一种制备2,4,5-三氯嘧啶的方法与流程
1.本发明属于农药技术领域,涉及一种制备2,4,5-三氯嘧啶的方法。
背景技术:
2.嘧啶类化合物是重要的生命物质,广泛的存在于生物体内,具有较强的生物活性而受到广泛关注,2,4,5-三氯嘧啶是一种新型活性染料中间体原料和抗菌消炎新化学药的合成原料,随着新型活性染料和抗菌消炎新药销量的不断增加,其合成所需的原料的需求也在不断增长,目前,对应2,4,5-三氯嘧啶的合成大多使用氯气、氯化亚砜、三氯氧磷和光气等,其合成过程繁琐且毒性大,给合成人员的身体健康带来一定的危害。
3.例如,cn102827085a公开了一种2,4,5-三氯嘧啶化合物的制备方法,在溶剂二氯乙烷中,投入5-氯尿嘧啶和氯化亚砜,保温回流反应,回流完毕,加入水,分去水层,然后进行蒸馏,蒸去二氯乙烷,得2,4,5-三氯嘧啶。
4.然而开发一条绿色环保、收率高兼具符合工业化生产的工艺仍为药化工作者孜孜追求的。
技术实现要素:
5.本发明提供了一种制备2,4,5-三氯嘧啶的方法,其包括:1)尿嘧啶与次氯酸钠在酸性条件下反应以形成5-氯尿嘧啶,2)5-氯尿嘧啶与双(三氯甲基)碳酸酯反应以形成2,4,5-三氯尿嘧啶。
6.在一些实施方案中,步骤1)中所用酸性试剂选自但不限于硫酸、盐酸或磷酸。在另一些实施方案中,步骤1)中所用酸性试剂为硫酸。
7.在另一些实施方案中,步骤1)中次氯酸钠的用量为尿嘧啶摩尔量的1.0至2.0当量,可以为1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9、2.0或任意两数之间值。在一些实施方案中,步骤1)中次氯酸钠的用量为尿嘧啶摩尔量的1.05-1.5当量。
8.在一些实施方案中,步骤1)反应所用溶剂为水。
9.另一方面,本发明中步骤2)中双(三氯甲基)碳酸酯的用量为5-氯尿嘧啶摩尔量的0.6至1.0当量,可以为0.6、0.7、0.8、0.9、1.0或任意两数之间值。在一些实施方案中,步骤2)中双(三氯甲基)碳酸酯的用量为5-氯尿嘧啶摩尔量的0.7-1.0当量。
10.在一些实施方案中,步骤2)所用溶剂为极性非质子溶剂。在一些实施方案中,步骤2)所用溶剂为三氯甲烷。在一些实施方案中,步骤2)所用溶剂为二氯甲烷。在一些实施方案中,步骤2)所用溶剂为四氢呋喃。
11.在另一些实施方案中,步骤2)还含有n,n-二甲基甲酰胺或n,n-二甲基乙酰胺。在
一些实施方案中,步骤2)中n,n-二甲基甲酰胺或n,n-二甲基乙酰胺的用量为5-氯尿嘧啶摩尔量的0.01至0.5当量,可以为0.01、0.02、0.03、0.04、0.05或任意两数之间值。
12.进一步地,在一些实施方案中,步骤2)反应温度为50-65℃(回流),优选60-65℃。
13.在一些实施方案中,制备2,4,5-三氯嘧啶的方法,其包括:1.1)向反应器中加入水,冷却,搅拌条件下加入98%硫酸,加完后,冷却至30-35℃,加入尿嘧啶;1.2)冷却、搅拌条件下加入次氯酸钠水溶液,5-25℃搅拌反应至脲嘧啶基本反应完全;1.3)加热95-100℃,然后冷却至5-25℃,过滤,干燥得5-氯尿嘧啶;2.1)向反应器中加入三氯甲烷,5-氯尿嘧啶和n,n-二甲基甲酰胺,搅拌溶解;2.2)搅拌条件下加入双(三氯甲基)碳酸酯,加热至60-65℃反应至5-氯尿嘧啶基本反应完全;2.3)冷却至室温,加入水,分液,浓缩得2,4,5-三氯嘧啶。
14.在另一些实施方案中,制备2,4,5-三氯嘧啶的方法,其包括:1.1)向反应器中加入水,冷却,搅拌条件下加入98%硫酸,加完后,冷却至30-35℃,加入尿嘧啶;1.2)冷却、搅拌条件下加入次氯酸钠水溶液,5-25℃搅拌反应至脲嘧啶基本反应完全(hplc检测,脲嘧啶含量不大于1%);1.3)加热至95-100℃,反应1-4h,然后冷却至5-25℃,过滤,干燥得5-氯尿嘧啶(hplc纯度不低于99%);2.1)向反应器中加入三氯甲烷,5-氯尿嘧啶和n,n-二甲基甲酰胺,搅拌溶解;2.2)搅拌条件下加入双(三氯甲基)碳酸酯,加热至60-65℃,反应至5-氯尿嘧啶基本反应完全(hplc检测,5-氯尿嘧啶含量不大于0.5%);2.3)冷却至室温,加入水,分液,浓缩得2,4,5-三氯嘧啶(hplc纯度不低于98%)。
15.与现有技术相比,本发明具有以下有益效果:1)用次氯酸钠代替氯气进一步氯化,避免了剧毒氯气的使用,大大降低了设备和场地的要求,提高了安全性,更容易工业化;同时将次氯酸钠氯化推向工业化,实现该步骤工艺的产业升级,整体工艺更为绿色环保。
16.2)整体2,4,5-三氯嘧啶工艺条件合理,操作简单安全,反应收率较高,生产成本低,易于实现工业化生产,具有较大社会效益和经济效益。
17.除非有相反陈述,否则下列用在说明书和权利要求书中的术语具有下述含义。
18.本发明“形成”并不特指两个底物间的转化反应为单步骤的,可为两个底物间的单步骤或多步骤的反应。
19.本公开中数值为仪器测量值,存在一定程度的误差,一般而言,正负10%均属于合理误差范围内。当然需要考虑该数值所用之处的上下文。在一些实施方案中,底物投料当量比误差变化不超过
±
10%,可以为
±
9%、
±
8%、
±
7%、
±
6%、
±
5%、
±
4%、
±
3%、
±
2%或
±
1%,优选
±
5%。
20.本发明化合物结构可通过核磁共振(nmr)或/和质谱(ms)来确定的。nmr位移(移r以10-6(ppm)的单位给出。nmr的测定是用bruker avance-400核磁仪,测定溶剂为氘代二甲
基亚砜 (dmso-d6)。
附图说明
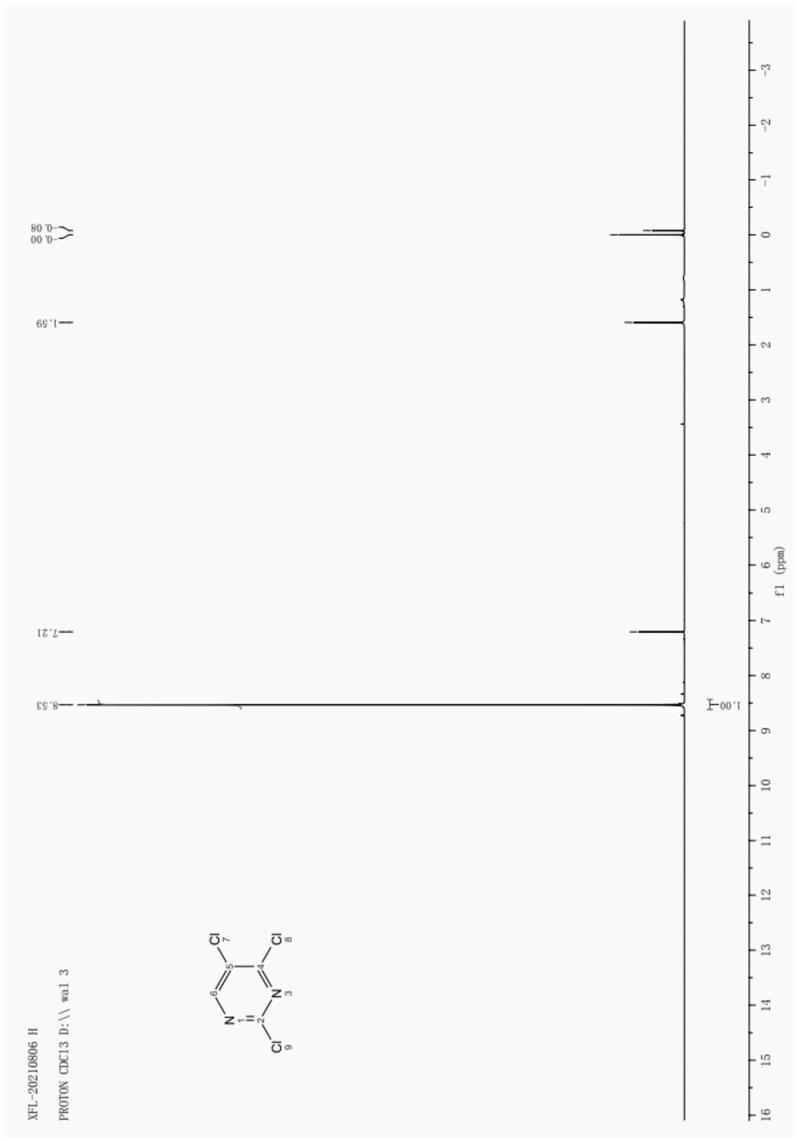
21.图1为样品的1h-nmr图;图2为样品的
13
c-nmr图。
具体实施方式
22.以下将结合实施例更详细地解释本发明,本发明的实施例仅用于说明本发明的技术方案,本发明的实质和范围并不局限于此。
23.实施例1步骤1:500ml四口瓶,加入240g稀硫酸(浓度9%),投入20g尿嘧啶,边冷却边搅拌20-30min;于10-15℃的温度条件下,加入110g次氯酸钠溶液,反应至hplc检测尿嘧啶基本反应完全(含量不大于1%);升温回流1-2h,然后冷却至20-30℃,过滤,用水洗涤,烘干,得白色粉末24.3g,收率92.8%,hplc纯度99.5%以上。
24.步骤2:向反应瓶中加入110g四氢呋喃,48g 5-氯尿嘧啶和0.6g dmf,搅拌溶解,然后滴加双(三氯甲基)碳酸酯的氯仿溶液,其中双(三氯甲基)碳酸酯的氯仿溶液为79g双(三氯甲基)碳酸酯溶解到150g四氢呋喃中;完成后升温至63-65℃,反应至hplc检测尿嘧啶基本反应完全(含量不大于1%), 加入70g水,搅拌分层;有机相先常压蒸馏至70-75℃,回收氯仿。再减压精馏浓缩,再减压精馏得橙色液体51.4g,收率85.7%,hplc纯度99%以上。
25.对本实施例得到的产品进行核磁共振,具体见图1和2。
26.实施例2步骤1:500l搪瓷釜,投270l水,冰盐水冷却下,滴加26kg 98%硫酸,滴完冷却至30-35℃,投30kg尿嘧啶;冷却至15-20℃,搅拌条件下缓慢加入次氯酸钠溶液161kg,搅拌反应至hplc检测
尿嘧啶基本反应完全(含量不大于1%);升温95~100℃反应,反应3~4小时后,冷却至20-25℃,离心,烘干得白色粉末37.4kg,收率95.3%,hplc纯度99.1%。
27.步骤2:将230kg 氯仿,100kg 5-氯尿嘧啶,1.1 kg dmf加入到500 l反应釜中,搅拌溶解,然后加入双(三氯甲基)碳酸酯的氯仿溶液(163kg双(三氯甲基)碳酸酯溶解到200kg氯仿中)。
28.加完成后升温至60-65℃反应2-2.5h,至hplc检测尿嘧啶基本反应完全(含量不大于1%), 加入150kg水,搅拌分层。
29.浓缩,再减压精馏得橙色液体109.5kg,收率87.5%,hplc纯度98.5%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1