一种修饰的核苷酸,组合物及试剂的制作方法
一种修饰的核苷酸,组合物及试剂
发明领域
1.本发明涉及生物检测领域,具体涉及一种修饰的核苷酸,组合物及试剂。
2.发明背景
3.单核苷酸多态性(snp)是指在基因组水平上由单个核苷酸的变异所引起的dna序列多态性,在人群中发生的频率高于1%,是人类可遗传的变异中最常见的一种,因此,snp的检测对遗传病的诊断、筛查以及用药等方面有及其重要的指导作用。
4.目前,snp检测的方法学主要包含实时荧光pcr法、pcr基因芯片法、pcr电泳法、pcr毛细电泳法分析法、pcr高分辨溶解曲线法、流式荧光杂交法、飞行时间质谱法,焦磷酸测序法、sanger测序法等。其中,实时荧光pcr法、pcr电泳法、pcr毛细电泳法分析法、pcr高分辨溶解曲线法和流式荧光杂交法的优点在于耗时较短、灵敏度较高,能够实现某些场景下的检测,但因其通量有限,所以无法方便快捷地满足临床对于数十个甚至数百个基因位点的检测需求。基因芯片法、焦磷酸测序法和sanger测序法,虽然检测较为准确,但检测成本较高,耗时较长,并不是snp检测的首要选择。而飞行时间质谱法,检测速度快,数据分析简单,通量较高,即弥补了传统方法学的不足,又降低了成本,相比前面几种方法是一种更好的选择,但却存在检测结果不够准确的问题,使其应用的进一步发展受到了限制。
5.核酸质谱snp检测主要基于pcr和引物延伸技术,其原理是首先通过pcr引物对待检测snp位点的目标片段进行扩增,产生的pcr产物经过虾碱性磷酸酶(shrimp alkaline phosphatase,sap)处理中和残留dntps。sap消化反应结束后,向反应液中加入缓冲液,延伸引物,dideoxynucleotide(ddntps)以及单碱基延伸酶等组分进行引物延伸反应。以dna扩增产物为模板,延伸引物可与待检测snp位点的5’端结合,延伸一个碱基。引物延伸完成后,向反应液中加入阳性数值进行脱盐处理,去除吸附在核酸片段上的金属离子。脱盐完成后,将样品与基质转移到靶板形成共结晶,通过质谱检测获得谱图。通过计算谱图中产物的分子量与延伸引物的分子量的差值可分析该样品snp位点的分型。
6.然而,以ddntp作为底物时,单碱基延伸酶的结合效率低,延伸效果差,影响谱图结果判断的准确性。另外一种可用于单碱基延伸的核苷酸底物为线性核苷酸(acyclonucleotides,acyntp),将dntp中常见的2
′‑
deoxyribofuranosyl sugar替换为2-hydroxyethoxymethyl group(如下所示)。dna聚合酶对于acyntp的识别效率为普通ddntp的30倍(参考文献:gardner,a.,and jack,w.(2002).acyclic and dideoxy terminator preferences denote divergent sugar recognition by archaeon and taq dna polymerases.nucleic acids res.30,605
–
613.doi:10.1093/nar/30.2.605)。
7.
技术实现要素:
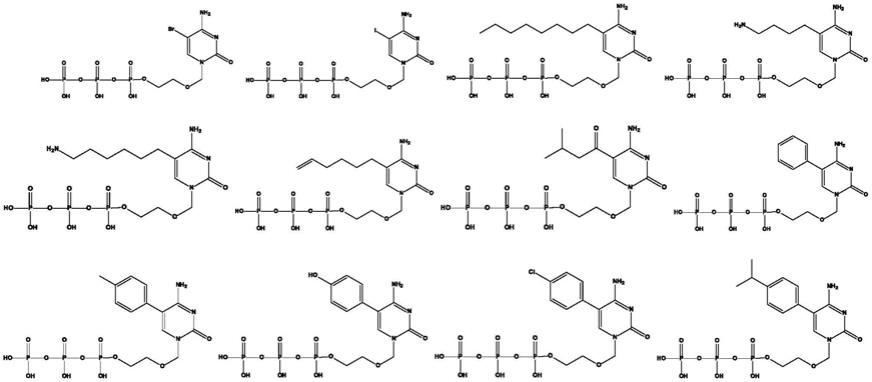
8.本发明为了解决核酸质谱snp检测中因为延伸效果差,带来的检测结果不准确的问题,利用分子量差值可分析snp位点分型的原理,公开了一种修饰的核苷酸,所述核苷酸的结构如下:
[0009][0010]
其中,r选自烷基;环烷基;-or1;-sr1;-so3h;-nr1r2和卤素;任选取代的芳基或杂环基;n为1-12整数;
[0011]
a选自ch2或o;
[0012]
所述核苷酸的分子量为474-924da。
[0013]
优选的,核苷酸的分子量为481-924da。
[0014]
现有技术中acyntp作为底物主要应用于测序,在核酸质谱snp检测中应用较少。acyntp作为底物时,acyctp、acyatp、acygtp和acyttp分子量分别为425.12da、449.12da、465.14da、440.10da。
[0015]
可以看出,acyatp与acyttp的分子量相隔较近,核苷酸之间分子量的差异决定了核酸质谱snp检测项目需要区分9da差异的峰信号,然而质谱仪很难有效区分9da差异的特征峰,尤其在大分子量检测区间如7000da-12000 da,从而导致部分snp类型无法精准区分,只有分辨率特别好的质谱仪,能够实现9da的分辨,而普通质谱仪,通常需要分子量相隔9da以上的峰信号,才能分隔开,相隔较近的特征峰会产生部分重叠,导致质谱仪器的分辨困难。而当分子量相差16da时,即使在普通的质谱仪上,峰信号也分隔得足够的远,能够轻易且准确的进行区分。
[0016]
所以本发明使用acyutp代替常规的acyttp,拉开acyatp与acyttp的分子量差距,acyutp的性能与acyttp接近,均能够与a进行碱基互补配对。
[0017]
acyctp、acyatp、acygtp和acyutp分子量分别为425.12da、449.12da、465.14da、426.10da。
[0018]
acyctp与acyutp的特征峰几乎重叠,无法区分,实际操作中可以选择改变其中一个的分子量,达到区分特征峰的目的。
[0019]
本发明选择在acyctp上进行修饰以改变其分子量,选择分子量474da以上,优选481da以上的核苷酸,保证了单碱基延伸酶对核苷酸底物高效识别的同时,使得各个核苷酸之间分子量可以区分,提高谱图分辨率与结果判读准确率。
[0020]
优选的,所述r选自c
1-c
10
烷基,进一步优选c
4-c
10
烷基;
[0021]
优选的,所述烷基上可以被取代,优选的取代基为-nh2。
[0022]
优选的,所述r1,r2独立选自h或c
1-c
20
烷基,优选c
1-c
10
烷基,进一步优选c
3-c
10
烷基;作为优选,卤素选自:f、cl、br或i,更优选cl、br或i;
[0023]
优选的,所述杂环基选自饱和杂环基,优选5到8元饱和杂环基;例如:四氢呋喃基、吗啉基、哌啶基、哌嗪基;或者所述杂环选自杂芳基,优选5到10元杂芳基,例如呋喃基、吡咯基、噻吩基、吡唑基、咪唑基、噁唑基、噻唑基、吡啶基、吡喃基、哒嗪基、嘧啶基、吡嗪基。所述杂环基独立任选被一个或多个c
1-c
10
烷基;卤素、-or1或-nr1r2取代。
[0024]
优选的,本发明式i化合物的举例性的、非限制性的具体实例如下所示:
[0025]
[0026]
[0027][0028]
另一方面,本发明公开了一种底物混合物,所述底物混合物包括式1。
[0029]
优选的,所述底物混合物还包括acyatp、acygtp和acy utp,结构式依次如下:
[0030][0031]
另一方面,本发明公开了一种用于引物延伸的试剂,包含上述底物混合物。
[0032]
另一方面,本发明公开了一种用于核酸质谱检测的试剂盒,包含上述用于引物延伸的试剂。
[0033]
有益效果:
[0034]
1、本发明公开的修饰的核苷酸,只存在一个位置的取代,方便合成。
[0035]
2、本发明创造性的将修饰的acyctp应用到质谱snp的检测中,保证单碱基延伸酶延伸效率的同时,使得产物分子量区分开,提高质谱分辨率,保证结果判读的准确性。
附图说明:
[0036]
图1:acy磺基-ctp作为底物检测突变位点281c>t的结果图;
[0037]
图2:acy磺基-ctp作为底物临床检测质谱图;
[0038]
图3:acy丁醚-ctp作为底物检测突变位点ivs-i-5(g》c)的结果图;
[0039]
图4:6种核苷酸的检测效果图。
具体实施方式:
[0040]
制备例核苷酸的修饰合成
[0041]
所有添加物质的用量相对于化合物i、ii、iii是均为过量的,不同物质合成的差别在于,得到的产物量不同。
[0042]
a:当a为o,n=1时
[0043][0044]
(1)化合物ii的合成
[0045]
称取30mmol化合物1,加入到洁净的250ml三口烧瓶中;加入150ml二氯甲烷后氮气置换三次;氮气保护;体系为非均相;称取阿昔洛韦侧链50mmol,一次性加入到上述三口烧瓶中;体系为非均相;量取7.5ml n,o-双三甲基硅基乙酰胺,一次性加入到上述三口烧瓶中;上述反应混合物室温搅拌过夜;体系仍然为非均相;量取4ml n,o-双三甲基硅基乙酰胺,一次性加入到上述三口烧瓶中;上述反应混合物再次在室温搅拌3小时后,体系变澄清;用冰水浴将反应体系冷却至0℃;量取1.2ml无水四氯化锡,一次性加入到上述三口烧瓶中;不撤冷浴,上述反应混合物自然升温至室温,然后在室温搅拌过夜;将上述反应混合物小心倒入250ml饱和碳酸氢钠水溶液中,用250ml二氯甲烷萃取3次,合并有机相后用无水硫酸钠干燥旋干;将残余的浅磺色固体用200ml甲基叔丁基醚打浆,得到白色固体;
[0046]
(2)化合物iii的合成
[0047]
将(1)中合成的化合物ii,加入到洁净的250ml单口瓶,用100ml甲醇溶解;氮气置换三次后氮气保护;称量纸称取5.2mmol叔丁醇钠,一次性加入到上述单口烧瓶中;将上述反应混合物室温搅拌过夜后将反应混合物直接旋干,所得残余物分散在50ml乙酸乙酯中,用1n盐酸酸化后,用50ml乙酸乙酯萃取4次,合并有机相后用无水硫酸钠干燥旋干得到白色固体。
[0048]
(3)化合物iv的合成
[0049]
称取0.2mmol化合物iii,加入到洁净的50ml茄型瓶中;加入2ml无水乙腈后氮气置换三次,氮气保护;称取0.65mmol干燥过的吡啶,一次性加入到上述茄型瓶中;用冰水浴将反应体系冷却至0℃;称取0.3mmol三氯氧磷,滴加到上述茄型瓶中;不撤冷浴,上述反应混合物在0℃搅拌30分钟后作为溶液a备用;称取0.55mmol焦磷酸三正丁胺,加入到另一洁净的50ml茄型瓶中;加入2ml无水乙腈后氮气置换三次,氮气保护;量取1.5ml干燥过的三正丁胺,一次性加入到上述茄型瓶中;用冰水浴将反应体系冷却至0℃;将溶液a滴加到上述反应
体系中;不撤冷浴,上述反应混合物在0℃搅拌30分钟;量取4ml去离子水,一次性加入到上述反应体系中,室温搅拌2小时;0℃浓缩掉有机溶剂后,残余水溶液用高压制备分离纯化系统(pre-hplc)分离纯化;所得洗脱液冻干后得到白色固体,复溶至1ml冷的去离子水中,-20℃冻存。
[0050]
其中,x的不同取代,化合物iv可以列举下述结构进行验证。
[0051]
当n=1,分子量如表1所示。
[0052]
表1化合物及质谱结构确认
[0053][0054]
b:当a为o,n大于1时
[0055]
需要在上述a的合成步骤中(a:当a为o,n=1时)加入长链合成的步骤,即在化合物3之后,首先合成长链,再修饰上三磷酸。
[0056]
所以,化合物i到化合物iii与上述步骤相同,当x取代为其他基团时,只需要替换化合物1,不需要更改合成的过程。
[0057]
化合物iii到化合物v,根据n数量的不同,添加不同量的环氧乙烷,控制合成的主要化合物,再通过pre-hplc纯化出所需化合物。化合物vi的合成过程与上述a的合成步骤中化合物iv的合成相同,区别在于,化合物iv是取合成的化合物iii进行合成,化合物vi是取合成的化合物v进行合成,其余过程均相同。
[0058]
本实施例以n=5为例。
[0059]
当n=5,除了化合物i、ii、iii、v的用量以外,所有添加物质的用量均为过量的,不同物质合成的差别在于,得到的产物量不同。
[0060][0061]
化合物v的合成
[0062]
称取10mmol化合物iii,加入到洁净的100ml水热合成反应釜中,用35ml n,n-二甲基甲酰胺溶解;加入60mmol环氧乙烷后密封;室温搅拌72小时后将反应混合物直接旋干,所得残余物柱层析纯化,然后再用高压制备分离纯化系统(pre-hplc)分离纯化;所得洗脱液冻干后得到白色固体。
[0063]
根据上述方法,变换不同的x取代基,制备得到一系列化合物vi,结构如表2所示;
[0064]
表2化合物及质谱结构确认
[0065]
[0066][0067]
c:当a为ch2,n=1时
[0068]
除了化合物i、vi、vii的用量以外,所有添加物质的用量均为过量的,不同物质合成的差别在于,得到的产物量不同。
[0069][0070]
(1)化合物vii的合成
[0071]
称取30mmol化合物i,加入到洁净的250ml三口烧瓶中;加入150ml n,n-二甲基甲酰胺后氮气置换三次;氮气保护;冰水浴冷却至0℃,称取65mmol钠氢(60%in oil)分批次加入到反应体系中;加完后升温至70℃搅拌2小时;然后再用冰水浴冷却至0℃;称取35mmol 4-溴丁基乙酸酯,滴加到上述反应体系中;加完后室温不撤冷浴,自然升温至室温,室温搅拌48小时;过滤,将滤液40℃水浴高真空旋干后柱层析(二氯甲烷:甲醇=9:1),得到白色固体;
[0072]
(2)化合物viii的合成
[0073]
将步骤(1)中的化合物vii加入到洁净的100ml单口瓶,用50ml甲醇溶解;氮气置换三次后氮气保护;称量纸称取1.5mmol叔丁醇钠,一次性加入到上述单口烧瓶中;将上述反
应混合物室温搅拌过夜后将反应混合物直接旋干,所得残余物分散在50ml乙酸乙酯中,用1n盐酸酸化后,用50ml乙酸乙酯萃取4次,合并有机相后用无水硫酸钠干燥旋干得到白色固体。
[0074]
(3)化合物ix的合成
[0075]
称取0.25mmol化合物viii,加入到洁净的50ml茄型瓶中;加入2ml无水乙腈后氮气置换三次,氮气保护;称取0.65mmol干燥过的吡啶,一次性加入到上述茄型瓶中;用冰水浴将反应体系冷却至0℃;称取0.3mmol三氯氧磷,滴加到上述茄型瓶中;不撤冷浴,上述反应混合物在0℃搅拌30分钟后作为溶液a备用;称取0.55mmol焦磷酸三正丁胺,加入到另一洁净的50ml茄型瓶中;加入2ml无水乙腈后氮气置换三次,氮气保护;量取1.5ml干燥过的三正丁胺,一次性加入到上述茄型瓶中;用冰水浴将反应体系冷却至0℃;将溶液a滴加到上述反应体系中;不撤冷浴,上述反应混合物在0℃搅拌30分钟;量取4ml去离子水,一次性加入到上述反应体系中,室温搅拌2小时;0℃浓缩掉有机溶剂后,残余水溶液用高压制备分离纯化系统(pre-hplc)分离纯化;所得洗脱液冻干后得到24.4mg白色固体,复溶至1ml冷的去离子水中,-20℃冻存。
[0076]
根据上述方法,变换不同的x取代基,制备得到一系列化合物ix,结构如表3所示;
[0077]
表3化合物及质谱结构确认
[0078]
[0079][0080]
d:当a为ch2,n大于1时
[0081][0082]
与当a为o的情况不同,当a为ch2时,需要先合成长链,再进行三磷酸修饰。
[0083]
本实施例以n=5,除了化合物i、x、xi的用量以外,所有添加物质的用量均为过量的,不同物质合成的差别在于,得到的产物量不同。
[0084]
具体步骤如下:
[0085]
(1)化合物x的合成
[0086]
称取30mmo化合物i,加入到洁净的250ml三口烧瓶中;加入150ml n,n-二甲基甲酰胺后氮气置换三次;氮气保护;冰水浴冷却至0℃,称取65mmol钠氢钠氢(60%in oil)分批次加入到反应体系中;加完后升温至70℃搅拌2小时;然后再用冰水浴冷却至0℃;称取25mmol 16-溴十六烷基乙酸酯,滴加到上述反应体系中;加完后室温不撤冷浴,自然升温至室温,室温搅拌48小时;过滤,将滤液40℃水浴高真空旋干后柱层析(二氯甲烷:甲醇=9:1),得到白色固体;
[0087]
(2)化合物xi的合成
[0088]
称取2.5mmol化合物x,加入到洁净的100ml单口瓶,用50ml甲醇溶解;氮气置换三次后氮气保护;称量纸称取1.2mmol叔丁醇钠,一次性加入到上述单口烧瓶中;将上述反应混合物室温搅拌过夜后将反应混合物直接旋干,所得残余物分散在50ml乙酸乙酯中,用1n盐酸酸化后,用50ml乙酸乙酯萃取4次,合并有机相后用无水硫酸钠干燥旋干得到0.86g白色固体。
[0089]
(3)化合物xii的合成
[0090]
称取0.15mmol化合物xi,加入到洁净的50ml茄型瓶中;加入2ml无水乙腈后氮气置
换三次,氮气保护;称取0.65mmol干燥过的吡啶,一次性加入到上述茄型瓶中;用冰水浴将反应体系冷却至0℃;称取0.3mmol三氯氧磷,滴加到上述茄型瓶中;不撤冷浴,上述反应混合物在0℃搅拌30分钟后作为溶液a备用;称取0.55mmol焦磷酸三正丁胺,加入到另一洁净的50ml茄型瓶中;加入2ml无水乙腈后氮气置换三次,氮气保护;量取1.5ml干燥过的三正丁胺,一次性加入到上述茄型瓶中;用冰水浴将反应体系冷却至0℃;将溶液a滴加到上述反应体系中;不撤冷浴,上述反应混合物在0℃搅拌30分钟;量取4ml去离子水,一次性加入到上述反应体系中,室温搅拌2小时;0℃浓缩掉有机溶剂后,残余水溶液用高压制备分离纯化系统(pre-hplc)分离纯化;所得洗脱液冻干后得到11.4mg白色固体,复溶至1ml冷的去离子水中,-20℃冻存。
[0091]
根据上述方法,变换不同的x取代基,制备得到一系列化合物xii,结构如表4所示;
[0092]
表4化合物及质谱结构确认
[0093]
[0094][0095]
效果例1
[0096]
a:snp检测试剂盒的组分如下:
[0097]
(1)提取试剂盒成分:裂解液、洗涤液i、洗涤液ii、洗脱液、蛋白酶k、磁珠溶液。
[0098]
(2)多重pcr试剂盒组分:扩增反应液(40mm tris-hcl,800μm dntp,200nm引物,8mm mgcl2),扩增酶液。
[0099]
上述提取试剂盒与多重pcr试剂盒均来自重庆中元汇吉生物技术有限公司。
[0100]
(3)虾碱性磷酸酶(sap酶)处理体系:1μl 45mm tris-hcl,1μl 2u/μl sap酶,1μl水。
[0101]
(4)延伸反应体系:4μl单碱基延伸反应液(45mm tris-hcl,16μm单碱基延伸引物,0.6mm acyntps混合液),3μl 1u/μl单碱基延伸酶。
[0102]
b:核酸质谱检测的步骤如下:
[0103]
(1)样品dna的提取:使用中元汇吉提取试剂盒成分提取dna,仪器:中元汇吉全自动核酸提取仪器exm6000。
[0104]
多重pcr反应(表5所示):
[0105]
表5 pcr反应体系
[0106]
试剂名称体积(μl)扩增反应液10扩增酶液5dna(上述提取的dna)5
[0107]
可以根据实验需求按照比例减少反应体积,以求可通过384pcr板进行高通量检测。将配制好的多重pcr反应体系上pcr仪进行扩增,pcr扩增反应如表6所示:
[0108]
表6 pcr扩增反应
[0109]
[0110][0111]
pcr扩增完成后进行磷酸酶37℃消化30min,65℃失活5min处理,可以根据实验需求按照比例减少反应体积,以求可通过384pcr板进行高通量检测。
[0112]
完成消化后,进行单碱基延伸反应。配制延伸反应体系,按照每孔7μl,将延伸反应体系加到经sap消化处理的产物中,进行反应。
[0113]
单碱基延伸反应设定如表7所示。
[0114]
表7单碱基延伸反应
[0115][0116]
树脂脱盐处理:每反应孔添加树脂20mg和30μl ddh2o。可以根据实验需求按照比例减少树脂与ddh2o的体积,以求可通过384pcr板进行高通量检测。上述树脂购买自市售产品。盖好反应管(如果用384pcr板则封好封口膜),放在旋转器上颠倒摇匀5分钟后短暂离心。
[0117]
脱盐完成后上机检测,质谱检测的仪器来自重庆中元汇吉exs3000质谱仪。
[0118]
当a为o时,检测效果如效果例2-3所示
[0119]
效果例2
[0120]
本效果例列举核苷酸的结构如下:
[0121]
分子量505.18,命名为acy磺基-ctp。
[0122]
检测前庭导水管扩大相关基因slc26a4的281c>t突变,slc26a4基因突变可以导致前庭导水管扩大——单纯性前庭导水管扩大或者是合并耳蜗畸形的前庭导水管扩大。前庭导水管扩大是内耳最常见的畸形,其在遗传性耳聋中占到1~8%。临床上主要表现为高频听力损失为主的感音神经性耳聋,听力损失程度多表现为重度或者是极重度聋。发病多在儿童时期,其发病前常有感冒、发烧、外伤等使颅内压增高的诱因。
[0123]
本发明分别使用无修饰的核苷酸底物(反应1)与修饰后的核苷酸底物(反应2)对受检测样本的281c>t位点进行分析。
[0124]
本效果例所述的无修饰的核酸底物为acyatp、acygtp、acyctp和acyttp混合液,修饰的核酸底物为acyatp、acygtp、acy磺基-ctp和acy ttp混合液。
[0125]
结果如表8和图1所示,结果显示,该分析样本为281c>t位点的突变基因携带患者,与测序结果保持一致。
[0126]
表8突变位点分析
[0127] 引物引物分子量(m/z)延伸碱基延伸产物分子量反应1/2primer1-281c>t5873.9c/t6069.02/6083.02反应3primer1-281c>t5873.9磺酸ctp/t6149.08/6083.02
[0128]
上述primer1-1174a》t的序列为seq.id.no.1:gtcatttcgggagttagta
[0129]
结果显示,修饰后的acy磺基-ctp与acyttp分隔更开,在谱图上判读更加容易。
[0130]
临床试验
[0131]
本发明利用修饰的核酸底物(acyatp、acygtp、acy磺基-ctp和acyttp混合液)对受检测样本进行耳聋基因20个snp位点突变分析。耳聋相关易感基因snp分型结果见表9-11及图2:
[0132]
表9多重扩增引物
[0133][0134]
表10单碱基延伸引物
[0135]
编号序列位点名称seq.id.no.22ctccacagtcaagca1975g》cseq.id.no.23aatcctgagaagatgt1174a》tseq.id.no.24caccactgctctttccc1226g》aseq.id.no.25tgttggagtgagatcac2027t》aseq.id.no.26cacgaagatcagctgca235delcseq.id.no.27gcagtagcaattatcgtcivs7-2a》gseq.id.no.28cgtacacaccgcccgtcac1494c》tseq.id.no.29acgtggactgctacattgcc538c》tseq.id.no.30cagcgtggccactagccca281c》tseq.id.no.31cagtgctctcctggacggcc1229c》tseq.id.no.32gatgaacttcctcttcttctc299_300delatseq.id.no.33ggattagataccccactatgct1095t》cseq.id.no.34tctgtagatagagtatagcatca2168a》gseq.id.no.35tgtctgcaacaccctgcagccag176_191del16seq.id.no.36tgccagtgccctgactctgctggtt589g》aseq.id.no.37acccctacgcatttatatagaggag1555a》gseq.id.no.38aaaacaaatttctagggataaaataivs15+5g》aseq.id.no.39gggcacgctgcagacgatcctggggg35delgseq.id.no.40ccatgaagtaggtgaagattttcttct547g》a
seq.id.no.41aaaggacacattctttttga2162c》t
[0136]
表11耳聋相关易感基因snp分型
[0137][0138][0139]
采用本发明提供的引物组能够对耳聋相关易感基因的20个snp位点进行分型,正确率100%,与基因检测金标准一代测序结果一致。
[0140]
效果例3
[0141]
本效果例列举核苷酸的结构如下:
[0142]
分子量673.44
[0143]
为了方便,命名为acy-丁醚-ctp(n=5)
[0144]
本效果例,探究n》1时的检测效果
[0145]
本效果例检测ivs-i-5(g》c),ivs-i-5(g》c)是β地中海贫血症的一个突变位点。
[0146]
表12突变位点分析
[0147] 引物引物分子量(m/z)延伸碱基延伸产物分子量反应1/2primer2-ivs-i-5(g》c)6172.10g/c6407.24/6367.22反应3primer2-ivs-i-5(g》c)6172.10g/丁醚-ctp6407.24/6615.54
[0148]
上述primer2-ivs-i-5(g》c)的序列为seq.id.no.42:gcaggttgaggctatcatta
[0149]
结果如图3显示,修饰后的acy-丁醚-ctp(n=5)与acygtp分隔很开,非常容易判读结果。
[0150]
效果例4当a为ch2的效果验证
[0151]
本效果例探究将a从o替换为ch2对检测效果的影响
[0152]
因为本发明保护的核苷酸种类较多,不能做到在实施例中完全列举,所以本效果例通过研究以下几个结构式,进行说明。
[0153]
本效果例中涉及的单碱基延伸引物分子量以及用不同核苷酸底物延伸后的产物分子量与效果例2中相同
[0154]
表13延伸产物分子量
[0155][0156][0157]
(1)
[0158]
分子量分别为551.20和549.04,命名为acy i-ctp
[0159]
(2)
[0160]
分子量分别为501.22和499.25,命名为acy苯-ctp
[0161]
(3)分子量分别为523.27和521.03,命名为acy 1,4-氨基哌啶-ctp
[0162]
结果如图4所示,图中谱线1、2、3、4、5、6分别代表的是acy i-ttp(a)、acy i-ttp(b)、acy苯-ttp(a)、acy苯-ttp(b)、acy 1,4-氨基哌啶-ctp(a)、acy 1,4-氨基哌啶-ctp(b)作为底物检测突变位点281c>t的结果图。
[0163]
从图中可以看出,就分子量而言,a为o或ch2相差并不大,所以在谱图上相隔较近,但相对于acyctp而言,分子量相差越大,越能够在谱图上分隔开。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1