一种基于高通量测序的假单胞菌种水平快速检测方法及其应用
1.本发明涉及一种基于高通量测序的假单胞菌种水平快速检测方法及其应用,属于分子生物学技术领域。
背景技术:
2.原料乳在生产过程中会受到一系列微生物污染。因此,冷链被广泛采用抑制原料乳中微生物的生长和繁殖,确保乳品质量。嗜冷菌(最适生长温度15-20℃,并能够在7℃以下普遍存在的细菌)作为冷藏原料乳中主要微生物,在冷链条件下生长并产生耐热的水解酶,成为制约现代乳制品质量的“短板”,因此对嗜冷菌的研究、检测和防控已成为现代乳企乃至食品企业关注的重点。假单胞菌属是牛奶嗜冷菌中最主要的属,在低温下生长,高表达耐受热处理的蛋白酶和脂肪酶。这些酶能够耐受热处理,成为巴氏杀菌和超高温瞬时灭菌(uht)后牛奶质量下降(蛋白凝胶化分层、脂肪上浮、腐败气味和苦味等)的主要因素。此外,由于假单胞菌生态位的多样性,不同种之间在生长条件、代谢特点、腐败潜力等方面不尽相同。因此,开发快速检测假单胞菌菌种的方法显得越发重要,甚至需要鉴定至种的水平,来评估其在原料乳中的多样性和组成,以了解其对牛奶品质的影响程度(doi:10.1017/s0022029919000645)。
3.基于传统培养的方法如cfc选择性培养基进行分类学鉴别的方法能够确定种类,但实验周期长,无法分析假单胞菌在原料乳中的相对丰度。在此基础上出现了一些对假单胞菌为主的嗜冷菌检测手段,如流式细胞术、直接荧光过滤法、atp生物荧光法、氨肽酶活性测定、elisa、电子阻抗法、生物传感器法。近年来,随着分子生物学方法的发展,实时定量pcr、荧光原位杂交被广泛用于菌种快速检测。特别是基于16s rdna基因的新一代测序技术的出现,使得大量的样品可以进行更深入的检测,但基于16s rdna的分型方法不足以对假单胞菌进行种水平区分,需要更高分辨率的分子标记才能满足不同种及亚种水平鉴定。
4.过往有研究通过系统基因组学中的多序列位点分型(mlst)的手段,通过串联4个管家基因(16srdna、gyrb、rpob、rpod)将227种假单胞菌分为荧光假单胞菌和铜绿假单胞菌两个谱系,并进一步分为14个物种群,(doi:10.1111/j.1462-2920.2010.02181.x,10.3390/genes11020139)。此外,adna、filc、aprx等基因也被用于针对假单胞菌属腐败潜力的预测分析。(doi:10.1111/1750-3841.13845,doi:10.1111/1750-3841.12645)最新的研究比较了基于16s rdna的高通量测序方法和maldi-tof ms方法鉴别牛奶中假单胞菌种的能力,两者在检测到的具体菌种上各有侧重,但是无法兼顾检测的精度和速度,揭示出两种方法在针对假单胞菌的检测中的不足之处,故仍无法作为完善的方法用于分析样品中的假单胞菌(doi:10.1016/j.idairyj.2019.06.001)。有研究通过建立荧光定量pcr(qpcr)和实时荧光环介导等温扩增检测技术(lamp)能够检测到猪肉中优势假单胞菌,能够在2.5h内产生结果(专利公开号cn109593868a)。另有研究提出了生鲜肉中假单胞菌属细菌的定量检测方法,用于区分假单胞菌与其他属(专利公开号cn109593868a)。
5.此外,根据调查,国内有关假单胞菌种检测或者方法报道的专利只集中在常见的几种。铜绿假单胞菌(专利号cn110470839a)、恶臭假单胞菌(专利号cn104374914a)、牛乳中假单胞菌属(专利号cn101798591b)、变形假单胞菌(专利号cn107400650a)、椰毒假单胞菌(专利号cn112695075a)、鱼香假单胞菌(专利号cn103276093b)),国际上有关于假单胞菌种水平检测的报导也非常少。还没有一种方法能够同时对牛奶等其他复杂样品中假单胞菌实现快速、灵敏、特异性的检测。
技术实现要素:
6.本发明通过分子生物学方法获取假单胞菌种的核心基因序列,在此基础上,对这些核心基因进行比对,选取一段具有高度分辨率的核心基因片段mreb基因为筛选标记,并结合illumina miseq高通量测序检测技术的使用,首次开发了一种假单胞菌种的快速检测技术。为解决现有技术存在的上述缺陷,本发明选取mreb基因(细胞骨架蛋白合成基因,一种编码细胞结构形成的管家基因10.1111/j.1365-2958.2010.07132.x)上的一段片段,以该片段为基础设计引物,并通过高通量测序技术在种水平进行假单胞菌的检测和鉴定。该方法可用于快速、灵敏和特异地检测复杂样品中不同假单胞菌的组成。
7.本发明提供了一种鉴定假单胞菌种的方法,所述方法是以mreb基因片段为标记物,以seq id no.1和seq id no.2所示序列为引物,扩增获得标记物基因片段的菌株为假单胞菌。
8.在一种实施方式中,所述标记物基因片段的大小为410~462bp bp。
9.在一种实施方式中,所述扩增是对含有微生物基因组的对象进行扩增。
10.在一种实施方式中,所述对象包括基因组dna提取物、菌悬液或单菌落。
11.在一种实施方式中,所述基因组dna包括但不限于从食品、空气、水、土壤、人体中提取获得。
12.在一种实施方式中,所述扩增是以含待测样品的基因组dna为模板进行扩增。
13.在一种实施方式中,所述扩增是以单菌落为模板进行扩增。
14.本发明提供了一种鉴定样品中假单胞菌种水平组成的方法,所述方法包括:(1)构建假单胞菌mreb基因标准文库;(2)以seq id no.1和seq id no.2所示序列为引物,对复杂样品基因组中mreb基因序列进行扩增;(3)将步骤(2)扩增结果进行回收、建库、测序;(4)将测序结果与步骤(1)构建的假单胞菌mreb基因标准文库比对,鉴定、分析复杂样品中假单胞菌种水平的组成特征。
15.在一种实施方式中,所述假单胞菌mreb基因标准文库含有genbank数据库中可检索到的mreb基因序列。
16.在一种实施方式中,所述方法包含以下步骤:
17.(1)提取待测样品的基因组dna;
18.(2)设计引物,正向引物如seq id no.1所示,反向引物如seq id no.2所示:
19.正向引物(seq id no.1):5
’‑
acccttatttacgtgcgcga-3’20.反向引物(seq id no.2):5
’‑
atrtcsacsaccatcgarc-3’21.(3)采用步骤(2)中所述的正向引物和反向引物,以步骤(1)中所提取的基因组dna为模板进行pcr扩增,然后对pcr产物进行纯化;
22.(4)对步骤(3)中纯化后pcr产物进行定量,并等量混样,构建miseq测序标准文库,然后进行高通量测序;
23.(5)根据步骤(4)中测序的结果,得到待测复杂样品或者混菌样品的mreb基因序列信息,以此为基础对待测样品中不同种假单胞菌进行检测。
24.本发明提供了一种鉴定假单胞菌种的试剂盒,所述试剂盒包含seq id no.1和seq id no.2所示的引物对。
25.在一种实施方式中,所述试剂盒中含有如seq id no.1所示的正向引物和如seq id no.2所示的反向引物。
26.本发明提供了一种筛选假单胞菌的方法,将待筛选的样品制成菌悬液,稀释、涂布于固体培养基上,培养至形成单菌落,再以seq id no.1和seq id no.2所示序列为引物,对mreb基因的序列进行扩增,扩增获得大小为410~462bp基因片段的菌株为假单胞菌。
27.在一种实施方式中,所述固体培养基包括但不限于cfc选择性培养基或nb营养肉汁培养基。
28.本发明的有益效果:与现有方法比较,该方法可以检测到复杂样品中除少数罕见菌种外的全部假单胞菌种(100余种),检测准确率达到100%。本发明的方法可实现高通量鉴定,批量处理菌株信息,无需分离纯化即可全面鉴定复杂样品中的假单胞菌组成,并且可以鉴定至种水平,分辨率高于16srdna方法。因此,基于本方法对于假单胞菌进行检测,结果较常规鉴定方法更精准。
29.传统的16s rrna条带为1500bp,要双向测序且需要拼接,而通过基于mreb基因的特异性引物扩展目的条带410~462bp,满足illumina miseq的测序读长要求,经过单向鉴定即可完成假单胞菌的鉴定;此外,16s rrna在细菌基因组中是非单克隆拷贝基因(即在基因组中不止一条),因此在扩增过程中同一条基因组可能会产生多个拷贝序列;而mreb基因在革兰氏阴性菌中均为单克隆拷贝基因(即在基因组中只有一条),在扩增过程中同一条基因组只产生一条拷贝序列,与菌落数有相关性。所以,二代测序进行测序的时候,用mreb基因作为引物针对复杂样品的鉴定可以节约50%以上的假单胞菌鉴定时间和鉴定成本,检测速度和效率更快,检测结果也更准确。
附图说明
30.图1是以mreb基因为基础的假单胞菌系统发育树。
31.图2是所选13株假单胞菌和10株非假单胞菌pcr扩增产物电泳图;其中,m代表marker(100bp),1-13分别为铜绿假单胞菌(pseudomonas aeruginosa)、荧光假单胞菌(pseudomonas fluorescens)、恶臭假单胞菌(pseudomonas putida)、青枯假单胞菌(pseudomonas solanacearum)、产碱假单胞菌(pseudomonas alcaligenes)、食油假单胞菌(pseudomonas oleovorans)、莓实假单胞菌(pseudomonas fragi)、变形假单胞菌(pseudomonas plecoglossicida)、耐冷假单胞菌(pseudomonas psychrotolerans)、猴假单胞菌(pseudomonas simiae)、隆德假单胞菌(pseudomonas ludensis)、草假单胞菌(pseudomonas poae)、丁香假单胞菌(pseudomonas syringae);14-23分别为地衣芽孢杆菌(bacillus licheniformis)、枯草芽孢杆菌(bacillus subtilis)、解淀粉芽孢杆菌(bacillus amyloliquefaciens)、黄杆菌(microbacterium maritypicum)、大肠杆菌
(escherichia coli)、发酵乳杆菌(lactobacillus fermentum)、粪肠球菌(enterococcus faecalis)、鲍曼不动杆菌(acinetobacter baumanii)、金黄葡萄球菌(staphylococcus aureus)、消化嗜冷杆菌(psychrobacter alimentarius)。
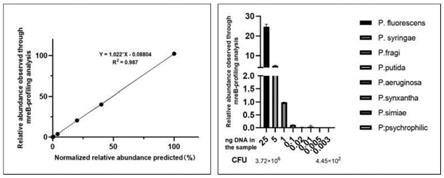
32.图3是以mreb基因为基础设计引物的准确度;通过将具有代表性的不同种假单胞菌以设计好的不同比例进行混合,然后以所设计引物扩增后进行高通量测序,最后将测序得到的结果与预先混合的不同假单胞菌的比例进行对比得到方法的准确度。
33.图4是实施例中牛奶样品中假单胞菌种水平构成图。
34.图5是实施例中待测牛奶样品中的pcr扩增产物的电泳检测图,1-8为不同样本。
35.图6是qpcr扩增样品基因组的溶解曲线。
36.图7是以16s rrna基因的v3-v4区为基础的常见假单胞菌系统发育树。
具体实施方式
37.实施例1:基于假单胞菌高分辨率核心基因构建假单胞菌特异性引物
38.1、收集并总结了在美国生物技术信息中心(national center for biotechnology information,ncbi)和欧洲分子生物学实验室(european molecular biology laboratory,embl)数据库中有文献报导过、完成了全基因组测序的142种假单胞菌,并按其测序完整度每种选取多条、总共下载了总计429条全基因组序列(仅有一株菌的种只能下载到一条)采用prokka软件对这些基因组进行重注释,并通过roary软件进行泛基因组分析。通过mega7软件将注释后的基因分别进行系统发育树构建,筛选得到mreb基因上一个具有区分不同种的能力的片段。如图1所示,由图可知,根据该基因构建的环形进化树中,各个种能够被分到不同的进化枝中。
39.2、构建mreb基因数据库,从ncbi和embl等数据库下载已知假单胞菌的mreb基因序列,利用所下载序列构建mreb基因比对数据库。所构建mreb基因序列数据库适用于目前已知的所有假单胞菌(表1)。
40.表1已知所有假单胞菌mreb基因序列信息
41.42.[0043][0044]
表2基于prokka重注释后得到的mreb基因(已上传ncbi)
[0045]
[0046]
[0047]
[0048]
[0049]
[0050]
[0051]
[0052]
[0053]
[0054]
[0055]
[0056]
[0057]
[0058]
[0059]
[0060]
[0061]
[0062]
[0063]
[0064]
[0065]
[0066]
[0067]
[0068]
[0069]
[0070]
[0071]
[0072]
[0073]
[0074]
[0075]
[0076]
[0077]
[0078]
[0079]
[0080]
[0081]
[0082]
[0083]
[0084]
[0085]
[0086]
[0087]
[0088]
[0089]
[0090]
[0091]
[0092]
[0093][0094]
3、对mreb基因保守序列和特异序列进行充分分析后,选取特定序列(以mreb全段基因为模板(全长1038bp),选择其中一段具有高度分辨率的片段)设计引物,并在这些引物对中挑选适合illumina miseq测序平台读长的引物对,最终确定seq id no.1所示的正向引物和seq id no.2所示的反向引物为最终测序引物。
[0095]
基于mreb基因设计并合成假单胞菌特异性引物序列:
[0096]
正向引物(seq id no.1):5
’‑
acccttatttacgtgcgcga-3’,
[0097]
反向引物(seq id no.2):5
’‑
atrtcsacsaccatcgarc-3’;
[0098]
注:上述引物中的r表示碱基a和g,s表示碱基g和c。
[0099]
利用primer-blast(https://www.ncbi.nlm.nih.gov/)进行模拟pcr,结果表明这对引物仅产生假单胞菌基因组的扩增子,这说明该引物具有较好的假单胞菌特异性,因此本发明所设计的引物确保了后续扩增的效率及鉴定的准确性。
[0100]
4、对于全部纳入的假单胞菌基因组数据库,采用blast方法验证特异性序列的真实性。完成blast验证后,对目的基因序列进行基于美国生物技术信息中心(national center for biotechnology information,ncbi)数据库中nr/nt库的微生物基因信息全库的blast检索,进一步验证识别的菌种特异性序列仅在目标菌中存在,而在其他已公布的微生物基因信息中缺失。
[0101]
实施例2:假单胞菌特异性引物在属水平上的验证
[0102]
选取原料乳中报导较多的13种假单胞菌,铜绿假单胞菌(pseudomonas aeruginosa)、荧光假单胞菌(pseudomonas fluorescens)、恶臭假单胞菌(pseudomonas putida)、青枯假单胞菌(pseudomonas solanacearum)、产碱假单胞菌(pseudomonas alcaligenes)、食油假单胞菌(pseudomonas oleovorans)、莓实假单胞菌(pseudomonas fragi)、变形假单胞菌(pseudomonas plecoglossicida)、耐冷假单胞菌(pseudomonas psychrotolerans)、猴假单胞菌(pseudomonas simiae)、隆德假单胞菌(pseudomonas ludensis)、草假单胞菌(pseudomonas poae)、丁香假单胞菌(pseudomonas syringae),以及10种非假单胞菌属原料乳常见污染菌地衣芽孢杆菌(bacillus licheniformis)、枯草芽孢杆菌(bacillus subtilis)、解淀粉芽孢杆菌(bacillus amyloliquefaciens)、黄杆菌(microbacterium maritypicum)、大肠杆菌(escherichia coli)、发酵乳杆菌(lactobacillus fermentum)、粪肠球菌(enterococcus faecalis)、鲍曼不动杆菌(acinetobacter baumanii)、金黄葡萄球菌(staphylococcus aureus)、消化嗜冷杆菌
(psychrobacter alimentarius)。
[0103]
分别提取上述菌株的基因组dna,微生物基因组提取参照细菌基因组dna提取试剂盒(天根生化科技(北京)有限公司,北京,中国)中说明书,用序列如seq id no.1和seq id no.2所示的引物,以提取的基因组为模板进行pcr扩增,扩增条件如下:
[0104]
①
pcr扩增反应体系的组成为:基因组dna模板1μl、2
×
taq mix 25μl(takara)、10um的正向引物和反向引物各lμl,加ddh2o至50μl。
[0105]
②
pcr扩增反应条件为:95℃预变性8min,然后以95℃变性40s、59℃退火40s、72℃延伸40s为一个循环,进行34个循环,最后72℃延伸8min。
[0106]
pcr结果如图3所示,地衣芽孢杆菌(bacillus licheniformis)、枯草芽孢杆菌(bacillus subtilis)、解淀粉芽孢杆菌(bacillus amyloliquefaciens)、黄杆菌(microbacterium maritypicum)、大肠杆菌(escherichia coli)、发酵乳杆菌(lactobacillus fermentum)、粪肠球菌(enterococcus faecalis)、鲍曼不动杆菌(acinetobacter baumanii)、金黄葡萄球菌(staphylococcus aureus)、消化嗜冷杆菌(psychrobacter alimentarius)均无法扩增出目的条带,铜绿假单胞菌(pseudomonas aeruginosa)、荧光假单胞菌(pseudomonas fluorescens)、恶臭假单胞菌(pseudomonas putida)、青枯假单胞菌(pseudomonas solanacearum)、产碱假单胞菌(pseudomonas alcaligenes)、食油假单胞菌(pseudomonas oleovorans)、莓实假单胞菌(pseudomonas fragi)、变形假单胞菌(pseudomonas plecoglossicida)、耐冷假单胞菌(pseudomonas psychrotolerans)、猴假单胞菌(pseudomonas simiae)、隆德假单胞菌(pseudomonas ludensis)、草假单胞菌(pseudomonas poae)、丁香假单胞菌(pseudomonas syringae),扩增出大小为410~462bp的目的条带。
[0107]
将pcr产物胶回收(胶回收试剂盒购自天根生化科技有限公司公司)、测序,采用美国thermo fisher科技公司varioskan lux多功能微孔板读数仪进行定量,并等量混样,用illuminatruseq dna lt sample preparation kit试剂盒进行标准文库构建,并在illumina miseq测序平台进行上机测序,并与实施例1构建的基因文库进行比对,对待测样品中假单胞菌的属进行检测。
[0108]
经验证,该比对结果与初始选定的菌株种属分类的信息一致。本发明的方法能够有效地将假单胞菌属的菌株与其它属的菌株区分,测序后的比对结果能够100%鉴定假单胞菌种。
[0109]
同时通过qpcr方法对样本基因组进行扩增,单一的溶解曲线证明该引物的特异性以及自身无错配和引物二聚体等影响扩增结果的现象发生,如图6。
[0110]
实施例3:假单胞菌快速检测方法的构建及其在种水平上的验证
[0111]
为了验证引物seq id no.1和seq id no.2在分析假单胞菌中的准确性,将铜绿假单胞菌(pseudomonas aeruginosa)、荧光假单胞菌(pseudomonas fluorescens)、恶臭假单胞菌(pseudomonas putida)、莓实假单胞菌(pseudomonas fragi)、变形假单胞菌(pseudomonas plecoglossicida)、丁香假单胞菌(pseudomonas syringae)、猴假单胞菌(pseudomonas simiae)共7种假单胞菌(如表3)基因组按照浓度0.003-25ng/μl进行混合,用于模拟复杂样品中不同种假单胞菌的检测。基于新设计的假单胞菌鉴定引物并结合二代测序仪,然后按照实施例2所述方法步骤进行模拟样品测序,最后对测序所得mreb基因序列
信息与所构建数据库(实施例1中构建)进行比对。由于每一种菌株dna在进行混合之前经过浓度检测,具体见表3,则每一株菌dna含量确定,经过假单胞菌检测方法进行检测,理论上每一种菌株dna浓度所能读取的数量应与其对应的混合样品中dna浓度具有一定的相关性。此外,由于菌株dna浓度测定,最终比对的结果应与对应浓度的菌株一致,因此这一步骤还可以检测本检测方法的准确性。
[0112]
表3假单胞菌快速检测方法的构建及其在种水平上的验证
[0113][0114]
将依据高通量测序方法得到的假单胞菌不同物种的含量与模拟样品中预先混的假单胞菌含量比较得到如图2的结果,从图2我们可以看出,利用引物seq id no.1和seq id no.2测序到的假单胞菌物种的含量与预先混入的假单胞菌物种含量具有很好的一致性(y=1.0076x-0.1012,r2=0.9974),因此,该高通量测序方法在检测假单胞菌物种上具有较高的准确性。
[0115]
因此本发明提供的基于高通量测序检测不同种假单胞菌的方法适用于对复杂样品的mreb基因序列信息与所构建数据库中已知物种的mreb基因序列比对,从而得出复杂样品中不同种的假单胞菌。
[0116]
实施例4:假单胞菌快速检测方法在原料乳样品中的应用
[0117]
具体实施方式参见实施例2,样品为取自奶厂的原料乳中提取的基因组dna,提取时采用美国mpbiomedicals公司的dna提取试剂盒进行,具体操作步骤以该试剂盒操作说明书为准。
[0118]
对测序所得mreb基因序列信息与所构建数据库(实施例1中构建)进行比对,检测结果如附图4所示,图4展示了样本中假单胞菌的组成结构。基于现有文库比对到99%以上的序列(assigned),荧光假单胞菌具有显著丰度优势,草假单胞菌、隆德假单胞菌、铜绿假单胞菌、恶臭假单胞菌丰度依次降低,另有16种被检测到少量存在,与文献报导相符。上述数据说明,所述方法可以快速、准确的检测原料乳内的假单胞菌组成。
[0119]
对照例1:16s rrna v3-v4对于假单胞菌鉴定的准确性验证
[0120]
采用16s rrna作为标记物,构建牛乳中常见假单胞菌的系统发育树(分别利用ncbi和embl数据库下载假单胞菌不同菌株16s rrna基因序列,通过mega软件利用neighbor-joining距离算法构建系统发育树)。从图7可以看出,16sr rna基因的v3-v4区鉴定分类结果不够准确,无法区分某些种,例如pseudomonas taiwanensis和pseudomonas veronii。
[0121]
虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权利要求书所界定的为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1