一种植物源7-酮基石胆酸异构体杂质的制备方法与流程
1.本发明属于有机化学合成/药物合成技术领域,具体涉及一种植物源7-酮基石胆酸异构体杂质的制备方法。
背景技术:
2.7-酮基石胆酸(3α-羟基7-酮-5β-胆甾烷-24-酸)化学式为c
24h38
o4,其结构式如下:
[0003][0004]
7-酮基石胆酸是一种重要的药物中间体,为制备鹅去氧胆酸、熊去氧胆酸、奥贝胆酸、牛磺熊去氧胆酸等多种胆酸类药物的主要起始原料。7-酮基石胆酸中5位氢原子为β位。
[0005]
从动物内脏中提取的鹅去氧胆酸及胆酸结构中5位氢原子为β位,但是以植物源发酵产物ba(21-羟基-20-甲基孕甾-4-烯-3-酮)经过一系列合成制备的3,7-二酮-5β-胆甾烷-24-酸存在异构体3,7-二酮-5α-胆甾烷-24-酸,其制备路线的最后几步如下所示:
[0006][0007]
该杂质与目标产物极性差别较小,仅仅是5位氢原子结构位置的区别,很难通过常规原料杂质分离纯化的方法得到纯净的对照品。
[0008]
在使用3,7-二酮-5β-胆甾烷-24-酸制备7-酮基石胆酸时,该杂质3,7-二酮-5α-胆甾烷-24-酸会生成7-酮基石胆酸的异构体,进而影响目标产物7-酮基石胆酸的质量。
[0009]
然而,在现有技术中,7-酮基石胆酸异构体(即3α-羟基7-酮-5α-胆甾烷-24-酸)的合成方法并没有相关报道,此异构体与7-酮基石胆酸非常类似,仅仅是5位氢原子位置差异,很难通过常规原料杂质分离纯化的方法得到纯净的对照品。
[0010]
也就是说,7-酮基石胆酸异构体(3α-羟基-7-酮-5α-胆甾烷-24-酸)在制备植物源7-酮基石胆酸的过程中可能出现,它作为7-酮基石胆酸的副产物会严重影响7-酮基石胆酸的质量。因此,7-酮基石胆酸中严格控制此杂质含量具有重要意义。但是,严格控制7-酮基石胆酸产品质量需要对产品杂质进行检测,需要此杂质作为研究对照品。
[0011]
因此,本领域需要一种合成7-酮基石胆酸异构体杂质的方法。
技术实现要素:
[0012]
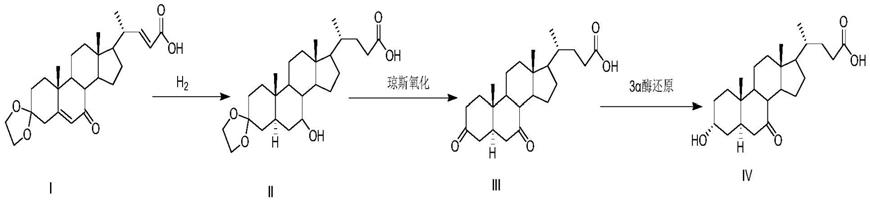
因此,本发明提供一种植物源7-酮基石胆酸异构体杂质的制备方法,所述7-酮基石胆酸异构体杂质即3α-羟基-7-酮-5α-胆甾烷-24-酸;所述制备方法包括选用如式i所示原料经加氢反应、氧化反应和还原反应得到所述7-酮基石胆酸异构体杂质。
[0013][0014]
在一种具体的实施方式中,所述制备方法包括如下步骤:
[0015]
第一步:选用如式i所示原料经加氢反应得到如式ii所示中间体;
[0016]
第二步:式ii所示中间体经氧化得到式iii所示中间体;
[0017]
第三步:式iii所示中间体经3α还原酶还原得到式iv所示的7-酮基石胆酸异构体杂质。
[0018][0019]
本发明中,因原料i化合物的选取,使得加氢反应、氧化反应及还原反应后形成该7-酮基石胆酸异构体杂质,而不是7-酮基石胆酸。也就是说,正因为该原料化合物i的选取,使得在加氢后,中间体ii的5位上形成α氢,而不是形成β氢。
[0020]
在一种具体的实施方式中,所述式i所示原料为如下式m所示化合物用氢氧化钠水溶液对侧链进行水解得到。
[0021][0022]
本发明中,式m所述化合物为由ba经一系列合成得到的一种公知化合物。
[0023]
在一种具体的实施方式中,第一步中式i所示原料经碱水、乙醇体系和在加氢催化剂存在下进行加氢反应得到如式ii所示中间体。
[0024]
在一种具体的实施方式中,第二步中式ii所示中间体在丙酮体系中滴加琼斯试剂
经氧化反应得到式iii所示中间体。
[0025]
在一种具体的实施方式中,第三步中式iii所示中间体经3α还原酶还原和在包含碱水的体系中反应得到式iv所示的7-酮基石胆酸异构体杂质。
[0026]
在一种具体的实施方式中,所述包含碱水的体系中包含使用氢氧化钠水溶液溶解式iii所述化合物;优选地,在反应器中加入式iii所述化合物和水,向其中加入氢氧化钠水溶液并调节溶液ph=13~14,溶清后加入盐酸调节ph=8~9,在温度为30~35℃时,加入葡萄糖、3α还原酶、葡萄糖脱氢酶、辅酶一和辅酶二,并用氢氧化钠水溶液调节溶液ph=7~8进行反应,得到式iv所示的7-酮基石胆酸异构体杂质。
[0027]
在一种具体的实施方式中,所述包含碱水的体系中包含使用甘油和氢氧化钠水溶液溶解式iii所述化合物;优选地,反应器中加入式iii所述化合物、甘油和水,向其中加入氢氧化钠水溶液并调节溶液ph=7~8,溶清后调节反应温度为30~35℃,加入葡萄糖、3α还原酶、葡萄糖脱氢酶、辅酶一和辅酶二,并用氢氧化钠水溶液调节溶液ph=7~8进行反应,得到式iv所示的7-酮基石胆酸异构体杂质。
[0028]
在一种具体的实施方式中,所述包含碱水的体系中包含使用2-甲基四氢呋喃和氢氧化钠水溶液溶解式iii所述化合物;优选地,反应器中加入式iii所述化合物、2-甲基四氢呋喃和水,向其中加入氢氧化钠水溶液并调节溶液ph=7~8,溶清后调节反应温度为30~35℃,加入葡萄糖、3α还原酶、葡萄糖脱氢酶、辅酶一和辅酶二,并用氢氧化钠水溶液调节溶液ph=7~8进行反应,得到式iv所示的7-酮基石胆酸异构体杂质。
[0029]
总的来说,本发明找到了一种合适的制备7-酮基石胆酸异构体杂质的方法,为进一步控制7-酮基石胆酸产品质量开辟了道路。同时,本发明的实验条件温和,生产过程的试剂均毒性小,污染小,生产成本低,可操作性强。
具体实施方式
[0030]
以如下实施例对本发明做出进一步说明,但本发明的保护范围并不仅限于下述实施例。
[0031]
本发明中,所述3α还原酶是3α羟基甾族化合物氧化还原酶的简称,购自南京都莱生物技术有限公司,该3α还原酶属于睾丸酮丛毛单胞菌来源。葡萄糖脱氢酶,购自上海麦克林生化科技有限公司。辅酶一(nad):烟酰胺腺嘌呤二核苷酸,购自上海跃腾生物技术有限公司。辅酶二(nadp),烟酰胺腺嘌呤二核苷酸磷酸盐,购自杭州唯泰生物药业有限公司。
[0032]
实施例1
[0033]
在合适高压罐中加入300.0ml水,降温至0~10℃,加入20.0g氢氧化钠,搅拌溶清后加入100.0g化合物i和100.0ml乙醇,升温至30~40℃搅拌溶清,加入15.0g 10%钯碳催化剂。将体系密封,先用氮气置换3次以上,彻底置换干净空气,再用氢气置换3次以上,彻底置换干净氮气,然后将体系氢气压力控制在0.6mpa,升温至50~55℃保温反应48h,hplc确定化合物i剩余0.1%以下。反应完成后,停止通气,用氮气置换氢气。将反应液过滤,过滤掉钯碳,将体系于50℃热水浴中减压浓缩,除尽乙醇。降温至0~10℃,缓慢滴加冰醋酸,调节ph=6~7,大量固体析出,保温搅拌30~40min,过滤,少量自来水淋洗,收集滤饼(尽可能抽干),得到化合物ii。
[0034]
将化合物ii滤饼溶解于1000ml丙酮中,降温至0~10℃,确保体系溶清。控温0-10
℃滴加105.0ml琼斯试剂(2.7mol/l,以氧化铬计算),滴加完成后,保温搅拌60~70min,滴加10%亚硫氢钠溶液300.0ml,然后加入3000.0ml水水析,控温0~10℃,保温搅拌50~60min,抽滤,自来水淋洗至中性,50℃鼓风干燥得化合物iii粗品,干重:88.3g,两步重量收率88.3%,摩尔收率97.4%。
[0035]
将88.3g化合物iii粗品溶解于450.0ml甲醇中,加入8.8g活性炭,升温至50~55℃,保温搅拌30~40min,热滤,收集滤液,有机相于50℃热水浴减压浓缩至粘稠状态为止。冰水浴降温至0~10℃,保温搅拌60~70min,抽滤,少量冰甲醇淋洗,50℃鼓风干燥得化合物iii精品,干重:80.5g。
[0036]
化合物iii精品制备化合物iv:在洁净的反应瓶中加入500ml水,搅拌下加入50.0g化合物iii精品,用5n氢氧化钠调节ph=13~14,待化合物iii全部溶清后,用3n盐酸调节ph=8~9。调节反应温度为30~35℃,加入120g葡萄糖、10g 3α还原酶、22g葡萄糖脱氢酶、1.5g辅酶一、1.5g辅酶二。搅拌均匀后,用2n氢氧化钠溶液调节ph=7-8反应,反应2小时,hplc监控化合物iii含量为0.1%为止。反应结束后,将体系升温至70~80℃,保温搅拌30min~60min。趁热抽滤,收集滤液,滤饼用100ml水再次升温至70~80℃,保温搅拌30min~60min,热滤后合并滤液。将滤液降温至0~10℃,缓慢滴加2n盐酸调节ph=2~3,大量固体析出,保温搅拌30min~60min,过滤,少量自来水淋洗,滤饼于50℃热风循环烘箱中干燥至合格。干重:49.5g,重量收率99.0%,液相色谱纯度:95.6%。
[0037]
实施例2
[0038]
由式i所述化合物制备式ii所述化合物,以及由式ii所述化合物制备式iii所述化合物的过程均与实施例1相同,但由式iii所述化合物制备式iv所示化合物的步骤与实施例1不同。具体地:
[0039]
化合物iii精品制备化合物iv:在洁净的反应瓶中加入500ml水,搅拌下加入50.0g化合物iii精品和50.0ml甘油,用2n氢氧化钠调节ph=7~8,待化合物i全部溶清后,调节反应温度30~35℃,加入120g葡萄糖、10g 3α还原酶、22g葡萄糖脱氢酶、1.5g辅酶一、1.5g辅酶二。搅拌均匀后,用2n氢氧化钠溶液调节ph=7~8反应,反应2小时,hplc监控化合物iii含量为0.1%为止。反应结束后,将体系升温至70~80℃,保温搅拌30min~60min。趁热抽滤,收集滤液,滤饼用100ml水再次升温至70~80℃,保温搅拌30min~60min,热滤后合并滤液。将滤液于50~60℃减压浓缩除尽甘油,降温至0~10℃,缓慢滴加2n盐酸调节ph=2~3,大量固体析出,保温搅拌30min~60min,过滤,少量自来水淋洗,滤饼于50℃热风循环烘箱中干燥至合格。干重:49.6g,重量收率99.2%,液相色谱纯度:98.7%。
[0040]
实施例3
[0041]
由式i所述化合物制备式ii所述化合物,以及由式ii所述化合物制备式iii所述化合物的过程均与实施例1相同,但由式iii所述化合物制备式iv所示化合物的步骤与实施例1不同。具体地:
[0042]
化合物iii精品制备化合物iv:在洁净的反应瓶中加入500ml水,搅拌下加入50.0g化合物iii精品和50.0ml 2-甲基四氢呋喃,用2n氢氧化钠调节ph=7~8,待化合物iii全部溶清后,调节反应温度30~35℃,加入120g葡萄糖、10g 3α还原酶、22g葡萄糖脱氢酶、1.5g辅酶一、1.5g辅酶二。搅拌均匀后,用2n氢氧化钠溶液调节ph=7~8反应,反应2小时,hplc监控化合物iii含量为0.1%为止。反应结束后,将体系升温至70~80℃,保温搅拌30min~
60min。趁热抽滤,收集滤液,滤饼用100ml水再次升温至70~80℃,保温搅拌30min~60min,热滤后合并滤液。将滤液于50~60℃减压浓缩除尽2-甲基四氢呋喃,降温至0~10℃,缓慢滴加2n盐酸调节ph=2~3,大量固体析出,保温搅拌30min~60min,过滤,少量自来水淋洗,滤饼于50℃热风循环烘箱中干燥至合格。干重:49.2g,重量收率98.4%,液相色谱纯度:97.8%。
[0043]
由上述实施例中液相色谱获得的产品纯度数据可见,单纯使用碱水体系会明显产生杂质,该杂质主要是强碱性体系溶解反应底物时聚合造成。本发明中,因酶不耐酸或碱的腐蚀,也不耐高温,发明人筛选出比氢氧化钠水溶液更好的两种弱碱体系,包括甘油体系和2-甲基四氢呋喃体系。弱碱体系下,甘油体系酶还原化合物iii得到的化合物iv纯度较2-甲基四氢呋喃体系更好。此外,酶法选择性还原工艺使用碱水体系,无污染,安全环保,适合大生产。
[0044]
上述实施例仅为清楚地说明本发明技术方案所作的举例,而并非是对本发明的实施方式的限定。在不改变本发明基本构思和实质的情况下,任何其它等同技术特征的变换或修改,都应属于本发明权利要求的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1