1.本发明属于新型非天然核苷酸类似物的合成技术领域,具体涉及一类非天然碱基三磷酸及其合成方法和应用。
背景技术:2.非天然碱基对dnam-dtpt3可以在体内完成遗传信息的存储和检索,并产生治疗性蛋白。但是非天然碱基的光敏性会对健康细胞的生长存在威胁,本发明通过化学方法改进dtpt3的结构来优化非天然碱基的稳定性。
3.
技术实现要素:4.本发明解决的技术问题是提供了一类非天然碱基三磷酸及其合成方法和应用,利用该方法合成了两种非天然碱基三磷酸类似物,这两种非天然碱基三磷酸类似物光稳定性好且体外扩增效率高,能够用于测序。
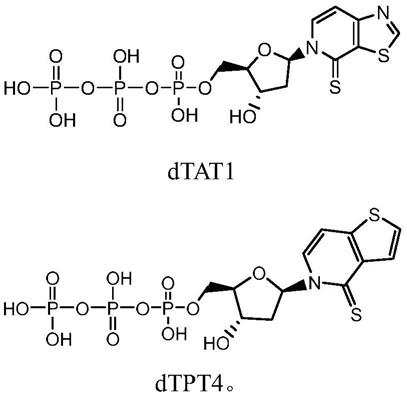
5.本发明为解决上述技术问题采用如下技术方案,一类非天然碱基三磷酸,其特征在于该非天然碱基三磷酸的结构式为:
[0006][0007]
本发明所述的非天然碱基三磷酸的合成方法,其特征在于具体步骤为:
[0008]
dtat1的合成:以噻唑-4-甲醛、丙二酸为原料合成化合物a;以化合物a、叠氮磷酸二苯酯、三乙胺为原料合成化合物b;化合物b以二苯醚为溶剂合成化合物c;以化合物c、n,o-双三甲硅基乙酰胺、氯代糖、四氯化锡为原料合成化合物d;以化合物d、劳森试剂为原料合成化合物e;以化合物e、甲醇钠为原料合成化合物f;以化合物f、三氯氧磷、焦磷酸盐为原
料合成目标产物dtat1,具体的合成路线为:
[0009][0010]
dtpt4的合成:以2-噻吩甲醛、丙二酸为原料合成化合物
①
;以化合物
①
、叠氮磷酸二苯酯和三乙胺为原料合成化合物
②
;化合物
②
以二苯醚为溶剂合成化合物
③
;以化合物
③
、n,o-双三甲硅基乙酰胺、氯代糖、四氯化锡为原料合成化合物
④
;以化合物
④
、劳森试剂为原料合成化合物
⑤
;以化合物
⑤
、甲醇钠为原料合成化合物
⑥
;以化合物
⑥
、三氯氧磷、焦磷酸盐为原料合成目标产物dtpt4,具体的合成路线为:
[0011][0012]
进一步限定,化合物d或化合物
④
与劳森试剂的反应以甲苯为溶剂,且反应过程在氮气保护下加热至120℃。
[0013]
进一步限定,化合物e或化合物
⑤
与甲醇钠的反应以甲醇为溶剂,且常温反应。
[0014]
进一步限定,化合物f或化合物
⑥
与三氯氧磷、焦磷酸盐的反应以磷酸三甲酯为溶剂,且反应过程在氮气保护下于-20℃冰盐浴反应,焦磷酸盐用n,n-二甲基甲酰胺作为溶剂溶解后加入。
[0015]
本发明所述的非天然碱基三磷酸的测序应用。
[0016]
本发明设计并合成了两个以tpt3的基本结构骨架为基础,但在不同位点修饰噻吩环的tpt3类似物,以提高非天然碱基对的光稳定性,并建立了包括光敏实验、动力学实验、体外pcr复制的综合筛选策略。一对新的dnam-dtat1在体内和体外与dnam-dtpt3对本身几乎具有同样的效率和保真度,且更具光稳定性和热稳定性。
附图说明
[0017]
图1a、图1b、图1c分别是dtpt3/dtpt4/dtat1三种样品的光稳定性曲线;
[0018]
图2是dtpt3/dtpt4/dtat1三种样品的hieff酶测序图;
[0019]
图3是dtpt3/dtpt4/dtat1三种样品的one tap酶测序图;
[0020]
图4是dtpt3/dtpt4/dtat1三种样品的tap酶测序图;
[0021]
图5是dtpt3/dtpt4/dtat1三种样品的插入胶图;
[0022]
图6是dtpt3/dtpt4/dtat1三种样品的延伸胶图。
具体实施方式
[0023]
以下通过实施例对本发明的上述内容做进一步详细说明,但不应该将此理解为本发明上述主题的范围仅限于以下的实施例,凡基于本发明上述内容实现的技术均属于本发明的范围。
[0024]
实施例1
[0025][0026]
具体合成步骤:
[0027][0028]
在氮气保护下,将化合物c(100mg,0.66mmol)、干燥的二氯甲烷2ml、n,o-双(三甲
基硅基)乙酰胺(146.7mg,0.72mmol)依次加入到25ml圆底烧瓶中,室温下搅拌1h。将3,5-二邻(对甲苯基)-2-脱氧-呋喃核糖酰氯(280mg,0.72mmol)加入到反应体系中,将反应体系移入到冰水浴中并加入无水四氯化锡(85.4mg,0.33mmol),继续室温搅拌。用tlc监测反应,待化合物3c不再变化,加入饱和碳酸氢钠溶液淬灭反应,用二氯甲烷和水萃取,合并有机相并用无水硫酸钠干燥,用减压装置除去溶剂。通过柱层析分离得到白色固体化合物d(α,β)混合物且质量为215.5mg,产率为65%。βanomer 1
h nmr(600mhz,cdcl3)δ9.11(s,1h),7.96(d,j=8.2hz,4h),7.63(d,j=8.2hz,1h),7.28(d,j=4.6hz,4h),7.11(d,j=8.0hz,1h),6.78(dd,j=8.0,5.7hz,1h),5.68
–
5.62(m,2h),4.78(dd,j=12.2,3.2hz,1h),4.72(dd,j=12.2,3.6hz,1h),2.96
–
2.92(m,1h),2.42(s,3h),2.39(s,3h),2.38
–
2.36(m,1h).
13
c nmr(151mhz,cdcl3)δ166.20,166.14,160.19,159.19,158.04,144.52,144.39,129.91,129.63,129.39,129.33,126.66,126.45,124.51,103.82,85.93,83.22,75.00,64.16,39.36,21.79,21.74。
[0029][0030]
在氮气保护下,将化合物d(α,β)(110mg,0.22mmol)、干燥的甲苯5ml、劳森试剂(105.2mg,0.26mmol)依次加入到25ml圆底烧瓶中,装上回流管并用塞子密封,放置于120℃油浴。用tlc监测反应,待化合物d(α,β)不再变化,用减压装置除去溶剂。通过柱层析分离得到淡黄色固体化合物e(α,β)混合物且质量为62.4mg,产率为55%。βanomer 1
h nmr(400mhz,cdcl3)δ9.16(s,1h),8.26(d,j=7.6hz,1h),8.00
–
7.96(m,4h),7.62(d,j=8.2hz,1h),7.35(s,1h),7.29(t,j=7.2hz,4h),5.67
–
5.63(m,2h),4.86(d,j=9.4hz,1h),4.78(t,j=6.2hz,1h),3.34
–
3.29(m,1h),2.44(s,6h),2.33
–
2.29(m,1h).
13
cnmr(151mhz,cdcl3)δ173.85,166.25,166.22,163.15,151.83,144.67,144.64,141.52,132.20,130.01,129.69,129.52,126.58,126.40,109.41,91.36,83.92,74.42,64.00,39.08,29.81,21.87,21.82。
[0031][0032]
将化合物e(α,β)(90mg,0.17mmol)、甲醇5ml、甲醇钠(37.8mg,0.7mmol)依次加入到25ml圆底烧瓶中,室温下搅拌。用tlc监测反应,待化合物e(α,β)反应完全,停止反应,用减压装置除去溶剂。通过柱层析分离得到化合物f(α)和f(β)的混合物,然后通过高效液相色谱分离,得到化合物淡黄色固体f(α)和f(β)且质量分别为14.8mg和22.3mg,产率分别是30%和45%。βanomer 1
h nmr(400mhz,cdcl3)δ9.16(s,1h),8.26(d,j=7.6hz,1h),8.00
–
7.96(m,4h),7.62(d,j=8.2hz,1h),7.35(s,1h),7.29(t,j=7.2hz,4h),5.67
–
5.63(m,2h),4.86(d,j=9.4hz,1h),4.78(t,j=6.2hz,1h),3.34
–
3.29(m,1h),2.44(s,6h),2.33
–
2.29(m,1h).
13
c nmr(151mhz,cdcl3)δ173.85,166.25,166.22,163.15,151.83,144.67,
144.64,141.52,132.20,130.01,129.69,129.52,126.58,126.40,109.41,91.36,83.92,74.42,64.00,39.08,29.81,21.87,21.82。
[0033][0034]
将化合物f(β)(10mg,0.035mmol)、磷酸三甲酯160μl、1,8-双二甲氨基萘(10.1mg,0.046mmol)依次加入到10ml圆底烧瓶中,并放置于-15℃的冰盐浴中,然后加入三氯氧磷(7.1mg,0.046mmol)并在-15℃保持3h。将三(四丁基铵)氢焦磷酸(162.4mg,0.18mmol溶于350μl dmf)、三正丁胺(38.9mg,0.21mmol)依次加入到反应体系中,并在-10℃和-5℃分别保持10分钟,在0℃保持5分钟,最后加入0.5m的teab缓冲液淬灭反应。先用deae sephadex a25分离得到粗产物,再通高效液相色谱分离得到淡黄色固体化合物g且质量为1.8mg,产率为10%。
31
p nmr(243mhz,d2o)δ-10.45(s),-11.32(d,j=18.7hz),-22.72(s)。ms(maldi(m/z):[m-h]-calcd for c11h14n2o12p3s2-,523.28,found,522.896。
[0035]
实施例2
[0036][0037]
具体合成步骤:
[0038][0039]
在氮气保护下,将化合物
③
(300mg,1.98mmol)、干燥的二氯甲烷3ml、n,o-双(三甲基硅基)乙酰胺(444mg,2.19mmol)依次加入到50ml圆底烧瓶中,室温下搅拌40min。将3,5-二邻(对甲苯基)-2-脱氧-呋喃核糖酰氯(850mg,2.18mmol)加入到反应体系中,将反应体系移入到冰水浴中并加入无水四氯化锡(258mg,0.99mmol),继续室温搅拌。用tlc监测反应,待化合物
③
不再变化,加入饱和碳酸氢钠溶液淬灭反应,用二氯甲烷和水萃取,合并有机相并用无水硫酸钠干燥,用减压装置除去溶剂。通过柱层析分离得到白色固体化合物
④
质量
为280mg,产率为28%。βanomer 1
h nmr(400mhz,cdcl3)δ7.85(dd,j1=8hz,j2=30,4h),7.56(d,j=5.6hz,2h),7.46(d,j=7.6hz,2h),7.19-7.16(m,3h),7.11(d,j=8hz,2h),6.73(dd,j1=5.6,j2=8hz,1h),6.53(d,j=4hz,1h),5.55(d,j=6.4hz,1h),4.65(ddd,j1=3.2,j2=12.4,j3=20.4hz,2h),4.52(d,j=2.8hz,1h),2.86(dd,j1=3.2,j2=13.6hz,1h),2.32(d,j=12.8hz,6h),2.23(dd,j1=7.6,j1=14.4hz,1h).
13
c nmr(101mhz,cdcl3)δ166.24,166.19,158.60,147.69,144.46,144.29,130.56,130.17,129.92,129.83,129.66,129.36,129.32,129.19,129.12,126.74,126.54,126.11,125.16,124.74,102.48,85.66,83.01,75.22,64.41,39.27,21.79,21.75。
[0040][0041]
在氮气保护下,将化合物
④
(250mg,0.5mmol)、干燥的甲苯25ml、劳森试剂(300mg,0.75mmol)依次加入到100ml圆底烧瓶中,装上回流管并用塞子密封,放置于120℃油浴。用tlc监测反应,待化合物
④
不再变化,用减压装置除去溶剂。通过柱层析分离得到淡黄色固体化合物
⑤
混合物且质量为101mg,产率为39%。βanomer 1
h nmr(600mhz,cdcl3)δ8.04(t,j=3.6hz,2h),7.99(d,j=8.4hz,2h),7.89(d,j=7.8hz,2h),7.48(dd,j1=6,j2=7.2hz,1h),7.40(d,j=5.4hz,1h),7.28(d,j=8.4hz,2h),7.21(d,j=7.8hz,2h),6.99(d,j=7.8hz,1h),5.64(m,1h),4.83(dd,j1=3,j2=12hz,1h),4.76(dd,j1=3,j2=12hz,1h),4.68(q,j=3,1h),3.36(ddd,j1=2.4,j2=5.4,j3=14.4hz,1h),2.24(d,j=20.4hz,6h),2.27(quint,j=7.2hz,1h).
13
c nmr(151mhz,cdcl3)δ175.82,166.29,144.60,144.52,142.86,142.27,130.04,129.72,129.48,129.42,129.27,128.18,126.84,126.69,126.51,108.06,91.14,83.67,74.66,64.21,38.97,21.88,21.83。
[0042][0043]
将化合物
⑤
(101mg,0.19mmol)、甲醇5ml、甲醇钠(37.8mg,0.7mmol)依次加入到25ml圆底烧瓶中,室温下搅拌。用tlc监测反应,待化合物
⑤
反应完全,停止反应,用减压装置除去溶剂。通过柱层析分离得到淡黄色固体
⑥
质量为46mg,产率分别是83%。βanomer 1
h nmr(400mhz,meod)δ8.40(d,j=7.2hz,1h),7.91(dd,j1=0.8,j2=6.4hz,1h),7.57(d,j=5.6hz,1h),7.35(m,2h),4.42(m,1h),4.06(q,j=4hz,1h),3.92(dd,j1=3.2,j2=12hz,1h),3.84(dd,j1=4,j2=12hz,1h),2.80-2.74(m,1h),2.14-2.07(m,1h).
13
c nmr(101mhz,meod)δ176.18,144.64,142.88,130.82,129.55,128.23,109.03,92.12,89.44,71.35,62.31,42.53。
[0044][0045]
将化合物
⑥
(10mg,0.035mmol)、磷酸三甲酯160μl、1,8-双二甲氨基萘(10.1mg,0.046mmol)依次加入到10ml圆底烧瓶中,并放置于-15℃的冰盐浴中,然后加入三氯氧磷(7.1mg,0.046mmol)并在-15℃保持3h。将三(四丁基铵)氢焦磷酸(162.4mg,0.18mmol溶于350μl dmf)、三正丁胺(38.9mg,0.21mmol)依次加入到反应体系中,并在-10℃和-5℃分别保持10分钟,在0℃保持5分钟,最后加入0.5m的teab缓冲液淬灭反应。先用deae sephadex a25分离得到粗产物,再通高效液相色谱分离得到淡黄色固体化合物dtpt4且质量为1.8mg,产率为10%。
31
p nmr(243mhz,d2o)δ-10.82,-10.91,-11.42,-11.51,-23.22,-23.30,-23.38。ms(maldi(m/z):[m-h]-calcd for c12h15no12p3s2-,521.9254,found,521.961。
[0046]
光稳定性
[0047]
将dtpt3/dtpt4/dtat1的样品分别放置于365nm、紫光、蓝光、绿光和红光分别照射0.25h、0.5h、0.75h、1h、1.25h,通过hplc观察其降解情况从而判断合成的两种非天然碱基的光稳定性,得知tat1的光稳定性优于tpt3,结果如图1所示。
[0048]
与非天然碱基dnam配对体外扩增测序结果
[0049]
将dtpt3/dtpt4/dtat1分别和dnam以及不同的dna聚合酶加入pcr体系,运行36个循环之后测序,并对测序结果进行保真度计算,计算得出dtat-1和dtpt4都可以在不同的dna聚合酶中扩增并且具有较好的保真性。而dtat1的保真度优于dtpt3,结果如下:
[0050][0051]
动力学测定dtat1和dtpt4体外扩增效率和保真度结果
[0052]
在引物上加上荧光标记,以dnam为模板,加入非天然碱基三磷酸和模板的下一个天然碱基dctp,通过荧光强度来评估非天然碱基的掺入和延伸效率,可以看出在相近的掺入和延伸效率下,dtat1和dtpt4的错误率更低。
[0053][0054]
以上实施例描述了本发明的合成路线、合成方法以及该化合物的光稳定性和扩增效率,本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中
描述的只是说明本发明的原理,在不脱离本发明原理的范围下,本发明还会有各种变化和改进,这些变化和改进均落入本发明保护的范围内。