一种新的丁内酯衍生物的合成方法及其在抗过敏药物中的应用与流程
1.本发明涉及药物化合物技术领域,具体涉及一种新的丁内酯衍生物的制合成方法及其在抗过敏药物中的应用。
背景技术:
2.近年来对丁烯酸内酯类化合物的研究越来越多,发现丁烯酸内酯环结构普遍存在于各类生物的代谢产物中,而且此类化合物具有多种显著的生物活性,包括抗菌、抗疟疾、抗肿瘤、抗炎、抗氧化、降糖等,具有很高的开发价值。其中,丁内酯
‑
i(butyrolactone i)具有很强的抗过敏活性,是一个非常有开发应用价值的抗过敏候选药物。但丁内酯
‑
i在体内生物利用度底,导致其在推广使用过程中受到极大的限制。
技术实现要素:
3.针对上述背景技术中提到的问题,本发明旨在提供一种以丁内酯
‑
i为原料合成新的丁内酯衍生物的方法及其应用,该丁内酯衍生物具有和丁内酯
‑
i类似的脱颗粒抑制活性和更高的代谢稳定性,极大程度提高了其应用范围。
4.为了实现上述目的,本发明采用如下技术方案:
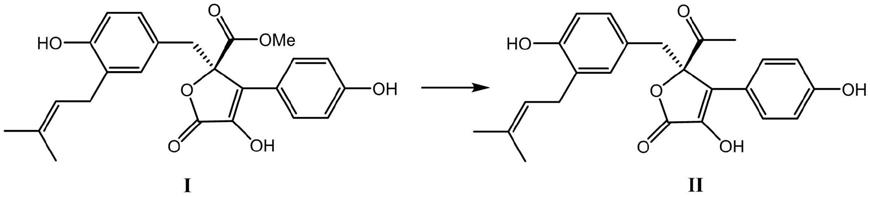
5.第一方面,本发明提供了一种新的丁内酯衍生物的合成方法,所述合成方法以丁内酯
‑
i为原料,在合适的有机溶剂反应体系中,经过多步反应,将呋喃环上的2
‑
甲酸甲酯还原为2
‑
甲基酮;所述多步反应过程包括取代反应、还原反应和/或氧化反应中的一步或多步。
6.如下所示:
[0007][0008]
其中:i为丁内酯
‑
i;ii为丁内酯
‑
i衍生物。
[0009]
优选的技术方案,所述合成方法包括以下步骤:
[0010]
(1)将丁内酯
‑
i(化合物i)溶解在有机溶剂1中,按物质的量之比1:(3.5~4.5)加入tbscl和咪唑,搅拌反应3~6h后,缓慢加入饱和的氯化铵溶液反应一定时间,然后淬灭反应,经分离、干燥、浓缩、提纯,得到化合物2;
[0011][0012]
(2)将化合物2溶解在有机溶剂2中,按物质的量之比1:(1.2~2.5)加入硼氢化锂,反应1~5h,缓慢加入饱和的氯化铵溶液反应一定时间,然后淬灭反应,经分离、干燥、浓缩、提纯,得到化合物3;
[0013][0014]
(3)将化合物3溶解在有机溶剂3中,按物质的量之比1:(1.2~2.5)加入戴斯马丁,反应10~60min,缓慢加入饱和的亚硫酸钠溶液和碳酸氢钠溶液反应一定时间,然后淬灭反应,经分离、干燥、浓缩、提纯,得到化合物4;
[0015][0016]
(4)将化合物4溶解在有机溶剂4中,加入缓慢加入三甲基铝的甲苯溶液,反应1~5h后,缓慢加入饱和的氯化铵溶液,然后分离、干燥、洗涤、蒸发浓缩,再加入有机溶剂4,按物质的量之比1:(2~4)加入戴斯马丁,反应0.5~1h,缓慢加入饱和的亚硫酸钠溶液和碳酸氢钠溶液,淬灭反应后,经分离、干燥、浓缩、提纯,得到化合物5;
[0017][0018]
(5)将化合物5溶解在有机溶剂5中,按物质的量之比1:(3~5)加入四正丁基氟化铵,反应0.5~1h,缓慢加入饱和的氯化铵溶液,反应一定时间,然后淬灭反应,经分离、干燥、浓缩、提纯,得到化合物ii,即目标产物。
[0019][0020]
优选的技术方案,所述有机溶剂1或有机溶剂2或有机溶剂3或有机溶剂4或有机溶剂5选自二氯甲烷、甲醇、四氢呋喃中的一种。
[0021]
优选的技术方案,所述分离方法为萃取,萃取选用的溶剂为二氯甲烷、乙酸乙酯中的至少一种。
[0022]
第二方面,本发明提供了一种采用上述的合成方法制备得到的新的丁内酯衍生物。
[0023]
第三方面,本发明还提供了一种丁内酯衍生物在抗过敏药物中的应用。
[0024]
优选的技术方案,所述丁内酯衍生物的给药方式包括经皮注射或口服。
[0025]
与现有技术相比,本发明的有益效果为:
[0026]
本发明合成方法制备得到的新的丁内酯衍生物具有和丁内酯
‑
i类似的抗过敏活性,且抗过敏活性显著。与丁内酯
‑
i相比,新的丁内酯衍生物在体内平均驻留时间得到了更显著的延长,体内达峰浓度也明显提高,药物半衰期也明显延长,生物利用度明显增加。因此,新的丁内酯衍生物能够明显提高体内的代谢稳定性,说明新的丁内酯衍生物经过结构改造之后,可应用于抗过敏药物的开发中。
附图说明
[0027]
图1是本发明实施例1中化合物ii的1h
‑
nmr谱图;
[0028]
图2是本发明实施例1中化合物ii的
13
c
‑
nmr谱图;
[0029]
图3是本发明实施例1中化合物ii的hsqc谱图;
[0030]
图4是本发明实施例1中化合物ii的1h
‑1h cosy谱图;
[0031]
图5是本发明实施例1中化合物ii的hmbc谱图。
具体实施方式
[0032]
为使本发明具体实施方式的目的和技术方案更加清楚,下面将结合本发明的附图和具体实施方式的实施实例,对本发明具体实施方式的技术方案进行清楚、完整地描述。显然,所描述的具体实施方式是本发明的一部分具体实施方式,而不是全部的具体实施方式。
[0033]
实施例1丁内酯衍生物的制备
[0034]
一种新的丁内酯衍生物的合成方法,包括如下步骤:
[0035]
(1)将4.17g化合物i溶于25ml的二氯甲烷中,加入咪唑2.67g,然后再加入tbscl5.9g,搅拌反应4h后,然后缓慢加入饱和的氯化铵溶液,搅拌30min后淬灭反应,使用乙酸乙酯萃取混合物,重复萃取操作多次,干燥,减压蒸发浓缩,利用硅胶色谱柱分离提纯产物,减压蒸发浓缩得到化合物2,经过计算,化合物2的产率为74%;
[0036]
(2)取431.3mg化合物2溶于20ml超干甲醇中,然后加入硼氢化锂的四氢呋喃溶液0.56ml,反应3h后,缓慢加入饱和的氯化铵溶液,然后用乙酸乙酯萃取混合物多次,干燥后
过滤,经过减压浓缩,利用硅胶色谱柱分离提纯产物后,经过减压蒸发浓缩得到化合物3,经计算,化合物3的产率为74%;
[0037]
(3)取296.1mg的化合物3溶于二氯甲烷中,然后加入戴斯马丁氧化剂339.3mg,反应半小时后,缓慢加入饱和的亚硫酸钠溶液和碳酸氢钠溶液,淬灭反应,然后用二氯甲烷萃取混合物多次,除水干燥后过滤洗涤,经过减压蒸发浓缩,利用硅胶色谱柱分离提纯,经过经减压蒸发浓缩得到化合物4,经计算其产率为57%。
[0038]
(4)将500.0mg的化合物4溶于二氯甲烷中,缓慢加入三甲基铝的甲苯溶液0.85ml,在25℃反应两小时后,缓慢加入饱和的氯化铵溶液,然后用二氯甲烷萃取混合物多次,除水干燥后过滤洗涤,经过减压蒸发浓缩后,加入二氯甲烷,加入戴斯马丁试剂865.2mg,反应半小时后,缓慢加入饱和nahco3和饱和na2so3,淬灭反应,用二氯甲烷萃取混合物多次,合并浓缩液,利用硅胶色谱柱分离提纯,获得化合物5,经计算,其产率为19%。
[0039]
(5)将92.5mg的化合物5溶于20ml的四氢呋喃中,然后加入四正丁基氟化铵的四氢呋喃溶液0.6ml,反应半小时后,缓慢加入饱和的氯化铵溶液,然后用乙酸乙酯萃取混合物多次,合并浓缩液,利用硅胶色谱柱分离提纯,获得化合物ii,经计算,其产率为80%。
[0040]
实施例2合成样品的结构分析
[0041]
由高分辨质谱确定化合物ii的分子式为c
24
h
24
o6,具有13个不饱和度。根据其1h
‑
nmr(附图1)和
13
c
‑
nmr(附图2),结合hsqc(附图3),得出式ii含有1个abx苯环,1个对位取代苯环,1个异戊二烯,1个甲基酮,以及典型的丁内酯片段。根据1h
‑1h cosy(附图4)和hmbc(附图5)的相关,可将上述五个片段连接在一起,从而确定化合物ii的平面结构。结合其比旋值,确定化合物ii为6
‑
脱氧
‑
丁内酯i,其理化数据如下:
[0042]
白色粉末,(c 1.0,ch3oh);1h和
13
c
‑
nmr数据,见表1;hresims(m/z)431.1471[m+na]
+
(calcd.for c
24
h
24
o6na,431.1463)。
[0043]
表1化合物ii的1h和
13
c nmr数据
[0044][0045]
实施例3丁内酯衍生物的体内代谢稳定性测试
[0046]
1、稳定性测试方法
[0047]
将sd大鼠6只(雌雄各半,体重160~240g)随机分成2组,每组3只雌性大鼠和3只雄性大鼠,分别灌胃(40mg/kg)给予化合物i和化合物ii。
[0048]
给药制剂按以下方法进行配制:
[0049]
化合物i和化合物ii分别用dmso配制成100mg/ml的dmso储备液;
[0050]
取dmso储备液和吐温80等体积混匀后,加入适量的生理盐水稀释至给药浓度,灌胃给药后0.08、0.25、0.5、1、2、4、6、8、10、12和24小时采集全血。将采集的全血样品于4℃离心分离得到血浆,血浆样品于
‑
70℃保存,待测。
[0051]
2、测试结果
[0052]
表2化合物i和ii药代动力学参数比较
[0053][0054]
通过表2可以看出,通过结构改造,口服药物衍生物体内达峰浓度达6655+2535.85ng/ml,相较blt
‑
1提高超过100倍,达峰时间更快tmax为0.08,说明药物可以更快吸收。药时曲线下峰面积增加大于130倍,药物半衰期也明显延长1倍。生物利用度较blt
‑
1明显增加,由8.29增加值9.88。体内吸收更快,代谢稳定性更好,生物利用度更高。静脉给药数据同时表明,该化合物代谢稳定性更好,auc较原化合物提高了近18倍,同时血浆中游离态药物更多,药物表观分布容积达到5858l/kg,药物的吸收效率更高。同时药物体内清除率也有所提高,半衰期更长达到了4.94,生物利用度更好。
[0055]
实施例4丁内酯衍生物的应用效果
[0056]
本实施例选择ige介导的rbl
‑
2h3细胞模型,检测细胞致敏后的脱颗粒效率,计算化合物对细胞脱颗粒效率的抑制率,并进一步计算化合物的抗过敏活性。
[0057]
本实施例分为以下4组:
[0058]
(1)阴性对照组(未激活组);
[0059]
(2)空白对照组(dnp
‑
bsa激活组);
[0060]
(3)阳性对照组(氯雷他定组);
[0061]
(4)化合物实验组:化合物i和ii。
[0062]
测试方法如下:
[0063]
(1)敏化细胞:胰酶消化回收rbl
‑
2h3细胞,加入96孔板,同时添加anti
‑
dnp
‑
ige,培养箱(37℃,5%co2)孵育过夜;
[0064]
(2)预保护细胞:将上述化合物i和化合物ii和阳性对照药氯雷他分别定溶于pbs中,分别取5μl样品,加95μltyrode’s缓冲液混匀;阴性对照组及空白对照组添加5μlpbs+95μltyrode’s缓冲液。每组分别取95μl加到培养板中,继续培养1h。
[0065]
(3)刺激细胞:空白对照组、阳性对照组及化合物实验组用dnp
‑
bsa刺激rbl
‑
2h3细胞1h;阴性对照组添加5μlpbs继续培养1h。
[0066]
(4)裂解细胞:回收细胞培养上清液后,向培养板中加tyrode’s缓冲液裂解细胞得到细胞裂解液。
[0067]
(5)β
‑
氨基己糖苷酶的活性测定:用25μl上清液或细胞裂解液分别入96孔荧光板,
每孔加4
‑
methylumbellife
‑
ryl
‑
n
‑
acetyl
‑
β
‑
d
‑
glucosaminide试剂,反应30min,用酶标仪获取每孔溶液360nm激发、450nm发射的荧光值。
[0068]
(6)脱颗粒效率计算公式:
[0069][0070]
(7)抗过敏抑制率计算公式
[0071][0072]
表3化合物i和ii的抗过敏活性结果
[0073][0074]
结果如表3所示,化合物ii具有和化合物i类似的抗过敏活性,其ic
50
分别为36和34μm,明显优于阳性对照药氯雷他定(92μm)。也说明化合物ii经过结构改造之后,具有很强的抗过敏活性,是一个非常有开发应用价值的抗过敏候选药物。
[0075]
在本说明书的描述中,所公开的具体特征、结构、材料或特点可以在任何一个或多个实施方案或示例中以任何合适的方式组合。尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定,都属于本发明保护的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1