一种含有三嗪、二苯并呋喃及咔唑结构的化合物及有机电致发光器件的制作方法
1.本发明涉及有机电致发光技术领域,具体涉及一种含有三嗪、二苯并呋喃及咔唑结构的化合物及有机电致发光器件。
背景技术:
2.有机电致发光器件(organic light-emitting devices,oled)是利用如下原理的自发性发光器件:当施加电场时,荧光物质通过正极注入的空穴和负极注入的电子的重新结合而发光。这种自发光器件,具有电压低、亮度高、视角宽、响应快、温度适应性好等特性,并且超薄,能制作在柔性面板上等优点,广泛应用于手机、平板电脑、电视、照明等领域。
3.相较于led(发光二极管)或lcd(液晶显示)的晶体层,oled的有机塑料层更薄、更轻而且更富于柔韧性;
4.相较于led(发光二极管)或lcd(液晶显示)的晶体层,oled的有机塑料层更薄、更轻而且更富于柔韧性;
5.oled的发光层比较轻,因此它的基层可使用富于柔韧性的材料,而不会使用刚性材料,oled基层为塑料材质,而led和lcd则使用玻璃基层;
6.oled属于一种电流型的有机发光器件,是通过载流子的注入和复合而致发光的现象,发光强度与注入的电流成正比。oled在电场的作用下,阳极产生的空穴和阴极产生的电子就会发生移动,分别向空穴传输层和电子传输层注入,迁移到发光层。当二者在发光层相遇时,产生能量激子,从而激发发光分子最终产生可见光。
7.由于oled的外量子效率与内量子效率之间存在巨大的差距,极大的制约了oled的发展,因此,如何提高oled的光取出效率也成为研究的热点。ito薄膜和玻璃衬底的界面以及玻璃衬底与空气的界面处会发生全反射,出射到oled器件前向外部空间的光约占有机材料薄膜el总量的20%,其余约80%的光主要以导波形式限制在有机材料薄膜、ito薄膜和玻璃衬底中,严重制约了oled的发展与应用,如何减少oled器件中的全反射效应、提高光耦合到器件前向外部空间的比例引,进而提高器件的性能起了人们的广泛关注。
8.oled并不需要采用lcd中的逆光系统,lcd工作时会选择性地阻挡某些逆光区域,从而让图像显现出来,而oled则是靠自身发光,因为oled不需逆光系统,所以它们的耗电量小于lcd(lcd所耗电量中的大部分用于逆光系统)这一点对于靠电池供电的设备来说,尤其重要。
技术实现要素:
9.本发明的目的是针对上述技术问题,本发明提供了一种含有三嗪、二苯并呋喃及咔唑结构的化合物及有机电致发光器件。
10.本发明的目的可以通过以下措施达到:
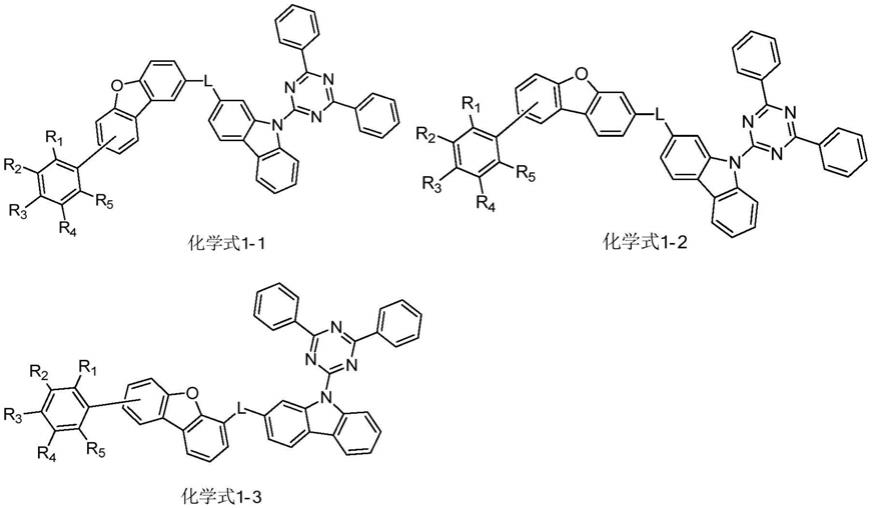
11.一种含有三嗪、二苯并呋喃及咔唑结构的化合物,其结构式由化学式1和化学式2
组合而成:
[0012][0013]
其中,化学式1的a*、b*和c*中的一个与化学式2的d*键结,且a*、b*和c*中未键结的两个是氢;
[0014]
l为单键、氘代或未氘代的亚苯基、氘代或未氘代的亚联苯基或者氘代或未氘代的亚奈基,r1、r2、r3、r4或r5各自独立的为氢或氘。
[0015]
在一种优选方案中,本技术的化合物可以由以下的化学式1-1、化学式1-2或化学式1-3表示:
[0016][0017][0018]
在化学式1-1、化学式1-2或化学式1-3中,l为单键、氘代或未氘代的亚苯基、氘代或未氘代的亚联苯基或者氘代或未氘代的亚奈基,r1、r2、r3、r4或r5各自独立的为氢或氘。
[0019]
优选的,本发明中的l为单键或者氘代或未氘代的亚苯基。
[0020]
优选的,本发明中的l为单键或者氘代或未氘代的下述基团:
[0021][0022]
优选的,r1、r2、r3、r4和r5同时为氢,或者同时为氘。
[0023]
优选的,r1、r2、r3、r4和r5所处的苯基与二苯并呋喃的1位、2位、3位或4位键结。
[0024]
更进一步的,本技术中的有机化合物为以下化合物的任意一种:
[0025]
[0026]
[0027]
[0028]
[0029]
[0030]
[0031]
[0032]
[0033]
[0034]
[0035]
[0036][0037]
化学式1-1反应历程如下(化学式1-2、化学式1-3类似):
[0038][0039]
一种有机电致发光器件,包括第一电极、第二电极以及在第一电极和第二电极之间形成的有机层,其中在有机层中含有上述本发明的化合物。
[0040]
进一步地,有机层包含空穴注入层、第一空穴传输层、第二空穴传输层、发光层、空穴阻挡层、电子传输层和电子注入层;在空穴注入层、第一空穴传输层、第二空穴传输层、发光层、空穴阻挡层、电子传输层、电子注入层中的至少一层含有上述本发明的化合物。
[0041]
进一步地,在发光层中含有本发明的上述的化合物。
[0042]
更进一步,在发光层中还含有以下化合物g1-g60中的至少一种:
[0043]
[0044][0045][0046]
一种含有上述有机电致发光器件的电子显示设备。
[0047]
一种含有上述有机电致发光器件的oled照明设备。
[0048]
本发明所指室温均为25
±
5℃。
[0049]
本发明所指的键结是指两个基团通过共价键结合在一起,例如化学式1中的a*与化学式2中的d*键结后形成化学式1-3。
[0050]
本发明的有益效果:
[0051]
本发明设计了一类全新的有机电致发光材料,其结构由咔唑及咔唑衍生物、二苯并呋喃及二苯并呋喃衍生物、三嗪及三嗪衍生物以特定的方式进行连接。本发明的化合物具有以下三个特点:
[0052]
1、咔唑的2号位与苯取代二苯并呋喃1-3位的连接,既避免了因咔唑1号位置空间位阻过大导致的反应困难、杂质多的问题,又可以保证材料具有较大的扭矩和三线态能级,进而避免了能量由掺杂材料向主体材料的反向传递,提高了器件的发光效率。
[0053]
2、咔唑的2号位置与苯取代二苯并呋喃1-3位相连接,增加了材料的扭矩,提高了材料的溶解性,进而提高了材料的量产性。同时,该类材料由于具备较大的扭矩,导致其具有相对较低的熔点,较低的熔点可以使得材料在蒸镀时呈现熔融状态,该状态能够有效避免材料在蒸镀过程中发生的堵孔情况,进而大幅度提高器件的良率。
[0054]
3、本发明中,咔唑及咔唑衍生物与三嗪以单键的形式直接连接,该种连接方式的优点在于,三嗪的吸电子特性,直接使得与之相连的咔唑及咔唑衍生物的电子云密度降低,进一步提高了材料分子的化学稳定性及热稳定性,进而提高器件的发光效率及寿命。
[0055]
经过器件验证,使用本发明化合物制备的器件均具有良好的发光效率及寿命。
附图说明
[0056]
图1为本发明有机电致发光器件的结构示意图;
[0057]
图中标号分别代表:1-阳极、2-空穴注入层、3-第一空穴传输层、4-第二空穴传输层、5-发光层、6-空穴阻挡层、7-电子传输层、8-电子注入层、9-阴极。
[0058]
图2为本发明实施例1中所制备的化合物14的hplc图。
[0059]
图3为本发明实施例1中所制备的化合物14的dsc图谱,由图3可知,化合物14的tm值为297.51℃。
[0060]
图4为本发明实施例1中所制备的化合物14的tga图谱,由图4可知,热失重温度td值为478.13℃。
[0061]
图5为本发明应用例1和对照例3中有机电致发光器件的寿命图;由图5可知,本发明应用例1和对照例3所制备的有机电致发光器件的t97%寿命分别为645h和606h。
具体实施方式
[0062]
以下进一步说明和描述了各个方面的实施例。应当理解,本文的描述并非旨在将权利要求书限制于所描述的特定方面。相反,旨在覆盖可包括在由所附权利要求书限定的本公开的精神和范围内的替代、修改和等同物。
[0063]
如本文所用,在“取代的”或“未取代的”中,术语“取代的”是指该基团中的至少一个氢与烃基、烃衍生物基、卤素或氰基(-cn)重新配位。术语“未取代的”是指该基团中的至少一个氢不与烃基、烃衍生物基、卤素或氰基(-cn)重新配位。烃基或烃衍生物基团的实例可包括c1至c20烷基、c2至c20烯基、c2至c20炔基、c6至c20芳基、c5至c20杂芳基、c1至c20烷氨基、c6至c20芳氨基、c6至c20杂芳氨基、c6至c20芳基杂芳氨基等,但不限于此。
[0064]
实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
[0065]
实施例1:
[0066][0067]
化合物14的合成方法如下:
[0068][0069]
氮气保护下,在1l三口瓶中加入1-a(10g,34.93mmol,1eq)、1-b(10.75g,36.67mmol,1.05eq)、碳酸钾(14.48g,104.78mmol,3eq)、pd(ppd3)4(四(三苯基膦)钯)(0.81g,0.7mmol,0.02eq)、甲苯400ml、乙醇200ml、水120ml,甲苯400ml、乙醇200ml、水120ml,加料完毕后升温至105℃搅拌反应,hplc监控反应进度,当化合物1-a≤1%时,停止反应。加入200ml水,降温搅拌析晶,待降到室温后,抽滤得滤饼,滤饼于85℃下鼓风干燥12小时,最终得到类白色固体目标产物化合物1-c(7.41g,17.88mmol,收率51.2%),esi-ms(m/z)(m+):理论值414.51,实测值415.34,元素分析结果(分子式c30h14d5no):理论值c,86.93;h,5.83;n,3.38;o,3.86;实测值c,86.90;h,5.86;n,3.42;o,3.82。
[0070]
在氮气保护下,于2l三口烧瓶中依次加入1-c(7.41g,17.88mmol,1eq)、1-d(8.61g,32.18mmol,1.8eq)、叔丁醇钠(3.44g,35.75mmol,2eq)、三叔丁基膦(0.14ml,0.72mmol,0.04eq)、pd2(dba)3(0.33g,0.36mmol,0.02eq)、甲苯650ml,加料完毕后升温至105℃搅拌反应,hplc监控。hplc监测化合物1-b反应完全后,停止反应,将反应液冷却至室温,加入300ml水,降温搅拌析晶,待温度降至室温后抽滤得滤饼。将滤饼加入至2l三口烧瓶中,加入800ml邻二氯苯后加热至产品溶解,趁热过硅胶粉和活性炭漏斗得滤液,滤液搅拌降温析晶,待降温至室温后抽滤得滤饼,滤饼使用800ml邻二氯苯重结晶5次后,降温抽滤得滤饼,滤饼于85℃下鼓风干燥,最终得到类白色固体目标产物化合物14(5.23g,8.1mmol,收率45.3%),esi-ms(m/z)(m+):理论值645.76,实测值646.33,元素分析结果(分子式
c45h23d5n4o):理论值c,83.70;h,5.15;n,8.68;o,2.48;实测值c,83.70;h,5.15;n,8.68;o,2.47。
[0071]
实施例2:
[0072][0073]
化合物25的合成方法如下:
[0074][0075]
制备方法与实施例1基本相同,区别在于将化合物1-c替换成化合物2-c,得最终目标产物化合物25,收率41.4%,esi-ms(m/z)(m+):理论值640.73,实测值640.54,元素分析结果(分子式c45h28n4o):理论值c,84.35;h,4.40;n,8.74;o,2.50;实测值c,84.32;h,4.44;n,8.78;o,2.46。
[0076]
实施例3:
[0077][0078]
化合物28的合成方法如下:
[0079][0080]
在氮气保护下,于2l三口烧瓶中,依次加入3-a(60g,244mmol,1eq)、3-b(42g,269mmol,1.1eq)、四三苯基膦钯(4.2g,3.6mmol,0.015eq),碳酸钾(85g,615mmol,2.75eq)、500ml甲苯、125ml乙醇和125ml水,加料完毕后升温至85℃搅拌反应,hplc监测反应进度,当化合物3-a≤1%时,停止反应,反应液自然降温至室温后,加入200ml水,搅拌10min,抽滤,水洗,乙醇淋洗后,于85℃干燥12小时后,得中间体3-c,62.9g,收率93%,esi-ms(m/z)(m+):理论值277.75,实测值277.52,元素分析结果(分子式c18h12cln):理论值c,77.84;h,4.35;n,5.04;cl,12.76;实测值c,77.82;h,4.38;n,5.02;cl,12.78。
[0081]
在氮气保护下,于2l三口烧瓶中加入中间体3-c(62.9g,226mmol,1eq)、联硼酸频那醇酯(86.1g,339mmol,1.5eq)、乙酸钾(67.0g,683mmol,3.0eq)、x-phos(6.5g,13.36mmol,0.06eq)、pd2(dba)3(12.23g,13.36mmol,0.06eq)、1,4-二氧六环700ml,加料完毕后升温至102℃下搅拌反应,hplc监测反应进度。当中间体3-c≤1%时,停止反应,反应液趁热过硅胶粉漏斗得滤液,滤液浓缩后,加入乙醇200ml,置于冰浴下搅拌10min后抽滤得滤饼,滤饼使用100ml乙醇淋洗,滤饼置于55℃下干燥12小时后,得淡黄色中间体3-d 70.4g,收率84%。esi-ms(m/z)(m+):理论值369.26,实测值369.18,元素分析结果(分子式c24h24bno2):理论值c,78.06;h,6.55;n,3.79;o,8.67;实测值c,78.03;h,6.57;n,3.80;o,8.71。
[0082]
在氮气保护下,于2l三口烧瓶中加入中间体3-d(65.0g,176.05mmol,1.06eq)、3-e(66.0g,166.08mmol,1eq)、四三苯基膦钯(3.8g,3.32mmol,0.02eq)、碳酸钾(58.08g,420.2mmol,2.53eq)、甲苯1000ml、乙醇250ml和水250ml,加料完毕后升温至85℃搅拌反应,hplc监测反应进度。当中间体3-e≤1%时,停止反应,将反应液自然降温至室温后,加入300ml水,搅拌20min后抽滤得滤饼,滤饼用50ml水淋洗2次后再用50ml乙醇淋洗2次得滤饼,将滤饼置于65℃下干燥12小时,得到灰色固体3-f,59.5g,收率73%。
[0083]
在氮气保护下,于2l三口烧瓶中加入3-f(59.5g,121.28mmol,1eq)、3-g(49.03g,183.13mmol,1.51eq)、pd2(dba)3(3.8g,3.27mmol,0.027eq)、叔丁醇钠(23.3g,242.56mmol,
2.0eq)、三叔丁基膦(0.98ml,4.85mmol 0.04eq)、甲苯1200ml,加料完毕后升温至102℃搅拌反应,hplc监测反应进度。当中间体3-e≤1%时,停止反应,反应液自然降温至室温后,加入300ml水,搅拌20min后抽滤得滤饼,滤饼用100ml水淋洗2次后使用100ml乙醇淋洗2次得到目标化合物3的粗品,将粗品置于85℃下干燥12小时,将干燥后的粗品加入到2l三口烧瓶中、加入1l邻二氯苯,升温至110℃使粗品完全溶解,溶液趁热过硅胶粉和活性炭漏斗后得滤液,滤液降至室温后析出晶体,抽滤得固体。将该固体使用800ml邻二氯苯重结晶三次,得最终目标产物化合物3(34.5g,收率39.4%),esi-ms(m/z)(m+):理论值721.86,实测值721.49,元素分析结果(分子式c51h27d5n4o):理论值c,84.86;h,5.17;n,7.76;o,2.22;实测值c,84.80;h,5.23;n,7.70;o,2.27。
[0084]
实施例4:
[0085][0086]
化合物29的合成方法如下:
[0087][0088]
制备方法与实施例1基本相同,区别在于将化合物1-c替换成化合物4-c,得最终目标产物化合物29,收率40.8%,esi-ms(m/z)(m+):理论值640.73,实测值640.46,元素分析结果(分子式c45h28n4o):理论值c,84.35;h,4.40;n,8.74;o,2.50;实测值c,84.39;h,4.45;n,8.71;o,2.45。
[0089]
实施例5:
[0090][0091]
化合物30的合成方法如下:
[0092][0093]
制备方法与实施例1基本相同,区别在于将化合物1-c替换成化合物5-c,得最终目标产物化合物30,收率55.2%,esi-ms(m/z)(m+):理论值645.76,实测值644.98,元素分析结果(分子式c45h23d5n4o):理论值c,83.70;h,5.15;n,8.68;o,2.48;实测值c,83.75;h,5.10;n,8.64;o,2.51。
[0094]
实施例6:
[0095][0096]
化合物42的合成方法如下:
[0097][0098]
制备方法与实施例3基本相同,区别在于将化合物3-f替换成化合物6-f,得最终目标产物化合物42,收率39.8%,esi-ms(m/z)(m+):理论值721.86,实测值722.48,元素分析结果(分子式c51h27d5n4o):理论值c,84.86;h,5.17;n,7.76;o,2.22;实测值c,84.89;h,5.13;n,7.79;o,2.19。
[0099]
实施例7:
[0100][0101]
化合物53的合成方法如下:
[0102][0103]
制备方法与实施例1基本相同,区别在于将化合物1-c替换成化合物7-c,得最终目标产物化合物53,收率46.3%,esi-ms(m/z)(m+):理论值640.73.,实测值640.16,元素分析结果(分子式c45h28n4o):理论值c,84.35;h,4.40;n,8.74;o,2.50;实测值c,84.37;h,4.45;n,8.73;o,2.45。
[0104]
实施例8:
[0105][0106]
化合物54的合成方法如下:
[0107][0108]
制备方法与实施例1基本相同,区别在于将化合物1-c替换成化合物8-c,得最终目标产物化合物54,收率48.1%,esi-ms(m/z)(m+):理论值645.76,实测值644.87,元素分析结果(分子式c45h23d5n4o):理论值c,83.70;h,5.15;n,8.68;o,2.48;实测值c,83.75;h,5.18;n,8.64;o,2.43。
[0109]
实施例9:
[0110][0111]
化合物63的合成方法如下:
[0112][0113]
制备方法与实施例3基本相同,区别在于将化合物3-f替换成化合物9-f,得最终目标产物化合物63,收率56.8%,esi-ms(m/z)(m+):理论值716.83,实测值716.92,元素分析结果(分子式c51h32n4o):理论值c,85.45;h,4.50;n,7.82;o,2.23;实测值c,85.48;h,4.54;n,7.78;o,2.20。
[0114]
实施例10:
[0115][0116]
化合物69的合成方法如下:
[0117][0118]
制备方法与实施例3基本相同,区别在于将化合物3-f替换成化合物10-f,得最终目标产物化合物69,收率47.2%,esi-ms(m/z)(m+):理论值716.83,实测值716.48,元素分析结果(分子式c51h32n4o):理论值c,85.45;h,4.50;n,7.82;o,2.23;实测值c,85.49;h,4.55;n,7.77;o,2.19。
[0119]
实施例11:
[0120][0121]
化合物75的合成方法如下:
[0122][0123]
制备方法与实施例3基本相同,区别在于将化合物3-f替换成化合物11-f,得最终目标产物化合物75,收率42.6%,esi-ms(m/z)(m+):理论值716.83,实测值716.19,元素分析结果(分子式c51h32n4o):理论值c,85.45;h,4.50;n,7.82;o,2.23;实测值c,85.52;h,4.58;n,7.75;o,2.15。
[0124]
实施例12:
[0125][0126]
化合物95的合成方法如下:
[0127][0128]
制备方法与实施例3基本相同,区别在于将化合物3-f替换成化合物12-f,得最终目标产物化合物95,收率39.2%,esi-ms(m/z)(m+):理论值716.83,实测值716.37,元素分析结果(分子式c51h32n4o):理论值c,85.45;h,4.50;n,7.82;o,2.23;实测值c,85.40;h,4.44;n,7.87;o,2.29。
[0129]
实施例13:
[0130][0131]
化合物107的合成方法如下:
[0132][0133]
制备方法与实施例3基本相同,区别在于将化合物3-f替换成化合物13-f,得最终目标产物化合物107,收率50.5%,esi-ms(m/z)(m+):理论值716.83,实测值716.54,元素分析结果(分子式c51h32n4o):理论值c,85.45;h,4.50;n,7.82;o,2.23;实测值c,85.46;h,4.52;n,7.80;o,2.22。
[0134]
实施例14:
[0135][0136]
化合物108的合成方法如下:
[0137][0138]
制备方法与实施例3基本相同,区别在于将化合物3-f替换成化合物14-f,得最终目标产物化合物108,收率42.1%,esi-ms(m/z)(m+):理论值721.86,实测值722.39,元素分析结果(分子式c51h27d5n4o):理论值c,84.86;h,5.17;n,7.76;o,2.22;实测值c,84.84;h,5.14;n,7.78;o,2.24。
[0139]
实施例15:
[0140][0141]
化合物119的合成方法如下:
[0142]
[0143]
制备方法与实施例3基本相同,区别在于将化合物3-f替换成化合物15-f,得最终目标产物化合物119,收率47.5%,esi-ms(m/z)(m+):理论值716.83,实测值716.05,元素分析结果(分子式c51h32n4o):理论值c,85.45;h,4.50;n,7.82;o,2.23;实测值c,85.48;h,4.54;n,7.78;o,2.20。
[0144]
实施例16:
[0145][0146]
化合物147的合成方法如下:
[0147][0148]
制备方法与实施例3基本相同,区别在于将化合物3-f替换成化合物16-f,得最终目标产物化合物147,收率54.1%,esi-ms(m/z)(m+):理论值720.85,实测值720.66,元素分析结果(分子式c51h28d4n4o):理论值c,84.98;h,5.03;n,7.77;o,2.22;实测值c,84.92;h,5.00;n,7.80;o,2.28。
[0149]
实施例17:
[0150][0151]
化合物165的合成方法如下:
[0152][0153]
制备方法与实施例3基本相同,区别在于将化合物3-f替换成化合物17-f,得最终目标产物化合物165,收率38.7%,esi-ms(m/z)(m+):理论值720.85,实测值720.28,元素分析结果(分子式c51h28d4n4o):理论值c,84.98;h,5.03;n,7.77;o,2.22;实测值c,84.95;h,5.06;n,7.73;o,2.26。
[0154]
实施例18:
[0155][0156]
化合物171的合成方法如下:
[0157][0158]
制备方法与实施例3基本相同,区别在于将化合物3-f替换成化合物18-f,得最终目标产物化合物171,收率48.5%,esi-ms(m/z)(m+):理论值720.85,实测值720.94,元素分析结果(分子式c51h28d4n4o):理论值c,84.98;h,5.03;n,7.77;o,2.22;实测值c,84.91;h,5.10;n,7.83;o,2.16。
[0159]
实施例19:
[0160]
[0161]
化合物191的合成方法如下:
[0162][0163]
制备方法与实施例3基本相同,区别在于将化合物3-f替换成化合物19-f,得最终目标产物化合物191,收率54.7%,esi-ms(m/z)(m+):理论值720.85,实测值720.36,元素分析结果(分子式c51h28d4n4o):理论值c,84.98;h,5.03;n,7.77;o,2.22;实测值c,84.92;h,5.09;n,7.75;o,2.24。
[0164]
实施例20:
[0165][0166]
化合物203的合成方法如下:
[0167][0168]
制备方法与实施例3基本相同,区别在于将化合物3-f替换成化合物20-f,得最终目标产物化合物203,收率36.2%,esi-ms(m/z)(m+):理论值720.85,实测值720.43,元素分析结果(分子式c51h28d4n4o):理论值c,84.98;h,5.03;n,7.77;o,2.22;实测值c,84.99;h,5.07;n,7.73;o,2.21。
[0169]
材料性质测试:
[0170]
测试本发明化合物14、25、28、29、30、42、53、54、63、69、75、95、107、108、119、147、165、171、191、203的热失重温度td和熔点tm,测试结果如下表1所示。
[0171]
注:热失重温度td是在氮气气氛中失重5%的温度,在tga n-1000热重分析仪上进行测定,测定时氮气流量为10ml/min,熔点tm由示差扫描量热法(dsc,新科dsc n-650)测
定,升温速率10℃/min。
[0172]
表1:
[0173]
项目材料td/℃tm/℃实施例0114478.13297.51实施例0225460.59278.14实施例0328473.25290.37实施例0429485.73282.40实施例0530487.06300.06实施例0642475.44305.43实施例0753466.17286.18实施例0854475.26293.04实施例0963459.09306.15实施例1059475.22303.46实施例1175483.01279.48实施例1295494.73286.57实施例13107468.07301.25实施例14108479.12298.14实施例15119462.28285.37实施例16147470.49291.55实施例17165495.10307.43实施例18171483.58302.86实施例19191471.36278.18实施例20203460.28286.93
[0174]
由上述数据可知,本发明所合成的化合物的热稳定性优良,说明符合本发明结构通式的化合物都具有优良的热稳定性,可以满足有机电致发光材料使用的要求。
[0175]
器件性能测试:
[0176]
应用例1:
[0177]
采用ito作为反射层阳极基板材料,并依次用水、丙酮、n2等离子对其进行表面处理;
[0178]
在ito阳极基板上方,沉积10nm掺杂有3%ndp-9的ht-1,形成空穴注入层(hil);
[0179]
在空穴注入层(hil)上方蒸镀100nm的ht-1形成第一空穴传输层(htl);
[0180]
在第一空穴传输层(htl)上方真空蒸镀gp,形成厚度为30nm的第二空穴传输层(gpl);
[0181]
将本发明设计的化合物14与g1按照5:5的质量比例作为绿色主体材料进行共同蒸镀,gd-1作为掺杂材料(gd-1用量为化合物14与g1总质量的8%)蒸镀在第二空穴传输层(gpl)上形成厚度为30nm的发光层;
[0182]
将hb-1蒸镀到发光层上得到厚度为20nm的空穴阻挡层(hbl);
[0183]
将et-1与liq按照5:5的质量比例进行共同蒸镀到空穴阻挡层(hbl)上得到厚度为30nm的电子传输层(etl);
[0184]
将镁(mg)和银(ag)以9:1的质量比例混合蒸镀到电子传输层(etl)上方,形成厚度为50nm的电子注入层(eil);
[0185]
此后将银(ag)蒸镀到电子注入层上方,形成厚度为100nm的阴极,在上述阴极封口层上沉积50nm厚度的dntpd,此外,在阴极表面以uv硬化胶合剂和含有除湿剂的封装薄膜(seal cap)进行密封,以保护有机电致发光器件不被大气中的氧气或水分所影响至此制备获得有机电致发光器件。
[0186][0187][0188]
应用例2-20
[0189]
分别以本发明实施例2-20中的化合物25、28、29、30、42、53、54、63、69、75、95、107、108、119、147、165、171、191、203作为绿光主体材料,其他部分与应用例1一致,据此制作出应用例2-20的有机电致发光器件。
[0190]
对照例1-4:
[0191]
与应用例1的区别在于,分别用cn113105437a中h1-55、h1-67以及cn113480527a中的化合物2和3代替本技术化合物14作为绿光主体材料,其余与应用例1相同。
[0192]
上述应用例制造的有机电致发光器件及对照例制造的有机电致发光器件的特性是在电流密度为10ma/cm2的条件下测定的,结果如表2所示。
[0193]
表2:
[0194][0195][0196]
由如上表2可知,本发明化合物应用于有机电致发光器件中,在相同电流密度下,发光效率得到了较大幅度的提升,器件的启动电压有所下降,器件的功耗相对降低,使得器件的寿命相应提高。
[0197]
分别将对照例1-4、应用例1-10所制备的有机电致发光器件进行发光寿命测试,得
到发光寿命t97%数据(发光亮度降低至初始亮度97%的时间),测试设备为teo发光器件寿命测试系统。结果如表3所示:
[0198]
表3:
[0199][0200][0201]
由上表3可知,将本发明化合物应用于有机电致发光器件中,在相同电流密度下,使用寿命得到较大程度提升,具有广阔的应用前景。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1