一种达泊西汀杂质对照品的制备方法与流程
1.本发明属于医药领域,具体而言涉及一种达泊西汀杂质对照品的制备方法。
背景技术:
2.达泊西汀是一种选择性的5
‑
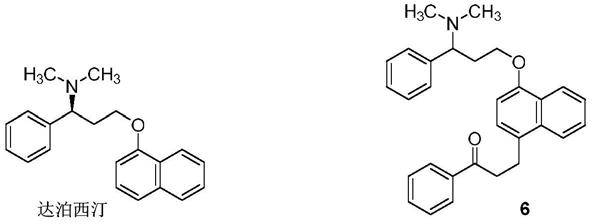
羟色胺再吸收抑制剂,用于男性早泄的治疗,其结构式如下,其中可能含杂质(式6)。
[0003][0004]
在达泊西汀的合成过程中,中间体(式3)的一种制备方法是使用3
‑
氯
‑1‑
苯基丙
‑1‑
酮(式1)经过还原得到3
‑
氯
‑1‑
苯基丙
‑1‑
醇(式2),然后与1
‑
萘酚进行成醚反应,得到中间体(式3)。
[0005][0006]
其中,在从3
‑
氯
‑1‑
苯基丙
‑1‑
酮(式1)还原至中间体(式2)的过程中,会有少量的1未被还原,在后续的与1
‑
萘酚的反应过程中,会与生成的中间体(式3)反应,在萘环的4位发生傅克烷基化反应,生成化合物(式4),然后与二甲胺(式5)反应,生成化合物(式6)。
[0007]
[0008]
化合物(式6)作为达泊西汀中潜在的一种杂质,在达泊西汀的质量研究过程中,需要获得其对照品。其合成方法尚未见公开报道。
技术实现要素:
[0009]
发明目的:本发明所要解决的技术问题是针对现有技术的不足,提供一种如式6所示的达泊西汀杂质对照品的制备方法。
[0010]
本发明还要解决的技术问题是提供式6所示的达泊西汀杂质对照品盐酸盐的制备方法。
[0011]
为了解决上述第一个技术问题,本发明公开了一种如式6所示的达泊西汀杂质对照品(图1)的制备方法,包括如下步骤:
[0012]
(1)使用对甲氧苄基将3
‑
(萘
‑1‑
基氧)
‑1‑
苯基丙
‑1‑
醇中的羟基进行保护,即将式3所示的3
‑
(萘
‑1‑
基氧)
‑1‑
苯基丙
‑1‑
醇与对甲氧苄基试剂反应,得到化合物7;
[0013]
(2)化合物7与式1所示的3
‑
氯
‑1‑
苯基丙
‑1‑
酮反应,得到化合物8;
[0014]
(3)使用氧化剂脱除化合物8中对甲氧苄基保护基,得到化合物4;
[0015]
(4)化合物4与烷基磺酰氯反应后,再与二甲胺反应,得到式6所示的达泊西汀杂质对照品;
[0016][0017]
式中,x选自cl或br;pmb代表对甲氧苄基;r选自甲基或对甲基苯基。
[0018]
其中,起始原料化合物3可以使用市售化学品,也可以按照journal of pharmaceutical and biomedical analysis 96,272
‑
277,2014中公开的方法的制备。
[0019]
步骤(1)中,所述对甲氧苄基试剂为对甲氧氯苄和/或对甲氧溴苄。
[0020]
步骤(1)中,所述式3所示的3
‑
(萘
‑1‑
基氧)
‑1‑
苯基丙
‑1‑
醇与对甲氧苄基试剂的摩尔比为1:(1.0~1.8),优选为1:1.2。
[0021]
步骤(1)中,所述反应的溶剂为二氯甲烷。
[0022]
步骤(1)中,所述反应优选为将式3所示的3
‑
(萘
‑1‑
基氧)
‑1‑
苯基丙
‑1‑
醇溶解在溶剂中,加入三乙胺,降温至10℃以下,加入对甲氧苄基试剂反应,即得含有化合物7的反应液;所述反应结束后,向反应液加入10%硫酸氢钾溶液,搅拌分层,收集下层有机相,加入正庚烷,降温析晶,过滤,干燥,即得化合物7。
[0023]
其中,所述式3所示的3
‑
(萘
‑1‑
基氧)
‑1‑
苯基丙
‑1‑
醇与三乙胺的摩尔比为1:(1.5
~3.5),优选为1:2.5。
[0024]
步骤(2)中,所述化合物7与3
‑
氯
‑1‑
苯基丙
‑1‑
酮的摩尔比为1:(2~5),优选为1:2.5。
[0025]
步骤(2)中,所述反应的溶剂为硝基苯和/或氯苯。
[0026]
步骤(2)中,所述反应在路易斯酸催化下进行;其中,所述路易斯酸为三氯化铝、三氯化铁和三氟化硼中的任意一种或几种组合;其中,当所述路易斯酸为三氟化硼时,其以三氟化硼乙醚溶液存在,优选为46.5%三氟化硼乙醚溶液。
[0027]
其中,所述化合物7与路易斯酸的摩尔比为1:(2~3)。
[0028]
步骤(2)中,所述反应为将化合物7溶解在溶剂中,加入路易斯酸,加热至100~110℃,加入式1所示的3
‑
氯
‑1‑
苯基丙
‑1‑
酮与溶剂的混合溶液,得到反应的混合物,继续反应,tlc监控至反应完全,停止加热,冷却至室温。
[0029]
其中,所述加入式1所示的3
‑
氯
‑1‑
苯基丙
‑1‑
酮与溶剂的混合溶液的加入方式为滴加和/或流加,加入的时间为1~5h,优选为3h。
[0030]
其中,所述tlc监控件为gf254硅胶板,展开剂为乙酸乙酯:石油醚(v/v)=1:20,254nm紫外灯观察。
[0031]
其中,所述反应结束后,将反应液滴加到稀盐酸溶液中,分出下层有机相,用饱和氯化钠溶液洗涤三次,减压浓缩至干,硅胶柱层析纯化,洗脱剂为乙酸乙酯:石油醚(v/v)=1:10至1:2,收集主产物点,蒸出溶剂,残余物加入乙酸乙酯和正庚烷析晶,得到化合物8。
[0032]
步骤(3)中,所述氧化剂为硝酸铈铵(can)和/或二氯二氰基苯醌(ddq)。
[0033]
步骤(3)中,反应的溶剂为乙腈与水的混合溶剂。
[0034]
步骤(3)中,所述氧化剂与化合物8的摩尔比为(1~3):1,优选为2:1。
[0035]
步骤(3)中,所述反应的温度为40~50℃。
[0036]
步骤(3)中,将化合物8溶解于溶剂中,加入氧化剂,加热,搅拌反应,tlc监控至反应完全;所述反应结束后,将反应液冷却至10℃以下,滴加到5%的硫代硫酸钠溶液中,加入乙酸乙酯搅拌,分出上层有机相,用饱和氯化钠溶液洗涤,有机相用硫酸钠干燥,滤除干燥剂,减压浓缩至干,残余物用硅胶柱层析纯化,洗脱剂为乙酸乙酯:石油醚(v/v)=1:10至1:1,分出主产物点,浓缩至干,得到白色固体,为化合物4。
[0037]
其中,tlc监控条件为gf254硅胶板,展开剂为乙酸乙酯:石油醚(v/v)=1:5,254nm紫外灯观察。
[0038]
步骤(4)中,所述烷基磺酰氯为甲基磺酰氯和/或对甲苯磺酰氯。
[0039]
步骤(4)中,将化合物4、三乙胺、4
‑
二甲氨基吡啶溶解在溶剂中,降温,加入烷基磺酰氯,在0~5℃进行第一反应;加入二甲胺四氢呋喃溶液,在20~30℃搅拌进行第二反应,减压浓缩,到化合物6。
[0040]
其中,化合物4、三乙胺和4
‑
二甲氨基吡啶的摩尔比为1:(0.5~1.5):(0.05~0.15),优选为1:1.5:0.1。
[0041]
其中,所述溶剂为四氢呋喃。
[0042]
其中,所述降温为降温至0~5℃。
[0043]
其中,化合物4和烷基磺酰氯的摩尔比为1:(1.0~1.8),优选为1:1.2。
[0044]
其中,所述第一反应的时间为0.5~1.5h,优选为1h。
[0045]
其中,所述第二反应的温度为20~30℃。
[0046]
其中,所述第二反应的时间为10~20h,优选为15h。
[0047]
为了解决上述第二个技术问题,本发明公开了式6所示的达泊西汀杂质对照品盐酸盐的制备方法,即按照上述方法制备得到式6所示的达泊西汀杂质,将式6所示的达泊西汀杂质溶解在乙酸乙酯中,通入氯化氢气体,将析出的白色固体滤出,得到化合物6的盐酸盐。
[0048]
如无特殊说明,本发明中所述%为质量百分比。
[0049]
有益效果:与现有技术相比,本发明具有如下优势:
[0050]
本发明首次制备出式6所示的达泊西汀杂质对照品。
附图说明
[0051]
下面结合附图和具体实施方式对本发明做更进一步的具体说明,本发明的上述和/或其他方面的优点将会变得更加清楚。
[0052]
图1为化合物6的结构式。
[0053]
图2为化合物6质谱图。
[0054]
图3为化合物6氢谱图。
具体实施方式
[0055]
下述实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂和材料,如无特殊说明,均可从商业途径获得。
[0056]
实施例1:使用对甲氧氯苄制备化合物7
[0057][0058]
将化合物3(200g,0.718mol,1eq.)溶解在二氯甲烷(2000ml)中,加入三乙胺(181.6g,248.8ml,1.795mol,2.5eq.),将反应液降温至10℃以下,滴加对甲氧氯苄(134.9g,0.862mol,1.2eq),反应结束后,加入10%的硫酸氢钾溶液(1000ml),搅拌分层,收集下层有机相,加入正庚烷(1000ml),降温至5
±
5℃析晶,过滤,40
±
5℃真空干燥,得到类白色固体260.4g,收率91%,为化合物7。
[0059]
esi
‑
ms(+):399.2[m+h]
+
.1h
‑
nmr(400mhz,cdcl3):8.31(d,j=5.5hz,1h),8.08(d,j=5.5hz,1h),7.62~7.59(m,3h),7.40~7.27(m,6h),6.98(d,j=5.5hz,2h),6.91(d,j=5.5hz,2h),6.42(d,j=5.0hz,1h),4.71(s,2h),4.43(t,j=7.0hz,1h),3.92(t,j=7.5hz,2h),3.70(s,3h),2.17(t,j=7.5hz,2h).
[0060]
实施例2:使用对甲氧溴苄制备化合物7
[0061]
[0062]
将化合物3(200g,0.718mol,1eq.)溶解在二氯甲烷(2000ml)中,加入三乙胺(181.6g,248.8ml,1.795mol,2.5eq.),将反应液降温至10℃以下,滴加对甲氧溴苄(173.3g,0.862mol,1.2eq),反应结束后,加入10%的硫酸氢钾溶液(1000ml),搅拌分层,收集下层有机相,加入正庚烷(1000ml),降温至5
±
5℃析晶,过滤,40
±
5℃真空干燥,得到类白色固体262.7g,收率91.8%,为化合物物7。
[0063]
实施例3:制备化合物8
[0064][0065]
将化合物7(50g,0.125mol)溶解在硝基苯(300ml)中,加入三氯化铝(33.46g,0.25mol,2eq.),将化合物1(42.31g,0.251mol,2eq.)溶解在硝基苯(200ml),将反应体系加热至100~110℃,将化合物1的溶液滴加进入,滴加用时3小时,得到的反应混合物,继续在100~110℃搅拌反应2小时,使用tlc监控反应(tlc监控条件为,gf254硅胶板,展开剂为乙酸乙酯:石油醚(v/v)=1:20,254nm紫外灯观察)至化合7基本消失,停止加热,冷却至20℃以下。
[0066]
将反应液滴加到10%稀盐酸溶液(1000ml)中,分出下层有机相,用饱和氯化钠溶液(300ml
×
3)洗涤,有机相减压浓缩至干,残余物使用硅胶柱层析纯化,洗脱剂为乙酸乙酯:石油醚(v/v)=1:10至1:2,收集主产物点,蒸除溶剂,残余物加入乙酸乙酯(50ml)和正庚烷(100ml)在10
±
5℃析晶,得到化合物8,共16.11g,收率24.2%。
[0067]
esi
‑
ms(+):531.3[m+h]
+
.1h
‑
nmr(400mhz,cdcl3):8.33(d,j=5.5hz,1h),7.95~7.90(m,3h),7.67~7.62(m,1h),7.32~7.25(m,6h),6.99(d,j=5.0hz,2h),6.90(d,j=5.0hz,2h),6.80(d,j=4.5hz,1h),6.30(d,j=5.0hz,1h),4.62(s,2h),4.48(t,j=7.5hz,1h),3.92(t,j=7.0hz,2h),3.81(s,3h),3.15(t,j=7.0hz,2h),2.90(t,j=7.0hz,2h),2.17~2.14(m,2h).
[0068]
实施例4:制备化合物8
[0069]
将化合物7(50g,0.125mol)溶解在硝基苯(300ml)中,加入三氯化铝(41.82g,0.312mol,2.5eq.),将化合物1(52.89g,0.314mol,2.5eq.)溶解在硝基苯(260ml),将反应体系加热至100~110℃,将化合物1的溶液滴加进入,滴加用时3小时,得到的反应混合物,继续在100~110℃搅拌反应2小时,使用tlc监控反应(tlc监控条件为,gf254硅胶板,展开剂为乙酸乙酯:石油醚(v/v)=1:20,254nm紫外灯观察)至化合7基本消失,停止加热,冷却至20℃以下。
[0070]
将反应液滴加到10%稀盐酸溶液(1000ml)中,分出下层有机相,用饱和氯化钠溶液(300ml
×
3)洗涤,有机相减压浓缩至干,残余物使用硅胶柱层析纯化,洗脱剂为乙酸乙酯:石油醚(v/v)=1:10至1:2,收集主产物点,蒸除溶剂,残余物加入乙酸乙酯(50ml)和正庚烷(100ml)在10
±
5℃析晶,得到化合物8,共18.31g,收率27.5%。
[0071]
实施例5:制备化合物8
[0072]
将化合物7(50g,0.125mol)溶解在硝基苯(300ml)中,加入三氯化铝(41.82g,0.312mol,2.5eq.),将化合物1(84.63g,0.502mol,4eq.)溶解在硝基苯(420ml),将反应体系加热至100~110℃,将化合物1的溶液滴加进入,滴加用时3小时,得到的反应混合物,继续在100~110℃搅拌反应2小时,使用tlc监控反应(tlc监控条件为,gf254硅胶板,展开剂为乙酸乙酯:石油醚(v/v)=1:20,254nm紫外灯观察)至化合7基本消失,停止加热,冷却至20℃以下。
[0073]
将反应液滴加到10%稀盐酸溶液(1000ml)中,分出下层有机相,用饱和氯化钠溶液(300ml
×
3)洗涤,有机相减压浓缩至干,残余物使用硅胶柱层析纯化,洗脱剂为乙酸乙酯:石油醚(v/v)=1:10至1:2,收集主产物点,蒸除溶剂,残余物加入乙酸乙酯(50ml)和正庚烷(100ml)在10
±
5℃析晶,得到化合物8,共23.44g,收率35.2%。
[0074]
实施例6:制备化合物8
[0075]
将化合物7(50g,0.125mol)溶解在硝基苯(300ml)中,加入三氯化铝(41.82g,0.312mol,2.5eq.),将化合物1(105.78g,0.627mol,5eq.)溶解在硝基苯(530ml),将反应体系加热至100~110℃,将化合物1的溶液滴加进入,滴加用时3小时,得到的反应混合物,继续在100~110℃搅拌反应2小时,使用tlc监控反应(tlc监控条件为,gf254硅胶板,展开剂为乙酸乙酯:石油醚(v/v)=1:20,254nm紫外灯观察)至化合7基本消失,停止加热,冷却至20℃以下。
[0076]
将反应液滴加到10%稀盐酸溶液(1000ml)中,分出下层有机相,用饱和氯化钠溶液(300ml
×
3)洗涤,有机相减压浓缩至干,残余物使用硅胶柱层析纯化,洗脱剂为乙酸乙酯:石油醚(v/v)=1:10至1:2,收集主产物点,蒸除溶剂,残余物加入乙酸乙酯(50ml)和正庚烷(100ml)在10
±
5℃析晶,得到化合物8,共27.70g,收率41.6%。
[0077]
实施例7:制备化合物8
[0078]
将化合物7(50g,0.125mol)溶解在硝基苯(300ml)中,加入三氯化铁(50.88g,0.312mol,2.5eq.),将化合物1(52.89g,0.314mol,2.5eq.)溶解在硝基苯(260ml),将反应体系加热至100~110℃,将化合物1的溶液滴加进入,滴加用时3小时,得到的反应混合物,继续在100~110℃搅拌反应2小时,使用tlc监控反应(tlc监控条件为,gf254硅胶板,展开剂为乙酸乙酯:石油醚(v/v)=1:20,254nm紫外灯观察)至化合7基本消失,停止加热,冷却至20℃以下。
[0079]
将反应液滴加到10%稀盐酸溶液(1000ml)中,分出下层有机相,用饱和氯化钠溶液(300ml
×
3)洗涤,有机相减压浓缩至干,残余物使用硅胶柱层析纯化,洗脱剂为乙酸乙酯:石油醚(v/v)=1:10至1:2,收集主产物点,蒸除溶剂,残余物加入乙酸乙酯(50ml)和正庚烷(100ml)在10
±
5℃析晶,得到化合物8,共24.90g,收率37.4%。
[0080]
实施例8:制备化合物8
[0081]
将化合物7(50g,0.125mol)溶解在硝基苯(300ml)中,加入三氯化铁(61.05g,0.375mol,3eq.),将化合物1(52.89g,0.314mol,2.5eq.)溶解在硝基苯(260ml),将反应体系加热至100~110℃,将化合物1的溶液滴加进入,滴加用时3小时,得到的反应混合物,继续在100~110℃搅拌反应2小时,使用tlc监控反应(tlc监控条件为,gf254硅胶板,展开剂为乙酸乙酯:石油醚(v/v)=1:20,254nm紫外灯观察)至化合7基本消失,停止加热,冷却至20℃以下。
[0082]
将反应液滴加到10%稀盐酸溶液(1000ml)中,分出下层有机相,用饱和氯化钠溶液(300ml
×
3)洗涤,有机相减压浓缩至干,残余物使用硅胶柱层析纯化,洗脱剂为乙酸乙酯:石油醚(v/v)=1:10至1:2,收集主产物点,蒸除溶剂,残余物加入乙酸乙酯(50ml)和正庚烷(100ml)在10
±
5℃析晶,得到化合物8,共19.11g,收率28.7%。
[0083]
实施例9:制备化合物8
[0084]
将化合物7(50g,0.125mol)溶解在硝基苯(300ml)中,加入46.5%三氟化硼乙醚溶液(40.36ml,0.31mol,2.5eq.),将化合物1(52.89g,0.314mol,2.5eq.)溶解在硝基苯(260ml),将反应体系加热至100~110℃,将化合物1的溶液滴加进入,滴加用时3小时,得到的反应混合物,继续在100~110℃搅拌反应2小时,使用tlc监控反应(tlc监控条件为,gf254硅胶板,展开剂为乙酸乙酯:石油醚(v/v)=1:20,254nm紫外灯观察)至化合7基本消失,停止加热,冷却至20℃以下。
[0085]
将反应液滴加到10%稀盐酸溶液(1000ml)中,分出下层有机相,用饱和氯化钠溶液(300ml
×
3)洗涤,有机相减压浓缩至干,残余物使用硅胶柱层析纯化,洗脱剂为乙酸乙酯:石油醚(v/v)=1:10至1:2,收集主产物点,蒸除溶剂,残余物加入乙酸乙酯(50ml)和正庚烷(100ml)在10
±
5℃析晶,得到化合物8,共18.38g,收率27.6%。
[0086]
实施例10:制备化合物8
[0087]
将化合物7(50g,0.125mol)溶解在硝基苯(300ml)中,加入46.5%三氟化硼乙醚溶液(32.55ml,0.25mol,2eq.),将化合物1(52.89g,0.314mol,2.5eq.)溶解在硝基苯(260ml),将反应体系加热至100~110℃,将化合物1的溶液滴加进入,滴加用时3小时,得到的反应混合物,继续在100~110℃搅拌反应2小时,使用tlc监控反应(tlc监控条件为,gf254硅胶板,展开剂为乙酸乙酯:石油醚(v/v)=1:20,254nm紫外灯观察)至化合7基本消失,停止加热,冷却至20℃以下。
[0088]
将反应液滴加到10%稀盐酸溶液(1000ml)中,分出下层有机相,用饱和氯化钠溶液(300ml
×
3)洗涤,有机相减压浓缩至干,残余物使用硅胶柱层析纯化,洗脱剂为乙酸乙酯:石油醚(v/v)=1:10至1:2,收集主产物点,蒸除溶剂,残余物加入乙酸乙酯(50ml)和正庚烷(100ml)在10
±
5℃析晶,得到化合物8,共18.04g,收率27.1%。
[0089]
实施例11:制备化合物8
[0090]
将化合物7(50g,0.125mol)溶解在氯苯(300ml)中,加入三氯化铝(41.82g,0.312mol,2.5eq.),将化合物1(52.89g,0.314mol,2.5eq.)溶解在氯苯(260ml),将反应体系加热至100~110℃,将化合物1的溶液滴加进入,滴加用时3小时,得到的反应混合物,继续在100~110℃搅拌反应2小时,使用tlc监控反应(tlc监控条件为,gf254硅胶板,展开剂为乙酸乙酯:石油醚(v/v)=1:20,254nm紫外灯观察)至化合7基本消失,停止加热,冷却至20℃以下。
[0091]
将反应液滴加到10%稀盐酸溶液(1000ml)中,分出下层有机相,用饱和氯化钠溶液(300ml
×
3)洗涤,有机相减压浓缩至干,残余物使用硅胶柱层析纯化,洗脱剂为乙酸乙酯:石油醚(v/v)=1:10至1:2,收集主产物点,蒸除溶剂,残余物加入乙酸乙酯(50ml)和正庚烷(100ml)在10
±
5℃析晶,得到化合物8,共14.98g,收率22.5%。
[0092]
实施例12:制备化合物8
[0093]
将化合物7(50g,0.125mol)溶解在氯苯(300ml)中,加入三氯化铁(50.88g,
0.312mol,2.5eq.),将化合物1(63.47g,0.376mol,3eq.)溶解在氯苯(320ml),将反应体系加热至100~110℃,将化合物1的溶液滴加进入,滴加用时3小时,得到的反应混合物,继续在100~110℃搅拌反应2小时,使用tlc监控反应(tlc监控条件为,gf254硅胶板,展开剂为乙酸乙酯:石油醚(v/v)=1:20,254nm紫外灯观察)至化合7基本消失,停止加热,冷却至20℃以下。
[0094]
将反应液滴加到10%稀盐酸溶液(1000ml)中,分出下层有机相,用饱和氯化钠溶液(300ml
×
3)洗涤,有机相减压浓缩至干,残余物使用硅胶柱层析纯化,洗脱剂为乙酸乙酯:石油醚(v/v)=1:10至1:2,收集主产物点,蒸除溶剂,残余物加入乙酸乙酯(50ml)和正庚烷(100ml)在10
±
5℃析晶,得到化合物8,共18.18g,收率27.3%。
[0095]
实施例13:制备化合物8
[0096]
将化合物7(50g,0.125mol)溶解在氯苯(300ml)中,加入46.5%的三氟化硼乙醚溶液(40.36ml,0.31mol,2.5eq.),将化合物1(74.05g,0.439mol,3.5eq.)溶解在氯苯(370ml),将反应体系加热至100~110℃,将化合物1的溶液滴加进入,滴加用时3小时,得到的反应混合物,继续在100~110℃搅拌反应2小时,使用tlc监控反应(tlc监控条件为,gf254硅胶板,展开剂为乙酸乙酯:石油醚(v/v)=1:20,254nm紫外灯观察)至化合7基本消失,停止加热,冷却至20℃以下。
[0097]
将反应液滴加到10%稀盐酸溶液(1000ml)中,分出下层有机相,用饱和氯化钠溶液(300ml
×
3)洗涤,有机相减压浓缩至干,残余物使用硅胶柱层析纯化,洗脱剂为乙酸乙酯:石油醚(v/v)=1:10至1:2,收集主产物点,蒸除溶剂,残余物加入乙酸乙酯(50ml)和正庚烷(100ml)在10
±
5℃析晶,得到化合物8,共19.51g,收率29.3%。
[0098]
实施例14:使用硝酸铈铵制备化合物4
[0099][0100]
将化合物8(15g,0.028mol)溶解在乙腈(100ml)和水(100ml)的混合溶剂中,加入硝酸铈铵(30.7g,0.056mol,2eq.),混合物加热至45
±
5℃,搅拌反应,使用tlc监控反应(tlc监控条件为,gf254硅胶板,展开剂为乙酸乙酯:石油醚(v/v)=1:5,254nm紫外灯观察),至反应完全,将反应液冷却至10℃以下,滴加到5%的硫代硫酸钠溶液(200ml)中,加入乙酸乙酯(200ml
×
3)萃取,合并有机相,用饱和氯化钠溶液(100ml
×
3)洗涤,有机相用硫酸钠干燥,滤除干燥剂,减压浓缩至干,残余物用硅胶柱层析纯化,洗脱剂为乙酸乙酯:石油醚(v/v)=1:10至1:1,分出主产物点,浓缩至干,在45
±
5℃真空干燥,得到白色固体,共10.72g,收率93.3%,为化合物4。
[0101]
esi
‑
ms(+):411.2[m+h]
+
.1h
‑
nmr(400mhz,cdcl3):8.33(d,j=5.0hz,1h),7.95~7.90(m,3h),7.67~7.65(m,1h),7.53~7.49(m,3h),7.32~7.20(m,6h),6.73(d,j=5.0hz,1h),6.11(d,j=5.0hz,1h),4.70(brs,1h),4.41(t,j=7.0hz,1h),3.92(t,j=7.0hz,2h),3.15(t,j=7.0hz,2h),2.90(t,j=7.0hz,2h),2.19~2.17(m,2h).
[0102]
实施例15:使用ddq制备化合物4
[0103]
将化合物8(15g,0.028mol)溶解在乙腈(100ml)和水(100ml)的混合溶剂中,加入二氯二氰基苯醌(12.7g,0.056mol,2eq.),混合物加热至45
±
5℃,搅拌反应,使用tlc监控反应(tlc监控条件为,gf254硅胶板,展开剂为乙酸乙酯:石油醚(v/v)=1:5,254nm紫外灯观察),至反应完全,将反应液冷却至10℃以下,滴加到5%的硫代硫酸钠溶液(200ml)中,加入乙酸乙酯(200ml
×
3)萃取,合并有机相,用饱和氯化钠溶液(100ml
×
3)洗涤,有机相用硫酸钠干燥,滤除干燥剂,减压浓缩至干,残余物用硅胶柱层析纯化,洗脱剂为乙酸乙酯:石油醚(v/v)=1:10至1:1,分出主产物点,浓缩至干,在45
±
5℃真空干燥,得到白色固体,共9.18g,收率80.2%,为化合物4。
[0104]
实施例16:制备化合物6
[0105][0106]
将化合物4(8g,0.019mol)、三乙胺(2.96g,0.029mol,1.5eq.),4
‑
二甲氨基吡啶(0.232g,1.9mmol,0.1eq.),溶解在四氢呋喃(100ml)中,降温至0~5℃,滴加甲基磺酰氯(2.98g,0.0228mol,1.2eq),滴加完毕,在0~5℃反应1小时,加入二甲胺2mol/l的四氢呋喃溶液(10.45ml,0.021mol,1.1eq),在25
±
5℃搅拌反应15小时,减压浓缩,残余物加入水(200ml)、二氯甲烷(200ml),分出有机相,在45℃以下减压浓缩得到化合物6,为褐色油状物。共6.2g,收率74.6%,质谱和氢谱如图2和图3所示。
[0107]
实施例17:制备化合物6
[0108]
将化合物4(8g,0.019mol)、三乙胺(2.96g,0.029mol,1.5eq.),4
‑
二甲氨基吡啶(0.232g,1.9mmol,0.1eq.),溶解在四氢呋喃(100ml)中,降温至0~5℃,滴加对甲苯磺酰氯(4.35g,0.0228mol,1.2eq)溶解在四氢呋喃(40ml)里的溶液,滴加完毕,在0~5℃反应1小时,加入二甲胺2mol/l的四氢呋喃溶液(10.45ml,0.021mol,1.1eq),在25
±
5℃搅拌反应15小时,减压浓缩,残余物加入水(200ml)、二氯甲烷(200ml),分出有机相,在45℃以下减压浓缩得到化合物6,为褐色油状物。共5.6g,收率67.4%。
[0109]
实施例18:制备化合物6的盐酸盐
[0110]
将化合物6(5g,0.0114mol),溶解在50ml乙酸乙酯中,降温至0~5℃,向反应液通入干燥的氯化氢气体,室温搅拌1小时,过滤,得到类白色固体,在45
±
5℃真空干燥,共5.0g,收率为92.3%
[0111]
esi
‑
ms(+):438.2497[m+h]
+
.1h
‑
nmr(600mhz,dmso
‑
d6):11.41(s,1h),8.11(d,j=12hz,1h,),8.01(d,j=6hz,1h,),7.96(d,j=6hz,2h),7.65~7.56(m,4h),7.50~7.44(m,6h),7.20(d,j=12hz,1h),6.65(d,j=12hz,1h),4.71(t,j=6hz,1h),4.10~4.07(m,1h),3.67~3.63(m,1h),3.39~3.35(m,2h),3.28(t,2h,j=6hz),2,93(m,1h),2.92(s,3h),2.91~2.90(m,1h),2.84(s,3h).
[0112]
本发明提供了一种达泊西汀杂质对照品的制备方法的思路及方法,具体实现该技术方案的方法和途径很多,以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。本实施例中未明确的各组成部分均可用现有技术加以实现。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1