一种基于克隆和一代测序鉴定BCOR基因15号外显子串联重复的方法与流程
一种基于克隆和一代测序鉴定bcor基因15号外显子串联重复的方法
技术领域
1.本发明涉及分子生物学技术领域,特别涉及一种基于克隆和一代测序鉴定bcor基因15号外显子串联重复的方法。
背景技术:
2.目前,鉴定bcor基因15号外显子串联重复的方法主要有以下四种鉴定方法。
3.(1)rna测序
4.其中,rna测序的缺陷为:a.rna提取过程复杂,福尔马林固定、石蜡包埋(ffpe)样本提取质量较差,可能对检测准确度产生不良影响;b.提取、检测成本较高。
5.(2)rt-pcr法
6.其中,rt-pcr法的缺陷为:a.目前报道bcor-内串联重复第15外显子编码序列的3
′‑
部分有6种不同的变异形式,长度从89-114bp不等;rt-pcr方法检测突变类型受限。
7.(3)dna甲基化分析
8.其中,dna甲基化分析的缺陷为:a.平台开展有限,检测可及性较低;b.操作复杂,对技术人员专业要求高,检测成本较高。
9.(4)液滴pcr法
10.其中,液滴pcr法的缺陷为:a.平台开展有限,检测可及性较低;b.检测突变类型受限。
11.为此,提出一种基于克隆和一代测序鉴定bcor基因15号外显子串联重复的方法。
技术实现要素:
12.本发明的目的在于提供一种基于克隆和一代测序鉴定bcor基因15号外显子串联重复的方法,利用t-a克隆和一代测序等分子生物学常用技术,检测bcor基因15号外显子编码序列内串联重复(itd)变异情况。
13.为了实现上述目的,本发明的技术方案如下:
14.一种基于克隆和一代测序鉴定bcor基因15号外显子串联重复的方法,包括以下步骤:
15.s1:对待测ffpe样本进行基因组dna提取,使用天根石蜡包埋组织dna快速提取试剂盒dp330-02;
16.s2:设计并合成正向引物和反向引物;
17.s3:使用上述s2步骤中的引物对基因组dna进行pcr扩增,对扩增产物检测并回收;
18.s4:对pcr扩增产物进行ta克隆,得到阳性克隆,挑取单克隆进行测序;
19.s5:对测序结果去除两端的载体序列,得到待测样本bcor序列信息,与理论序列比对获得样本中bcor-内串联重复的变异情况。
20.具体的,所述s1步骤中的具体处理步骤如下:
21.s101:脱蜡:将蜡块切下的蜡卷或将石蜡切片上的组织刮下装到干净的ep管中,向ep管中加入1ml的二甲苯,静置五分钟后12000rpm离心10min;倒掉二甲苯,再向ep管中加入1ml的二甲苯,12000rpm离心10min;倒掉二甲苯,向ep管中加入1ml的乙醇溶液,12000rpm离心10min;倒掉乙醇溶液,再向ep管中加入1ml的乙醇溶液,12000rpm离心10min;倒掉上清液,保留底部沉淀,放置56℃金属浴挥发酒精;
22.s102:加200μl缓冲液ga,再加入20μl proteinase k,充分混匀;
23.s103:56℃消化过夜,直到样品完全裂解;
24.s104:90℃孵化1h;
25.s105:快速离心收集管壁上的液体,加入220μl缓冲液gb涡旋混匀,再加入250μl无水乙醇,涡旋振荡充分混匀,可能会产生白色絮状沉淀。放置56℃金属浴中,待白色絮状沉淀溶解后,快速离心;
26.s106:将样品12000rpm离心3min,取上清;
27.s107:将上清转移到一个吸附柱cr2中,8000rpm离心30sec,倒掉废液;重新将吸附柱放回收集管中;
28.s108:打开盖子,向吸附柱cr2中加入500μl缓冲液gd,6000
×
g(8000rpm)离心30sec,倒掉废液,将吸附柱cr2放回收集管中;
29.s109:打开盖子加入600μl漂洗液pw,6000
×
g(8000rpm)离心30sec,倒掉废液,将吸附柱cr2放回收集管中;
30.s110:重复操作s109步骤;
31.s111:12000rpm离心2min,以彻底甩干吸附材料中残余的漂洗液;打开盖子晾三分钟,待残余酒精挥发完毕;
32.s112:将吸附柱cr2转移到1.5ml干净的离心管中,打开柱盖,向吸附膜的中间部位悬空滴加30-100μl去离子水洗脱,室温放置2-5min,再12000rpm离心1min收集产物;
33.s113:可将离心得到的溶液再加入吸附柱cr2中,室温放置2min,12000rpm离心2min,即得到基因组dna。
34.具体的,所述s2步骤中的正向引物为bcor ex16f2:tcctcccgcatatttcgctg,所述s2步骤中的反向引物为bcor ex16r:acacactgtacatggtgggtcc。
35.具体的,所述s3步骤中的具体处理步骤如下:
36.s301:pcr扩增,pcr反应体系:premix taq 10μl;primer11μl;primer2 1μl;基因组dna 5μl;ddh2o 3μl;反应条件依次为:94℃5min;35cycles:94℃30s,58℃30s,72℃1min;72℃10min;
37.s302:pcr产物割胶纯化:pcr产物经2%琼脂糖凝胶电泳,分别割取大小条带,使用天根回收试剂盒dp214-03纯化回收。
38.具体的,所述s302步骤中的具体处理步骤如下:
39.s3021:切下含有目的dna的琼脂糖凝胶放入干净的离心管中,称取质量;
40.s3022:加3倍凝胶质量的buffer de-a,混合均匀后于75℃加热直至凝胶块完全熔化;
41.s3023:加0.5倍buffer de-a体积的buffer de-b,混合均匀;当分离的dna片段小于400bp时,加入1倍凝胶质量的异丙醇;
42.s3024:将混合液转移到dna制备管,12000rpm离心1min,弃滤液;
43.s3025:向制备管中加入500μl buffer w1,12000rpm g离心30s,弃滤液;
44.s3026:向制备管中加入700μl buffer w2,12000rpm离心30s,弃滤液;重复加入700μl buffer w2,12000rpm离心1min;
45.s3027:12000rpm再次离心1min;
46.s3028:将制备管置于洁净的1.5ml离心管中,在制备膜中央加25-30μl去离子水,室温静置1min;12000rpm离心1min洗脱dna,即回收得到pcr扩增产物。
47.具体的,所述s4步骤中的具体处理步骤如下:
48.s401:使用takara pmd19-t载体进行常规ta克隆,目的基因与质粒载体连接体系:缓冲液i 5μl;takara pmd19-t载体(0.1μg/μl)1μl;pcr产物(0.1μg/μl)4μl;连接条件:16℃连接过夜;
49.s402:大肠杆菌的转化;
50.s403:重组菌的鉴定,从平板上分别挑取转化子,用2ml相应抗性的lb培养过夜,用pcr法进行鉴定;
51.s404:sanger测序实验。
52.具体的,所述s402步骤中的具体处理步骤如下:
53.s4021:在100μl大肠杆菌感受态细胞的离心管内加入待转化的连接产物5μl,轻柔混匀,冰浴30min;
54.s4022:42℃热激90s,冰上冷却2~3min;
55.s4023:加入700μl lb,于37℃,200rpm,恢复培养80min;
56.s4024:离心弃去600μl培养基,混匀剩余的菌体和培养基均匀涂布在lb+amp抗性平板上,37℃倒置培养14~16h。
57.具体的,所述s404步骤中的具体处理步骤如下:
58.s4041:测序反应体系:测序用的试剂盒为:bigdye v3.1 chemistry kit(applied biosystems,foster city,ca);总体系5μl,测序primer2μl(3.2μm),纯化后pcr产物1-3μl,bigdye mix kit 1μl;
59.s4042:反应条件依次为:94℃1min;28cycles:94℃20s,50℃10s,60℃4min;4℃30min;
60.s4043:nh4ac.edta纯化:待测序反应结束后,将96孔板以3700rpm离心30s;加入1μl nh4ac.edta溶液,置于混匀器上震荡30s,3700rpm离心30s;
61.s4044:100%乙醇沉淀:加入18μl的100%乙醇溶液,-20℃沉淀30min,4500rpm离心30min,倒置离心400rpm,2min;
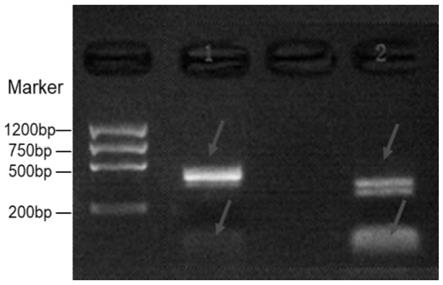
62.s4045:75%乙醇洗涤:加入50μl的75%乙醇,4500rpm离心10min,倒置离心400rpm,2min;
63.s4046:变性:加入6μl hi-di,3700rpm离心1min,-20℃存放待上3730xl测序仪;
64.s4047:测序:使用3730 xl dna测序仪进行测序,用dna sequencing analysis软件分析测序结果,用sequencing analysis 5.2.0软件进行解读。
65.具体的,所述s5步骤中的具体处理步骤如下:
66.s501:将测序所得序列在ebi water-align网站与理论序列进行比对;
67.s502:将测序所得序列在ncbi-blast网站与理论序列进行比对,查看dot plot图谱。
68.本发明的有益效果为:
69.(1)本发明的鉴定方法操作较简便、快捷,本领域的普通技术人员可以理解;
70.(2)本发明的检测平台可及性高,成本较低;
71.(3)本发明的鉴定方法可检测多种突变类型。
附图说明
72.图1为本发明实施例的pcr产物电泳图;
73.图2为本发明实施例的ebi water-align序列比对;
74.图3为本发明实施例的ncbi-blast dot plot图。
具体实施方式
75.下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
76.本发明是基于克隆和一代测序鉴定bcor-内串联重复bcor-itd的方法,包含下述步骤:
77.1.对待测ffpe样本进行基因组dna提取,使用天根石蜡包埋组织dna快速提取试剂盒(dp330-02),步骤如下:
78.(1)脱蜡:将蜡块切下的蜡卷或将石蜡切片上的组织刮下装到干净的ep管中,向ep管中加入1ml的二甲苯,静置五分钟后12000rpm离心10min;倒掉二甲苯,再向ep管中加入1ml的二甲苯,12000rpm离心10min;倒掉二甲苯,向ep管中加入1ml的乙醇溶液,12000rpm离心10min;倒掉乙醇溶液,再向ep管中加入1ml的乙醇溶液,12000rpm离心10min;小心倒掉上清液,保留底部沉淀,放置56℃金属浴挥发酒精;
79.(2)加200μl缓冲液ga,再加入20μl proteinase k,充分混匀;
80.(3)56℃消化过夜,直到样品完全裂解;
81.(4)90℃孵化1h;
82.(5)快速离心收集管壁上的液体,加入220μl缓冲液gb涡旋混匀,再加入250μl无水乙醇,涡旋振荡充分混匀,可能会产生白色絮状沉淀。放置56℃金属浴中,待白色絮状沉淀溶解后,快速离心;
83.(6)将样品12000rpm离心3min,小心取上清;
84.(7)将上清小心转移到一个吸附柱cr2中,8000rpm离心30sec,倒掉废液;重新将吸附柱放回收集管中;
85.(8)小心打开盖子,向吸附柱cr2中加入500μl缓冲液gd,6000
×
g(8000rpm)离心30sec,倒掉废液,将吸附柱cr2放回收集管中;
86.(9)小心打开盖子加入600μl漂洗液pw,6000
×
g(8000rpm)离心30sec,倒掉废液,将吸附柱cr2放回收集管中;
87.(10)重复操作步骤(9);
88.(11)12000rpm离心2min,以彻底甩干吸附材料中残余的漂洗液;打开盖子晾三分钟,待残余酒精挥发完毕;
89.(12)将吸附柱cr2转移到1.5ml干净的离心管中,小心打开柱盖,向吸附膜的中间部位悬空滴加30-100μl去离子水洗脱,室温放置2-5min,再12000rpm离心1min收集产物;
90.(13)可将离心得到的溶液再加入吸附柱cr2中,室温放置2min,12000rpm离心2min,即得到基因组dna。
91.2.设计并合成正向引物和反向引物;
92.bcor ex16f2:tcctcccgcatatttcgctg
93.bcor ex16r:acacactgtacatggtgggtcc
94.3.使用上述引物对基因组dna进行pcr扩增,对扩增产物检测并回收;
95.(1)pcr扩增:pcr反应体系:premix taq 10μl;primer1 1μl;primer2 1μl;基因组dna 5μl;ddh2o 3μl;反应程序:94℃5min;35cycles:94℃30s,58℃30s,72℃1min;72℃10min。
96.(2)pcr产物割胶纯化:pcr产物经2%琼脂糖凝胶电泳,分别割取大小条带参考附图1,使用天根回收试剂盒(dp214-03)纯化回收,步骤如下:
97.a.切下含有目的dna的琼脂糖凝胶放入干净的离心管中,称取质量;
98.b.加3倍凝胶质量的buffer de-a,混合均匀后于75℃加热直至凝胶块完全熔化;
99.c.加0.5倍buffer de-a体积的buffer de-b,混合均匀;当分离的dna片段小于400bp时,加入1倍凝胶质量的异丙醇;
100.d.将混合液转移到dna制备管,12000rpm离心1min,弃滤液;
101.e.向制备管中加入500μl buffer w1,12000rpm g离心30s,弃滤液;
102.f.向制备管中加入700μl buffer w2,12000rpm离心30s,弃滤液;重复加入700μl buffer w2,12000rpm离心1min;
103.g.12000rpm再次离心1min;
104.h.将制备管置于洁净的1.5ml离心管(试剂盒内提供)中,在制备膜中央加25-30μl去离子水,室温静置1min。12000rpm离心1min洗脱dna,即回收得到pcr扩增产物。
105.4.对pcr扩增产物进行ta克隆,得到阳性克隆,挑取单克隆进行测序;
106.(1)使用takara pmd19-t载体进行常规ta克隆,目的基因与质粒载体连接体系:缓冲液i 5μl;takara pmd19-t载体(0.1μg/μl)1μl;pcr产物(0.1μg/μl)4μl;连接条件:16℃连接过夜;
107.(2)大肠杆菌的转化
108.a.在100μl大肠杆菌感受态细胞的离心管内加入待转化的连接产物5μl,轻柔混匀,冰浴30min;
109.b.42℃热激90s,冰上冷却2~3min;
110.c.加入700μl lb,于37℃,200rpm,恢复培养80min;
111.d.离心弃去600μl培养基,混匀剩余的菌体和培养基均匀涂布在lb+amp抗性平板上,37℃倒置培养14~16h。
112.(3)重组菌的鉴定
113.从平板上分别挑取转化子,用2ml相应抗性的lb培养过夜,用pcr法进行鉴定。
114.(4)sanger测序实验
115.a.测序反应体系:测序用的试剂盒为:bigdye v3.1 chemistry kit(applied biosystems,foster city,ca);总体系5μl,测序primer2μl(3.2μm),纯化后pcr产物1-3μl,bigdye mix kit 1μl。
116.b.反应程序:94℃1min;28cycles:94℃20s,50℃10s,60℃4min;4℃30min。
117.c.nh4ac.edta纯化:待测序反应结束后,将96孔板以3700rpm离心30s;加入1μl nh4ac.edta溶液,置于混匀器上震荡30s,3700rpm离心30s。
118.d.100%乙醇沉淀:加入18μl的100%乙醇溶液,-20℃沉淀30min,4500rpm离心30min,倒置离心400rpm,2min。
119.e.75%乙醇洗涤:加入50μl的75%乙醇,4500rpm离心10min,倒置离心400rpm,2min。
120.f.变性:加入6μl hi-di,3700rpm离心1min,-20℃存放待上3730xl测序仪。
121.g.测序:使用3730 xl dna测序仪进行测序。用dna sequencing analysis软件分析测序结果,用sequencing analysis 5.2.0软件进行解读。
122.5.对测序结果去除两端的载体序列,得到待测样本bcor序列信息,与理论序列比对获得样本中bcor-内串联重复的变异情况。
123.(1)将测序所得序列在ebi water-align网站与理论序列进行比对,参考附图2;
124.(2)将测序所得序列在ncbi-blast网站与理论序列进行比对,查看dot plot图谱,参考附图3。
125.进一步的,bcor基因exon15理论序列:
126.gccacgaaactggctactgctttcggatgtccttaagaaattgaaaatgtcctcccgcatatttcgctgcaattttccaaacgtggaaattgtcaccattgcagaggcagaattttatcggcaggtttctgcaagtctcttgttctcttgctccaaagacctggaagccttcaaccctgaaagtaaggagctgttagatctggtggaattcacgaacgaaattcagactctgctgggctcctctgtagagtggctccaccccagtgatctggcctcagacaactactggtgagcaagctggacccaccatgtacagtgtgttatagtgttaatccttgtgcatatgtgtcataatacaactatttctgtaaagaaaggacactattacatatgaaaatatctcttctttatataagagaaattactccagtcagaaggacttagaaacatgtttttttccttttaaacttttaagtcagtttttatgaagttgttataatgtttctttacttttcaatgcacacatgctttgggatacgtttgtttttacttggaacatttgtttcttttcttttttaaggagaaaaaaaaatgagtaaaaggagctccacactttgacttaatttcatacaaagctctgatgacaggccatgactgtagagtggtcagaactgtgtggttggtttgagggagcgaattcggggaaggcacttggtgatataactttgttttgtttacagagtacctgctcgggccaggtaaatgctattggatgtaatccagtagtgtgtaatataaattcaaaccatatccacacacaacaactaattgtatgaaacttttatatcctaatttaaaagctgtgaaattagttttcacgcatcaaaccggattgtttatatgtttaaacattttatgctcttatttaaagaagactttgagctatttttttctgtaccctgtaaaatattgaaaactaacataatatgttgaggttgcttggaaatgtacataaaactaaaattttctgaatcgtgtgtttatgtttgaaatctgtgttttaactttgtaagtaaattctctgcctttgtatttatattttacaaaaattttcttaaaaggcaataaaactgttgaggaaaggagaaaa。
127.最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技
术方案进行修改或者等同替换,而不脱离本发明技术方案的宗旨和范围,其均应涵盖在本发明的权利要求范围当中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1