的现代药理研究与临床应用[j].中国中医药现代远程教育.2008,6(8):986
‑
987;park ks.catalpol reduces the production of inflammatory mediators via ppar
‑
γactivation in human intestinal caco
‑
2cells[j].j nat med.2016,70(3):620
‑
626)。在我国含梓醇的植物资源十分丰富,但中药材地黄中梓醇含量最为丰富,这为以梓醇作为活性成分的新药研发和商业化生产奠定了坚实基础。
[0004]
根据我国新药注册登记研究试验的质量要求和国际新药研究的普遍要求,梓醇在注册登记上市前需以放射性同位素碳
‑
14或氚标记梓醇作为示踪剂,借助其直观灵敏、特异性强、不受环境基质干扰等优点,深入地开展药物代谢动力学研究(尤其是组织分布、代谢产物结构鉴定、质量平衡等),进而为梓醇的安全性和有效性的研判提供必要、准确、可靠的试验数据和信息,以使其顺利通过注册登记而上市(国家药品监督管理局药品审评中心.药物非临床药代动力学研究技术指导原则.2014
‑
05
‑
13;国家药品监督管理局药品审评中心.化学药物临床药代动力学研究技术指导原则.2005
‑
03
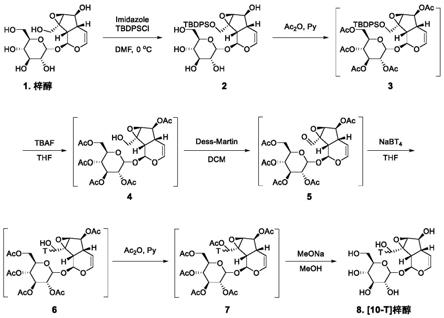
‑
18;roberts d,et al.radiochemistry,a vital role supporting drug development[j].drug discovery world,2004,fall issue:59
‑
64;maxwell b d,et al.radiosynthesis for adme studies[m].adme
‑
enabling technologies in drug design and development.john wiley&sons,inc.2012:461
‑
472)。
[0005]
药代动力学研究中最常用的低能核素是碳
‑
14和氚,但优先考虑采用碳
‑
14标记物[魏敏吉,赵明.创新药物药代动力学研究与评价[m].北京大学医学出版社,2008:354
‑
371;jens atzrodt.john allen.synthesis of radiolabeled compounds for clinical studies[m]//vogel h g,maas j,gebauer a.drug discovery and evaluation:methods in clinical pharmacology.springer berlin heidelberg.2011:105
‑
118]。梓醇系地黄等中药次生代谢过程中产生的环烯醚萜葡萄糖苷类化合物,化学结构复杂。迄今为止,文献中未见该化合物全合成的报道,因而制备梓醇中具有足够稳定性的环烯醚萜骨架碳
‑
14标记物比较困难。
技术实现要素:
[0006]
本发明的一个目的在于提供一种放射性同位素氚标记梓醇及其合成方法,其中所述放射性同位素氚标记梓醇结构骨架的稳定部位,放射性同位素氚标记牢固且不易掉落,完全满足药代动力学研究对标记物的要求。
[0007]
本发明的另一个目的在于提供一种放射性同位素氚标记梓醇及其合成方法,其中所述放射性同位素氚标记梓醇的合成方法中,采用反应叠缩技术,缩减了操作步骤,标记合成过程产生含放射性废物少;标记合成操作简单安全,放射性同位素原料成本低。
[0008]
为实现本发明以上至少一个目的,本发明提供一种放射性同位素氚标记梓醇的合成方法,其中所述方法包括:
[0009]
s1:梓醇与硅保护试剂反应,以获得梓醇10位羟基的硅保护基保护产物(2);
[0010]
s2:梓醇10位羟基的硅保护基保护产物(2)与乙酸酐反应获得乙酰基保护的产物5,2
′
,3
′
,4
′
,6
′‑
五乙酰氧基
‑
[10
‑
(叔丁基二苯基)氧基]梓醇(3);
[0011]
s3:以脱硅基试剂与乙酰基保护的产物5,2
′
,3
′
,4
′
,6
′‑
五乙酰氧基[10
‑
(叔丁基二苯基)氧基]梓醇(3)反应,脱去硅保护基,得到5,2
′
,3
′
,4
′
,6
′‑
五乙酰氧基梓醇(4);
[0012]
s4:以氧化剂氧化5,2
′
,3
′
,4
′
,6
′‑
五乙酰氧基梓醇(4),得到5,2
′
,3
′
,4
′
,6
′‑
五乙酰氧基
‑
10
‑
甲酰基梓醇(5);
[0013]
s5:以硼氚化钠作为还原剂,还原5,2
′
,3
′
,4
′
,6
′‑
五乙酰氧基
‑
10
‑
甲酰基梓醇(5),得到5,2
′
,3
′
,4
′
,6
′‑
五乙酰氧基[10
‑
t]梓醇(6);
[0014]
s6:以乙酸酐与5,2
′
,3
′
,4
′
,6
′‑
五乙酰氧基[10
‑
t]梓醇(6)反应,获得10位羟基的乙酰基保护产物5,10,2
′
,3
′
,4
′
,6
′‑
六乙酰氧基[10
‑
t]梓醇(7);
[0015]
s7:用强碱脱去10位羟基的乙酰基保护产物5,10,2
′
,3
′
,4
′
,6
′‑
六乙酰氧基[10
‑
t]梓醇(7)中全部乙酰基,得到放射性同位素氚标记梓醇(8,简称为[10
‑
t]梓醇)。
[0016]
根据本发明一实施例,在所述步骤s1中,所述硅保护基选自叔丁基二苯基氯硅烷、三甲基氯硅烷、三乙基氯硅烷、叔丁基二甲基氯硅烷中的至少一种。
[0017]
根据本发明一实施例,在所述步骤s3中,所述脱硅基试剂为四丁基氟化铵或氢氟酸。
[0018]
根据本发明一实施例,氧化剂选自戴斯马丁氧化剂、琼斯试剂、沙瑞特试剂、柯斯林试剂、pdc、ibx氧化剂中至少一种。
[0019]
根据本发明一实施例,在s4中,在s7中,强碱选自氢氧化钠、氢氧化钾中至少一种。
[0020]
根据本发明一实施例,在所述步骤s1中,在氩气保护和冰
‑
水浴冷却下,梓醇与硅保护试剂反应。
[0021]
根据本发明一实施例,在所述步骤s1中,在氩气保护和室温下,以脱硅基试剂与乙酰基保护的产物5,2
′
,3
′
,4
′
,6
′‑
五乙酰氧基[10
‑
(叔丁基二苯基)氧基]梓醇反应(3)。
[0022]
根据本发明一实施例,在冰
‑
盐浴冷却下,还原5,2
′
,3
′
,4
′
,6
′‑
五乙酰氧基
‑
10
‑
甲酰基梓醇(5)。
[0023]
根据本发明的另一个方面,本发明提供一种放射性同位素氚标记梓醇(8),放射性同位素氚标记在梓醇分子10位碳原子上,其结构式如下:
[0024][0025]
根据本发明的另一个方面,本发明提供一种放射性同位素氚标记梓醇的合成方法合成的中间体或含氚合成砌块。
[0026]
本发明的这些和其它目的、特点和优势,通过下述的详细说明,得以充分体现。
附图说明
[0027]
图1为本发明第一个实施例中,[10
‑
t]梓醇(8)的合成路线示意图;
[0028]
图2为本发明第一个实施例中,原料梓醇的核磁共振氢谱图;
[0029]
图3为本发明第一个实施例中,中间体(2)的核磁共振氢谱图;
[0030]
图4为本发明第一个实施例中,中间体(2)的核磁共振1h
‑1h cosy二维谱图;
[0031]
图5为本发明第一个实施例中,[10
‑
t]梓醇(8)的在线放射性高效液相色谱图;
具体实施方式
[0032]
以下描述中的优选实施例只作为举例,本领域技术人员可以想到其他显而易见的变型。在以下描述中界定的本发明的基本原理可以应用于其他实施方案、变形方案、改进方案、等同方案以及没有背离本发明的精神和范围的其他技术方案。
[0033]
参考图1至图5,依本发明一较佳实施例的放射性同位素氚标记梓醇的合成方法将在以下被详细地阐述。
[0034]
具体地,所述放射性同位素氚标记梓醇的合成方法,包括:
[0035]
s1:梓醇与硅保护试剂反应,以获得梓醇10位羟基的硅保护基保护产物(2);
[0036]
优选地,在所述步骤s1中,所述硅保护基选自叔丁基二苯基氯硅烷、三甲基氯硅烷、三乙基氯硅烷、叔丁基二甲基氯硅烷中的至少一种。
[0037]
值得一提的是,在所述步骤s1中,在防潮气体,如氩气保护和冰
‑
水浴冷却下,梓醇与硅保护试剂反应。
[0038]
所述放射性同位素氚标记梓醇(8)的合成方法,还包括:
[0039]
s2:梓醇10位羟基的硅保护基保护产物(2)与乙酸酐反应获得乙酰基保护的产物5,2
′
,3
′
,4
′
,6
′‑
五乙酰氧基
‑
[10
‑
(叔丁基二苯基)氧基]梓醇(3);
[0040]
s3:以脱硅基试剂与乙酰基保护的产物5,2
′
,3
′
,4
′
,6
′‑
五乙酰氧基[10
‑
(叔丁基二苯基)氧基]梓醇(3)反应,脱去硅保护基,得到5,2
′
,3
′
,4
′
,6
′‑
五乙酰氧基梓醇(4)。
[0041]
优选地,在所述步骤s3中,所述脱硅基试剂为四丁基氟化铵或氢氟酸。
[0042]
s4:以氧化剂氧化5,2
′
,3
′
,4
′
,6
′‑
五乙酰氧基梓醇(4),得到5,2
′
,3
′
,4
′
,6
′‑
五乙酰氧基
‑
10
‑
甲酰基梓醇(5)。
[0043]
值得一提的是,所述氧化剂选自戴斯马丁氧化剂、琼斯试剂、沙瑞特试剂、柯斯林试剂、pdc、ibx氧化剂中至少一种。
[0044]
所述放射性同位素氚标记梓醇(8)的合成方法,还包括:
[0045]
s5:以硼氚化钠作为还原剂,还原5,2
′
,3
′
,4
′
,6
′‑
五乙酰氧基
‑
10
‑
甲酰基梓醇(5),得到5,2
′
,3
′
,4
′
,6
′‑
五乙酰氧基[10
‑
t]梓醇(6)。
[0046]
值得一提的是,标记合成所用的起始原料为硼氚化钠,原料性质稳定,便于储存和运输。
[0047]
值得一提的是,在冰
‑
盐浴冷却下,还原5,2
′
,3
′
,4
′
,6
′‑
五乙酰氧基
‑
10
‑
甲酰基梓醇(5)。
[0048]
s6:以乙酸酐与5,2
′
,3
′
,4
′
,6
′‑
五乙酰氧基[10
‑
t]梓醇(6)反应,获得10位羟基的乙酰基保护产物5,10,2
′
,3
′
,4
′
,6
′‑
六乙酰氧基[10
‑
t]梓醇(7)。
[0049]
s7:用强碱脱去10位羟基的乙酰基保护产物5,10,2
′
,3
′
,4
′
,6
′‑
六乙酰氧基[10
‑
t]梓醇(7)中全部乙酰基,得到本发明目标物[10
‑
t]梓醇(8)。
[0050]
优选地,在s7中,强碱选自氢氧化钠、氢氧化钾中至少一种。
[0051]
可以理解的是,本发明实施例中,梓醇分子中环烯醚萜骨架上10位碳骨架具有一定的化学稳定性和代谢稳定性,且选择10位碳原子作为梓醇中的标记位点时,是先将10位羟甲基氧化为甲酰基,然后再通过氚化或氘化还原剂在还原甲酰基为羟甲基过程中,在10位碳上引入氚或氘,进而达到利用放射性同位素氚或氘定位标记梓醇的目的。这样一来,制作形成的所述放射性同位素氚标记梓醇化学稳定性和代谢稳定性都比较好。
[0052]
具体在本发明的第一个实施例中,通过本发明方法制备的氚标记梓醇(8,简称为[10
‑
t]梓醇)结构式如下:
[0053][0054]
参考图1至图5,具体地,氚标记的梓醇的合成方法包括以下步骤:
[0055]
s1a:在氩气保护和冰
‑
水浴冷却下,将叔丁基二苯基氯硅烷(缩写为tbdpscl,1.2mmol)的无水n,n
‑
二甲基甲酰胺(缩写为dmf,2ml)溶液缓慢加入梓醇(1,1.0mmol)和咪唑(2.5mmol)的无水dmf(10ml)溶液中。撤去冰浴,搅拌15h。
[0056]
反应完毕后,向反应体系中加水,经乙酸乙酯萃取、无水硫酸钠干燥、减压浓缩和制备型hplc纯化得目标物纯品(化学纯度>98%)。色谱条件:xbridge prep c18柱(5μm,250mm
×
19mm;waters co.,ma,usa),流速15.00ml/min,波长254nm,进样量400ul;梯度洗脱(min/%a)控制:1/60,45/100,49/100,50/60,60/60;a为甲醇,其余b为水。
[0057]
收集保留时间为14.59~15.64min组分。将收集液浓缩至干得白色固体(2,0.3mmol,180mg,30%)。熔点:110.5~113.9℃。1h nmr(400mhz,cd3od)δ7.72
‑
7.69(m,4h),7.44
‑
7.37(m,6h),6.32(dd,j=5.9,1.5hz,1h),5.07(d,j=6.8hz,1h),5.05(d,j=6.8hz,1h),4.70(d,j=7.6hz,1h),4.25(d,j=12.4hz,1h),4.17(d,j=12.4hz,1h),3.90(d,j=8.4hz,1h),3.87(dd,j=12.4,2.0hz,1h),3.56(s,1h),3.42(dd,j=11.6,7.6hz,1h),3.36
‑
3.31(m,2h),3.26(td,j=9.6,1.6hz,1h),3.07(dd,j=9.6,4.8hz,2h),2.48(t,j=9.2hz,1h),2.32(tdd,j=7.6,4.0,1.6hz,1h),1.04(s,9h).esi
‑
ms(+)m/z:623[m+na]
+
。
[0058]
s2a:在氩气保护和室温下,将醋酸酐(3.0mmol)滴入中间体(2,0.3mmol)的无水吡
啶(缩写为py,3ml)溶液中。室温搅拌10h。反应完毕后,加水淬灭,经乙酸乙酯萃取、无水硫酸钠干燥、减压浓缩得白色固体(3,0.3mmol)。esi
‑
ms(+)m/z:833[m+na]
+
。无需纯化而直接进行下一步反应。
[0059]
s3a:在氩气保护和冰
‑
水浴冷却下,将四丁基氟化铵(0.45mmol)的四氢呋喃(缩写为thf,0.45ml)溶液滴入中间体(3,0.3mmol)的无水thf(3ml)溶液中。维持此温度继续搅拌2h。反应完毕后,加水淬灭,经乙酸乙酯萃取、无水硫酸钠干燥、减压浓缩得白色固体(4,0.3mmol)。esi
‑
ms(+)m/z:595[m+na]
+
。无需纯化而直接进行下一步反应。
[0060]
s4a:在室温下,将戴斯马丁氧化剂(0.45mmol)加入中间体(4,0.3mmol)的二氯甲烷(缩写为dcm,3ml)溶液中,搅拌2h。反应完毕后,抽滤,浓缩滤液后得白色固体(5,0.16mmol)。esi
‑
ms(+)m/z:593[m+na]
+
。无需纯化而直接进行下一步反应。
[0061]
s5a:在冰
‑
盐浴冷却下,将硼氚化钠(0.15mmol,500mci/mmol,75mci)加入中间体(5,0.15mmol)的thf(1.5ml)溶液中,搅拌2h,此间反应体系自然升至室温。反应完毕后,加水淬灭反应,经乙酸乙酯萃取、无水硫酸钠干燥和减压浓缩得白色固体(6,0.15mmol),esi
‑
ms(+)m/z:595[m+na]
+
。无需纯化而直接进行下一步反应。
[0062]
s6a:在氩气保护和室温下,将醋酸酐(1.5mmol)滴入中间体(6,0.15mmol)的无水吡啶(1.5ml)溶液中,室温搅拌8h。反应完毕后,加水淬灭,经乙酸乙酯萃取、无水硫酸钠干燥和减压浓缩后得白色固体(7,0.15mmol)。esi
‑
ms(+)m/z:637[m+na]
+
。无需纯化而直接进行下一步反应。
[0063]
s7a:在氩气保护和室温下,将甲醇钠(0.45mmol)加入中间体(7,0.15mmol)的无水甲醇(1.5ml)溶液中,室温搅拌5h。反应完毕后,经过滤、减压浓缩和hplc制备纯化得白色固体目标物(8,35mg,0.096mmol,10mci,32%)。esi
‑
ms(+)m/z:385[m+na]
+
。
[0064]
经分析可知目标物(8)技术参数:比活度:120mci/mmol,化学纯度和放化纯度均>98%。
[0065]
hplc制备条件:xbridge prep c18柱(5μm,250
×
19mm;waters co.,ma,usa),流速10.00ml/min,波长210nm,进样量200ul;梯度洗脱(min/%a)控制:1/5,20/100,22/100,28/5,30/5;a为甲醇,其余b为水。收集保留时间为7.17~8.24min组分。
[0066]
在线放射性高效液相色谱条件:sugar ks
‑
804凝胶柱(6μm,8mm
×
300mm);流动相为纯水,流速为1.00ml/min;进样量5ul;流动液体闪烁测量仪用c型闪烁液,流速6.00ml/min;r
t
=12.117min。
[0067]
值得一提的是,在本实施例中,梓醇分子中环烯醚萜骨架上的10位碳原子具有一定的化学稳定性和代谢稳定性,因而可将其氧化为甲酰基,通过氚化还原剂在还原甲酰基为羟甲基过程中在10位碳上引入氚,进而达到利用放射性同位素氚定位标记梓醇的目的。
[0068]
具体在本发明的第二个实施例中,通过本发明方法制备的氘标记梓醇([10
‑
d]梓醇)的结构式如下:
[0069][0070]
具体地,其合成方法包括以下步骤:
[0071]
s1b:在氩气保护和冰
‑
水浴冷却下,将叔丁基二苯基氯硅烷(缩写为tbdpscl,1.2mmol)的无水n,n
‑
二甲基甲酰胺(缩写为dmf,2ml)溶液缓慢加入梓醇(1,1.0mmol)和咪唑(2.5mmol)的无水dmf(10ml)溶液中。撤去冰浴,搅拌15h。
[0072]
反应完毕后,向反应体系中加水,经乙酸乙酯萃取、无水硫酸钠干燥、减压浓缩和制备型hplc纯化得目标物纯品(化学纯度>98%)。色谱条件:xbridge prep c18柱(5μm,250mm
×
19mm;waters co.,ma,usa),流速15.00ml/min,波长254nm,进样量400ul;梯度洗脱(min/%a)控制:1/60,45/100,49/100,50/60,60/60;a为甲醇,其余b为水。
[0073]
收集保留时间为14.59~15.64min组分。将收集液浓缩至干得白色固体(2,0.3mmol,180mg,30%)。熔点:110.5~113.9℃。1h nmr(400mhz,cd3od)δ7.72
‑
7.69(m,4h),7.44
‑
7.37(m,6h),6.32(dd,j=5.9,1.5hz,1h),5.07(d,j=6.8hz,1h),5.05(d,j=6.8hz,1h),4.70(d,j=7.6hz,1h),4.25(d,j=12.4hz,1h),4.17(d,j=12.4hz,1h),3.90(d,j=8.4hz,1h),3.87(dd,j=12.4,2.0hz,1h),3.56(s,1h),3.42(dd,j=11.6,7.6hz,1h),3.36
‑
3.31(m,2h),3.26(td,j=9.6,1.6hz,1h),3.07(dd,j=9.6,4.8hz,2h),2.48(t,j=9.2hz,1h),2.32(tdd,j=7.6,4.0,1.6hz,1h),1.04(s,9h).esi
‑
ms(+)m/z:623[m+na]
+
。
[0074]
s2b:在氩气保护和室温下,将醋酸酐(3.0mmol)滴入中间体(2,0.3mmol)的无水吡啶(缩写为py,3ml)溶液中。室温搅拌10h。反应完毕后,加水淬灭,经乙酸乙酯萃取、无水硫酸钠干燥、减压浓缩得白色固体(3,0.3mmol)。esi
‑
ms(+)m/z:833[m+na]
+
。无需纯化而直接进行下一步反应。
[0075]
s3b:在氩气保护和冰
‑
水浴冷却下,将四丁基氟化铵(0.45mmol)的四氢呋喃(缩写为thf,0.45ml)溶液滴入中间体(3,0.3mmol)的无水thf(3ml)溶液中。维持此温度继续搅拌2h。反应完毕后,加水淬灭,经乙酸乙酯萃取、无水硫酸钠干燥、减压浓缩得白色固体(4,0.3mmol)。esi
‑
ms(+)m/z:595[m+na]
+
。无需纯化而直接进行下一步反应。
[0076]
s4b:在室温下,将戴斯马丁氧化剂(0.45mmol)加入中间体(4,0.3mmol)的二氯甲烷(缩写为dcm,3ml)溶液中,搅拌2h。反应完毕后,抽滤,浓缩滤液后得白色固体(5,0.16mmol)。esi
‑
ms(+)m/z:593[m+na]
+
。无需纯化而直接进行下一步反应。
[0077]
s5b:在冰
‑
盐浴冷却下,将硼氘化钠(0.15mmol)加入中间体(5,0.15mmol)的thf(1.5ml)溶液中,搅拌2h,此间反应体系自然升至室温。反应完毕后,加水淬灭,经乙酸乙酯萃取、无水硫酸钠干燥和减压浓缩得白色固体(9,0.15mmol),esi
‑
ms(+)m/z:596[m+na]
+
。无需纯化而直接进行下一步反应。
[0078]
s6b:在氩气保护和室温下,将醋酸酐(1.5mmol)滴入中间体(9,0.15mmol)的无水吡啶(1.5ml)溶液中,室温搅拌8h。反应完毕后,加水淬灭,经乙酸乙酯萃取、无水硫酸钠干燥和减压浓缩后得白色固体(10,0.15mmol)。esi
‑
ms(+)m/z:638[m+na]
+
。无需纯化而直接进行下一步反应。
[0079]
s7b:在氩气保护和室温下,将甲醇钠(0.45mmol)加入中间体(10,0.15mmol)的无水甲醇(1.5ml)溶液中,室温搅拌5h。反应完毕后,经过滤、减压浓缩和hplc制备纯化得白色固体(11,37mg,34%)。1h nmr(400mhz,cd3od)δ6.34(dd,j=6.0,1.2hz,1h),5.07(t,j=5.6hz,1h),5.04(d,j=10.0hz,1h),4.77(d,j=8.0hz,1h),4.13(d,j=15.6hz,0.5h),3.92(d,j=3.2hz,1h),3.89(s,1h),3.78(d,j=15.6hz,0.5h),3.63(dd,j=12.0,6.4hz,1h),3.43(s,1h),3.39(d,j=9.2hz,1h),3.31
‑
3.23(m,4h),2.53(dd,j=9.2,8.4hz,1h),
2.29
–
2.24(m,1h).esi
‑
ms(+)m/z:386[m+na]
+
。与文献中梓醇分析数据相比较,所得产物(11)的核磁共振氢谱在化学位移3.79和4.15两处峰面积积分之和减半,表明产物(11)中10位仅含1个氢原子,即产物(11)为[10
‑
d]梓醇(tran thi phuong thao,thanh q.bui,phan tu quy,et al.isolation,semi
‑
synthesis,docking
‑
based prediction,and bioassay
‑
based activity of dolichandrone spathacea iridoids:new catalpol derivatives as glucosidase inhibitors[j].rsc advances,2021,11(20):11959
‑
11975)。
[0080]
值得一提的是,通过氘标记的梓醇可以验证通过第一个实施例的方式能够合成所述氚标记梓醇(8)。
[0081]
本领域的技术人员应理解,上述描述所示的本发明的实施例只作为举例而并不限制本发明。本发明的目的已经完整并有效地实现。本发明的功能及结构原理已在实施例中展示和说明,在没有背离所述原理下,本发明的实施方式可以有任何变形或修改。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1