硝基噻吩甲胺类光学异构体及其医药用途
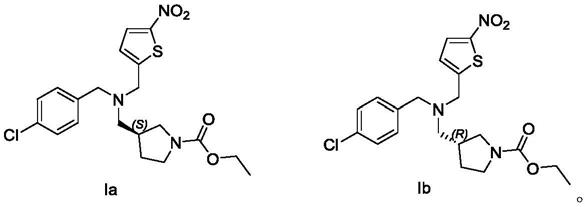
1.本发明属于药物化学领域,具体而言,涉及式ia和ib所示的光学异构体,即(s)-3-(((4-氯苄基)((5-硝基噻吩-2-基)甲基)氨基)甲基吡咯烷-1-甲酸乙酯和(r)-3-(((4-氯苄基)((5-硝基噻吩-2-基)甲基)氨基)甲基吡咯烷-1-甲酸乙酯,及其药学上可接受的盐,及其制备方法,以及ia和ib所示的光学异构体和它们的盐作为活性成分的药物组合物,以及在制备用于抗病毒、抗肿瘤、降血脂、减肥、美体、抗衰老、骨质酥松、失眠、倒时差等的药物中的用途。
2.
背景技术:
3.核受体是一大类转录调节因子,可直接将胞外激素信号和核内基因转录联系起来,广泛调节机体各种生理代谢过程。其中,rev-erbs包括rev-erbα(简称nr1d1)和rev-erbβ(简称nr1d2),是核受体亚家族1d组重要成员[molecular biology of the cell,1989,9(3):1128-1136;biochemistry,2009,48(29):7056-7071;cell growth&differentiation,1994,5(12):1357-1365]。人体中,超过50%的核受体被证明经其特异配体激活而发挥转录调节功能。rev-erbs与其内源性配体血红素/铁卟啉相互作用,调节目标基因的转录。
[0004]
rev-erbα、rev-erbβ相互协调共同来保护机体正常生物节律和代谢功能(genes&development,2012,26(6):657-667),比如,rev-erbs调节重要的时钟蛋白bmal1循环表达,对睡眠/唤醒周期具有关键作用(journal of biological rhythms,2005,20(5):391-403);rev-erbs通过调节载脂蛋白cⅲ(apocⅲ)的表达,以及与过氧化物酶增殖激活受体(pparα)竞争而抑制β-氧化途径的烯酰coa水解酶/3-羟化coa脱氢酶以及线粒体的细胞色素p450脂肪酸ω-羟化酶的表达等路径调控脂蛋白代谢(the journal of biological chemistry,2003,278(39):37672-37680);rev-erbs还可以通过靶向elovⅰ3和ⅰ型纤溶酶原激活物抑制物(pai-1),在机体脂肪生成及动脉粥样硬化病理过程中具有重要调节作用;同时,rev-erbs可能对机体维持糖代谢平衡起到关键的调节作用。
[0005]
rev-erbs在免疫调节也扮演重要的角色。比如,在血管平滑肌细胞中过量表达rev-erbα会诱导nf-κb介导的反式激活作用,前炎症细胞因子il-6和环氧化酶(cox)-2的表达量增高(febs letters,2004,561(1-3):69-74);单核细胞中rev-erbα的过度表达会对il10表达产生强烈的抑制作用;rev-erbα还通过与nlrp3和il1b的启动子区域结合,负责调节nlrp3炎症小体的表达;rev-erbα与th1 7细胞中的ror反应元件结合,抑制包括ror-γ在内的依赖于rorα的基因的表达等等。
甲酸乙酯和(r)-3-(((4-氯苄基)((5-硝基噻吩-2-基)甲基)氨基)甲基吡咯烷-1-甲酸乙酯。
[0012][0013]
根据本发明的另一个方面,本发明的另一个目的在于提供式ia和ib所示的硝基噻吩甲胺类光学异构体的药学上可接受的盐,所述盐包括无机酸盐和有机酸盐。
[0014]
根据本发明的另一个方面,本发明的另一个目的在于提供式ia和ib所示的硝基噻吩甲胺类光学异构体的制备方法,所述制备方法选自以下三种制备方法之一:
[0015]
制备方法一,如以下反应式1所示
[0016][0017]
反应式1
[0018]
步骤1)以5-硝基噻吩-2-甲醛(1)和对氯苄胺(2)为起始原料,在三乙酰氧基硼氢化钠存在的条件下,发生还原胺化反应得到化合物(3);
[0019]
步骤2)化合物(3)与光学纯度的化合物(4a)反应,再在三乙酰氧
基硼氢化钠存在条件下,还原胺化生成中间体(5a);
[0020]
步骤3)中间体(5a)在盐酸乙酸乙酯(hcl/ea)存在下,脱去boc保护基,得到中间体(6a);
[0021]
步骤4)中间体(6a)在三乙胺存在下,与氯甲酸乙酯反应得到光学纯的光学异构体(ia);
[0022]
步骤1’)以5-硝基噻吩-2-甲醛(1)和对氯苄胺(2)为起始原料,在三乙酰氧基硼氢化钠存在条件下,发生还原胺化反应得到(3);
[0023]
步骤2’)化合物(3)与光学纯度的化合物(4b)反应,再在三乙酰氧基硼氢化钠存在条件下,还原胺化生成中间体(5b);
[0024]
步骤3’)中间体5b)在盐酸乙酸乙酯(hcl/ea)存在下,脱去boc保护基,得到中间体(6b);
[0025]
步骤4’)中间体(6b)在三乙胺存在下,与氯甲酸乙酯反应得到光学纯的光学异构体(ib)。
[0026]
制备方法二,如以下反应式2所示
[0027][0028]
反应式2
[0029]
步骤1)以5-硝基噻吩-2-甲醛(1)和(s)-1-boc-3-氨甲基吡咯烷(7a)为起始原料,在三乙酰氧基硼氢化钠存在的条件下,发生还原胺化反应得到化合物(8a);
[0030]
步骤2)化合物(8a)与对氯苯甲醛反应,再在三乙酰氧基硼氢化钠存在条件下,还原胺化生成中间体(5a);
[0031]
步骤3)中间体(5a)在盐酸乙酸乙酯(hcl/ea)存在下,脱去boc保护基,得到中间体(6a);
[0032]
步骤4)中间体(6a)在三乙胺存在下,与氯甲酸乙酯反应得到光学纯的光学异构体(ia);
[0033]
步骤1’)以5-硝基噻吩-2-甲醛(1)和(r)-1-boc-3-氨甲基吡咯烷(7b)位起始原料,在三乙酰氧基硼氢化钠存在条件下,发生还原胺化反应得到(8b);
[0034]
步骤2’)化合物(8b)与光学对氯苯甲醛反应,再在三乙酰氧基硼氢化钠存在条件下,还原胺化生成中间体(5b);
[0035]
步骤3’)中间体(5b)在盐酸乙酸乙酯(hcl/ea)存在下,脱去boc保护基,得到中间体(6b);
[0036]
步骤4’)中间体(6b)在三乙胺存在下,与氯甲酸乙酯反应得到光学纯的光学异构体(ib)。
[0037]
制备方法三,如以下反应式3所示
[0038][0039]
反应式3
[0040]
步骤1)以对氯苯甲醛(9)和(s)-1-boc-3-氨甲基吡咯烷(7a)为起始原料,在三乙
酰氧基硼氢化钠存在的条件下,发生还原胺化反应得到化合物(10a);
[0041]
步骤2)化合物(10a)与5-硝基噻吩-2-甲醛(1)反应,再在三乙酰氧基硼氢化钠存在条件下,还原胺化生成中间体(5a);
[0042]
步骤3)中间体(5a)在盐酸乙酸乙酯(hcl/ea)存在下,脱去boc保护基,得到中间体(6a);
[0043]
步骤4)中间体(6a)在三乙胺存在下,与氯甲酸乙酯反应得到光学纯的光学异构体(ia);
[0044]
步骤1’)以对氯苯甲醛和(s)-1-boc-3-氨甲基吡咯烷(7b)位起始原料,在三乙酰氧基硼氢化钠存在条件下,发生还原胺化反应得到化合物(8b);
[0045]
步骤2’)化合物(8b)与5-硝基噻吩-2-甲醛(1)光学反应,再在三乙酰氧基硼氢化钠存在条件下,还原胺化生成中间体(5b);
[0046]
步骤3’)中间体(5b)在盐酸乙酸乙酯(hcl/ea)存在下,脱去boc保护基,得到中间体(6b);
[0047]
步骤4’)中间体6b)在三乙胺存在下,与氯甲酸乙酯反应得到光学纯的光学异构体(ib)。
[0048]
根据本发明的另一个方面,本发明的另一个目的在于提供一种药物组合物,所述药物组合物含有作为活性成分的式ia或ib所示的硝基噻吩甲胺类光学异构体及其药学上可接受的盐,以及药学上可接受的载体或辅料。
[0049]
根据本发明的另一个方面,本发明的另一个目的在于提供式ia或ib所示的硝基噻吩甲胺类光学异构体,其药学上可接受的盐,以及包含其作为活性成分的药物组合物在制备抗衰老、神经退行性疾病、抗肿瘤、减肥、降血脂、降血糖、抗骨质疏松、抗病毒的药物中的用途。
[0050]
优选地,所述神经退行性疾病包括老年痴呆、帕金森等病症。
[0051]
优选地,所述抗病毒的药物中所述病毒为流感病毒、黄病毒、丙肝病毒、库尼亚病毒、冠状病毒。
[0052]
优选地,所述肿瘤包括白血病、脑胶质瘤、肝癌、肺癌、胰腺癌、大肠癌、乳腺癌。
[0053]
有益效果
[0054]
通常而言,光学对映体之间存在的活性关系可能存在3种情况,第一种情况是两者药效相同;第二种情况是一个有效、另一个无效;第三种情况是二者药效相反。意料之外的是,本发明的ia和ib的细胞毒性远小于sr9009(消旋体),提示安全性方面ia和ib具有毒性协同叠加作用;而减肥和抗病毒实验显示ia不具有抗病毒和减肥药效,ib具有较好的减肥和抗病毒药效,sr9009的显示较弱药效(相当于ia和ib合用),提示ia拮抗了ib的部分药效。ia和ib之间,这种既存在一方面相互协同、又存在另一方面无效异构体拮抗有效异构体的关系是少见的。鉴于ib具有更好的安全性和更强的药效,因此,相对于sr9009而言,ib具有更好的开发前景和更低的安全风险。
附图说明
[0055]
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单的介绍,显而易见地,下面描述中的
附图是本发明的一些实施方式,对本领域普通技术人员而言,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0056]
图1为消旋体sr9009、式ia和ib的硝基噻吩甲胺类光学异构体细胞毒性活性测试结果;
[0057]
图2为消旋体sr9009、式ia和ib的硝基噻吩甲胺类光学异构体抗病毒活性测试结果;
[0058]
图3为消旋体sr9009、式ia和ib的硝基噻吩甲胺类光学异构体减肥药效测试结果;
[0059]
图4为式ia的硝基噻吩甲胺类光学异构体与空白溶媒的抗衰老药效实验对比图;
[0060]
图5为式ib的硝基噻吩甲胺类光学异构体与空白溶媒的抗衰老药效实验对比图。
[0061]
图6为消旋体sr9009、式ia和ib的硝基噻吩甲胺类光学异构体抗早幼粒白血病细胞实验对比图。
具体实施方式
[0062]
以下,将详细地描述本发明。在进行描述之前,应当理解的是,在本说明书和所附的权利要求书中使用的术语不应解释为限制于一般含义和字典含义,而应当在允许发明人适当定义术语以进行最佳解释的原则的基础上,根据与本发明的技术方面相应的含义和概念进行解释。因此,这里提出的描述仅仅是出于举例说明目的的优选实例,并非意图限制本发明的范围,从而应当理解的是,在不偏离本发明的精神和范围的情况下,可以由其获得其他等价方式或改进方式。
[0063]
本发明化合物或其药学上可接受的盐可以存在为其水合物,溶剂化物或前药的形式。因此,本发明化合物或其药学上可接受的盐的水合物,溶剂化物或前药也包括在本发明的范围内。
[0064]
本公开中所采用的术语“药学上可接受的”,是针对那些化合物、材料、组合物和/或剂型而言,它们在可靠的医学判断的范围之内,适用于与人类和动物的组织接触使用,而没有过多的毒性、刺激性、过敏性反应或其它问题或并发症,与合理的利益/风险比相称。
[0065]
术语“药学上可接受的盐”是指本发明化合物的盐,由本发明的式ia和ib的光学异构体与相对无毒的酸或碱制备。当本发明的化合物中含有相对酸性的功能团时,可以通过在纯的溶液或合适的惰性溶剂中用足够量的碱与其中性形式接触的方式获得碱加成盐。药学上可接受的碱加成盐包括钠、钾、钙、铵、有机氨或镁盐或类似的盐。当本发明的式ia和ib的光学异构体中含有相对碱性的官能团时,可以通过在纯的溶液或合适的惰性溶剂中用足够量的酸与其中性形式接触的方式获得酸加成盐(即药学上可接受的盐),实例包括无机酸盐以及有机酸盐,所述无机酸包括例如盐酸、氢溴酸、硝酸、碳酸,碳酸氢根,磷酸、磷酸一氢根、磷酸二氢根、硫酸、硫酸氢根、氢碘酸、亚磷酸等;所述有机酸包括如苯甲酸、2-羟基乙磺酸、甲磺酸、氨基磺酸、苯磺酸、苯乙酸、扁桃酸、丙二酸、丙酸、草酸、对氨基苯磺酸、对甲苯磺酸、多聚半乳糖醛、反丁烯二酸、泛酸、富马酸、谷氨酸、琥珀酸、甲烷磺酸、酒石酸、抗坏血酸、邻苯二甲酸、马来酸、柠檬酸、苹果酸、葡庚糖、葡糖酸、羟乙磺酸、乳酸、乳糖、十二烷基磺酸、双羟萘酸、水杨酸、辛二酸、亚磷酸等;亚乙酸、依地酸、乙醇酸、乙酸、乙烷磺酸、异丁酸、延胡索酸、枸橼酸、三氟醋酸、硬脂酸等类似的酸;还包括氨基酸(如精氨酸等)的盐,以及如葡糖醛酸等有机酸的盐。
[0066]
优选地,所述式ia和ib所示的光学异构体的药学上可接受的盐包括盐酸盐、氢溴酸盐、硫酸盐、磷酸盐、甲磺酸盐、琥珀酸盐、枸橼酸盐、草酸盐、富马酸盐、马来酸盐、乳酸盐、延胡索酸盐、醋酸盐、三氟醋酸盐等无机酸盐和有机酸盐。
[0067]
术语“药学上可接受的载体”是指能够递送本发明有效量活性物质、不干扰活性物质的生物活性并且对宿主或者患者无毒副作用的任何制剂或载体介质代表性的载体包括水、油、蔬菜和矿物质、膏基、洗剂基质、软膏基质等。这些基质包括悬浮剂、增粘剂、透皮促进剂等。它们的制剂为化妆品领域或局部药物领域的技术人员所周知。关于载体的其他信息,可以参考remington:the science and practice of pharmacy,21st ed.,lippincott,williams&wilkins(2005),该文献的内容通过引用的方式并入本文。
[0068]
针对药物或药理学活性剂而言,术语“有效量”或“治疗有效量”是指无毒的但能达到预期效果的药物或药剂的足够用量。对于本发明中的口服剂型,组合物中一种活性物质的“有效量”是指与该组合物中另一种活性物质联用时为了达到预期效果所需要的用量。有效量的确定因人而异,取决于受体的年龄和一般情况,也取决于具体的活性物质,个案中合适的有效量可以由本领域技术人员根据常规试验确定。
[0069]
以下实施例仅是作为本发明的实施方案的例子列举,并不对本发明构成任何限制,本领域技术人员可以理解在不偏离本发明的实质和构思的范围内的修改均落入本发明的保护范围。除非特别说明,以下实施例中使用的试剂和仪器均为市售可得产品。
[0070]
实施例1:式ia所示的硝基噻吩甲胺类光学异构体的合成
[0071]
步骤1)n-(4-氯苄基)-1-(5-硝基噻吩-2-基)甲胺(3)的合成
[0072][0073]
在150ml圆底烧瓶中加入对氯苄胺(2124mg,15mmol),加入1,2-二氯乙烷(15ml)溶解,并将5-硝基噻吩-2-甲醛(1571mg,10mmol)溶解于1,2-二氯乙烷(25ml)中,滴入上述溶液,室温搅拌2h;然后,将三乙酰氧基硼氢化钠分5批次加入反应液中,每次加635.8mg(3mmol)。加毕,tlc检测,待原料点消失。将反应液倒入水(60ml)中,用乙酸乙酯萃取(30ml
×
4),合并有机相,无水硫酸钠干燥,过滤滤除干燥剂,滤液浓缩,得深黄色油状物。将残留物通过柱层析分离纯化,以石油醚:乙酸乙酯=5:1至3:1进行梯度洗脱,收集所需组分,减压蒸干得到2.627g黄色油状产物(3),收率92.9%。
[0074]
步骤2)(s)-3-((((4-氯苄基)((5-硝基噻吩-2-基)甲基)氨基)甲基)吡咯烷-1-羧酸叔丁酯(5a)的合成
[0075][0076]
在圆底烧瓶(150ml)中加入化合物3(1414mg,5mmol)、(r)-3-甲酰基吡咯烷-1-羧
酸叔丁酯(1196mg,6mmol,北京金瑞梅香,光学纯度98%)、冰醋酸(600mg,10mmol),再加入1,2-二氯甲烷(30ml),室温反应0.5h;将三乙酰氧基硼氢化钠分5批加入反应液中,每次423.8mg(2mmol)。tlc检测反应完全。将反应液倒入40ml水中,用乙酸乙酯(25ml
×
4)萃取,合并有机相,无水硫酸钠干燥,过滤滤除干燥剂,滤液减压蒸馏除去有机溶剂,得黄色油状物。将残留物通过柱层析分离,以石油醚:乙酸乙酯=5:1至3:1进行梯度洗脱,收集所需组分,减压蒸干得黄色油状物5a(2.258g),收率96.9%。[a]
d24
=-12.7
°
(c=0.69g/100ml,ch3oh)。
[0077]
步骤3)(r)-n-(4-氯苄基)-1-(5-硝基噻吩-2-基)-n-(吡咯烷-3-基甲基)甲胺盐酸盐(6a)的合成
[0078][0079]
在圆底烧瓶(100ml)中加入盐酸/乙酸乙酯(1.9ml,7.5mmol,4m),加入乙酸乙酯(15ml),将化合物5a(1165mg,2.5mmol)溶于15ml乙酸乙酯,在低温下(≤5℃),逐滴缓慢滴入盐酸/乙酸乙酯中,得到类白色不溶固体。tlc检测无原料点。减压蒸馏除去有机溶剂得到1.057g类白色固体(6a),收率96.3%。
[0080]
步骤4)光学异构体(s)-3-((((4-氯苄基)((5-硝基噻吩-2-基)甲基)氨基)甲基)吡咯烷-1-甲酸乙酯(ia)的合成
[0081][0082]
在圆底烧瓶(50ml)中加入化合物6a(1053mg,2.4mmol),加入无水二氯甲烷(10ml),置于-5℃冰盐浴中,滴加728.6mg(3mmol)三乙胺,反应10min,将氯甲酸乙酯(520.8mg,2mmol)溶于二氯甲烷(3ml)中,缓慢滴入上述反应液。tlc检测反应完全。反应液倒入水(20ml)中,用二氯甲烷(15ml
×
4)萃取,合并有机相,无水硫酸钠干燥,过滤滤除干燥剂,滤液减压蒸馏浓缩,残留物通过柱层析分离,以石油醚:乙酸乙酯=5:1至2:1梯度洗脱,收集所需组分,减压蒸干得0.695g棕色油状物光学异构体(ia),收率66.1%,[a]
d24
=-10.4
°
(c=0.69g/100ml,ch3oh)。ms(esi,m/z):438.12(m+h)
+
,460.10(m+na)
+
,1h-nmr(600mhz,cdcl3):δ7.79(d,j=3.6hz,1h),7.32(m,4h),6.87(s,1h),4.12(p,j=21.6,13.8,6.6hz,2h),3.76(s,2h),3.62(m,2h),3.54(s,1h),2.48(s,3h),2.02(m,1h),1.70
–
1.51(m,2h),1.29
–
1.21(m,5h)。
[0083]
实施例2:式ib所示的硝基噻吩甲胺类光学异构体的合成
[0084]
步骤1)(r)-3-((((4-氯苄基)((5-硝基噻吩-2-基)甲基)氨基)甲基)吡咯烷-1-羧
酸叔丁酯(5b)的合成
[0085][0086]
在圆底烧瓶(150ml)中加入化合物3(918.8mg,3.25mmol)、(s)-3-甲酰基吡咯烷-1-羧酸叔丁酯(777.1mg,3.9mmol,北京金瑞梅香,光学纯度98%)、冰醋酸(390mg,6.5mmol),再加入1,2-二氯甲烷(20ml),室温反应0.5h;将三乙酰氧基硼氢化钠分5批加入反应液中,每次275.5mg(1.3mmol)。tlc检测反应完全。将反应液倒入水(40ml)中,用乙酸乙酯(25ml
×
4)萃取,合并有机相,无水硫酸钠干燥,过滤滤除干燥剂,滤液减压蒸馏除去有机溶剂,得黄色油状物。将残留物通过柱层析分离,以石油醚:乙酸乙酯=5:1至3:1进行梯度洗脱,收集所需组分,减压蒸干得1.497g黄色油状物(5b),收率98.9%。[a]
d24
=+13.8
°
(c=0.52g/100ml,ch3oh)。
[0087]
步骤2)(s)-n-(4-氯苄基)-1-(5-硝基噻吩-2-基)-n-(吡咯烷-3-基甲基)甲胺盐酸盐(6b)的合成
[0088][0089]
在圆底烧瓶(100ml)中加入盐酸/乙酸乙酯(2.43ml,9.72mmol,4m),加入乙酸乙酯(15ml),冰盐浴降温至零下5℃。另将化合物5b(1509mg,2.5mmol)溶于乙酸乙酯(15ml),逐滴缓慢滴入到上述的盐酸/乙酸乙酯中,得到类白色不溶固体。tlc检测无原料点。抽滤,得到类白色固体6b(1.095g),收率96.3%。
[0090]
步骤3)光学异构体(r)-3-((((4-氯苄基)((5-硝基噻吩-2-基)甲基)氨基)甲基)吡咯烷-1-甲酸乙酯(ib)的合成
[0091][0092]
在圆底烧瓶(50ml)中加入化合物6b(1097mg,2.5mmol),加入无水二氯甲烷(10ml),置于-5℃冰盐浴中,滴加三乙胺(759mg,7.5mmol),搅拌10min。另将氯甲酸乙酯(542.5mg,5mmol)溶于二氯甲烷(3ml)中,缓慢滴入到上述反应液中。tlc检测反应完全。反应液倒入20ml水中,用二氯甲烷(15ml
×
4)萃取,合并有机相,无水硫酸钠干燥,过滤滤除干燥剂,滤液减压蒸馏浓缩,残留物通过柱层析分离,以石油醚:乙酸乙酯=5:1至2:1(v:v)梯
度洗脱,收集所需组分,减压蒸干得棕色油状物式ib的光学异构体(0.840g),收率76.7%,[a]
d24
=+10.4
°
(c=0.69g/100ml,ch3oh)。ms(esi,m/z):438.11(m+h)
+
,460.09(m+na)
+
,1h-nmr(600mhz,cdcl3):δ7.72(d,j=3.6hz,1h),7.25(m,4h),6.80(s,1h),4.09
–
3.99(m,2h),3.69(s,2h),3.61
–
3.50(m,2h),3.47(s,1h),3.33(s,1h),2.41(s,3h),1.95(m,1h),1.19(m,6h)。光学纯度98%。
[0093]
实施例3:式ia所示的硝基噻吩甲胺类光学异构体的合成
[0094]
步骤1)(s)-3-(((5-硝基噻吩-2-基)甲基)氨基)甲基)吡咯烷-1-羧酸叔丁酯(8a)的合成
[0095][0096]
于50ml圆底烧瓶中加入600.9mg(3mmol)(s)-1-boc-3-氨甲基吡咯烷,加入10ml1,2-二氯乙烷溶解,并将471.3mg(3mmol)5-硝基噻吩-2-甲醛(1)溶解于5ml的1,2-二氯乙烷中,滴入上述溶液,室温搅拌2h;将三乙酰氧基硼氢化钠分五批次加入反应液中,每次254.2mg(1.2mmol),时间间隔1h。tlc检测反应完全。将反应液倒入20ml水中,用二氯甲烷萃取(15ml
×
4),合并有机相,无水硫酸钠干燥,过滤滤除干燥剂,滤液浓缩,得深黄色油状物。将残留物通过柱层析分离纯化,以石油醚:乙酸乙酯=3:1至2:1进行梯度洗脱,收集所需组分,减压蒸干得到922mg黄色油状产物(8a),收率90.0%。ms(esi,m/z):364.1(m+na)
+
,1h-nmr(500mhz,cdcl3)δ7.82(d,j=4.1hz,1h),6.91(dd,j=9.5,3.2hz,1h),4.07-4.01(m,2h),3.63-3.40(m,2h),3.32(td,j=18.2,9.0hz,1h),3.05(dd,j=18.7,11.0hz,1h),2.71(dd,j=14.9,7.2hz,2h),2.41-2.31(m,1h),2.06(s,1h),1.71-1.58(m,1h),1.47(s,9h)。
[0097]
步骤2)(s)-3-((((4-氯苄基)((5-硝基噻吩-2-基)甲基)氨基)甲基)吡咯烷-1-羧酸叔丁酯(5a)的合成
[0098][0099]
于50ml圆底烧瓶中加入883mg(2.58mmol)化合物8a、362.6mg(2.58mmol)对氯苯甲醛、309.6mg(5.16mmol)冰醋酸,再加入15ml的1,2-二氯甲烷,室温反应1h;将三乙酰氧基硼氢化钠分5批加入反应液中,每次218.6mg(1.03mmol),时间间隔1h。tlc检测反应完全。将反应液倒入20ml水中,用二氯甲烷(15ml
×
4)萃取,合并有机相,无水硫酸钠干燥,过滤滤除干燥剂,滤液减压蒸馏除去有机溶剂,得黄色油状物。将残留物通过柱层析分离,以石油醚:乙酸乙酯=5:1至4:1进行梯度洗脱,收集所需组分,减压蒸干得889mg黄色油状物(5a),收率74.0%,[a]
d20
=-13.4(c=0.1569g/100ml,ch3oh)。1h-nmr(500mhz,dmso)δ8.02(d,j=4.2hz,1h),7.40(dd,j=21.5,8.4hz,4h),7.13(d,j=4.1hz,1h),3.82(dt,j=20.5,16.3hz,2h),3.69
–
3.53(m,2h),3.38(dd,j=17.1,10.2hz,2h),3.13(t,j=7.2hz,2h),
2.87(dd,j=17.8,7.8hz,1h),2.38(t,j=8.1hz,2h),1.90(dd,j=12.2,6.1hz,1h),1.37(d,j=10.0hz,10h).ms(esi,m/z):488.1(m+na)
+
。
[0100]
步骤3)(r)-n-(4-氯苄基)-1-(5-硝基噻吩-2-基)-n-(吡咯烷-3-基甲基)甲胺盐酸盐(6a)的合成
[0101][0102]
于50ml圆底烧瓶中加入盐酸/乙酸乙酯1.41ml(5.64mmol,4m),加入10ml乙酸乙酯,将878mg(1.88mmol)化合物5a溶于15ml乙酸乙酯,逐滴缓慢滴入盐酸/乙酸乙酯中,得到类白色不溶固体。tlc检测无原料点。减压蒸馏除去有机溶剂得到789.8mg类白色固体(6a),收率95.7%。
[0103]
步骤4)光学异构体(s)-3-((((4-氯苄基)((5-硝基噻吩-2-基)甲基)氨基)甲基)吡咯烷-1-甲酸乙酯(ia)的合成
[0104][0105]
于50ml圆底烧瓶中加入778mg(1.77mmol)化合物6a,加入5ml无水二氯甲烷,置于-5℃冰盐浴中,滴加537.4mg(5.31mmol)三乙胺,反应20min,将384.1mg(3.54mmol)氯甲酸乙酯溶于3ml二氯甲烷中,缓慢滴入上述反应液。tlc检测反应完全。反应液倒入15ml水中,用二氯甲烷(10ml
×
4)萃取,合并有机相,无水硫酸钠干燥,过滤滤除干燥剂,滤液减压蒸馏浓缩,残留物通过柱层析分离,以石油醚:乙酸乙酯=5:1至3:1梯度洗脱,收集所需组分,减压蒸干得0.667g黄色油状物(ia),收率86.1%。[a]
d20
=-10.9(c=0.1581g/100ml,ch3oh)。
[0106]
实施例4:式ib所示的硝基噻吩甲胺类光学异构体的合成
[0107]
步骤1)(r)-3-(((5-硝基噻吩-2-基)甲基)氨基)甲基)吡咯烷-1-羧酸叔丁酯(8b)的合成
[0108][0109]
于50ml圆底烧瓶中加入600.9mg(3mmol)(r)-1-boc-3-氨甲基吡咯烷,加入10ml1,
2-二氯乙烷溶解,并将471.3mg(3mmol)5-硝基噻吩-2-甲醛(1)溶解于5ml的1,2-二氯乙烷中,滴入上述溶液,加入冰醋酸360mg,室温搅拌2h;将三乙酰氧基硼氢化钠分五批次加入反应液中,每次254.2mg(1.2mmol),时间间隔1h。tlc检测反应完全。将反应液倒入20ml水中,用二氯甲烷萃取(15ml
×
4),合并有机相,无水硫酸钠干燥,过滤滤除干燥剂,滤液浓缩,得深黄色油状物。将残留物通过柱层析分离纯化,以石油醚:乙酸乙酯=3:1至2:1进行梯度洗脱,收集所需组分,减压蒸干得到924mg黄色油状产物(8b),收率90.2%。ms(esi,m/z):364.1(m+na)
+
,1h-nmr(500mhz,dmso)δ8.02(d,j=4.2hz,1h),7.09(d,j=4.2hz,1h),3.95(s,2h),3.39(dd,j=10.7,7.4hz,1h),3.28(dt,j=14.9,5.6hz,1h),3.20
–
3.12(m,1h),2.96(ddd,j=18.5,10.5,7.5hz,1h),2.56(dd,j=11.7,5.1hz,1h),2.27(dt,j=13.5,7.0hz,1h),1.92(dd,j=11.4,6.9hz,1h),1.62
–
1.47(m,1h),1.39(s,10h).
[0110]
步骤2)(r)-3-((((4-氯苄基)((5-硝基噻吩-2-基)甲基)氨基)甲基)吡咯烷-1-羧酸叔丁酯(5b)的合成
[0111][0112]
于100ml圆底烧瓶中加入877mg(2.56mmol)化合物8b、359.8mg(2.56mmol)对氯苯甲醛、307.2mg(6.5mmol)冰醋酸,再加入20ml1,2-二氯甲烷,室温反应1h;将三乙酰氧基硼氢化钠分5批加入反应液中,每次216.4mg(1.02mmol),时间间隔1h。tlc检测反应完全。将反应液倒入20ml水中,用乙酸乙酯(20ml
×
4)萃取,合并有机相,无水硫酸钠干燥,过滤滤除干燥剂,滤液减压蒸馏除去有机溶剂,得黄色油状物。将残留物通过柱层析分离,以石油醚:乙酸乙酯=5:1至4:1进行梯度洗脱,收集所需组分,减压蒸干得0.824g黄色油状物(5b),收率69.1%。ms(esi,m/z):488.1(m+na)
+
,1h nmr(500mhz,dmso)δ8.02(d,j=4.2hz,1h),7.41(dd,j=23.1,8.4hz,4h),7.13(d,j=4.2hz,1h),3.82(dt,j=16.0,10.3hz,2h),3.71
–
3.53(m,2h),3.39(d,j=10.1hz,1h),3.13(t,j=7.2hz,2h),2.87(dd,j=17.8,7.6hz,1h),2.38(dd,j=10.5,5.6hz,2h),1.90(dt,j=11.9,5.9hz,1h),1.55
–
1.43(m,1h),1.37(d,j=9.8hz,10h).[a]
d20
=+13.0(c=0.1154g/100ml,ch3oh)。
[0113]
步骤3)(s)-n-(4-氯苄基)-1-(5-硝基噻吩-2-基)-n-(吡咯烷-3-基甲基)甲胺盐酸盐(6b)的合成
[0114]
[0115]
于100ml圆底烧瓶中加入盐酸/乙酸乙酯0.75ml(3mmol,4m),加入10ml乙酸乙酯,将466mg(1mmol)化合物5b溶于15ml乙酸乙酯,逐滴缓慢滴入盐酸/乙酸乙酯中,得到类白色不溶固体。tlc检测无原料点。减压蒸馏除去有机溶剂得到0.338g类白色固体(6b),收率77%。
[0116]
步骤4)光学异构体(r)-3-((((4-氯苄基)((5-硝基噻吩-2-基)甲基)氨基)甲基)吡咯烷-1-甲酸乙酯(ib)的合成
[0117][0118]
于50ml圆底烧瓶中加入1097mg(2.5mmol)化合物6b,加入10ml无水二氯甲烷,置于-5℃冰盐浴中,滴加759mg(7.5mmol)三乙胺,反应10min,将542.5mg(5mmol)氯甲酸乙酯溶于3ml二氯甲烷中,缓慢滴入上述反应液。tlc检测反应完全。反应液倒入20ml水中,用二氯甲烷(15ml
×
4)萃取,合并有机相,无水硫酸钠干燥,过滤滤除干燥剂,滤液减压蒸馏浓缩,残留物通过柱层析分离,以石油醚:乙酸乙酯=5:1至3:1梯度洗脱,收集所需组分,减压蒸干得0.909g黄色油状物光学异构体(ib),收率83.0%。ms(esi,m/z):438.11(m+h)
+
,460.09(m+na)
+
,1h-nmr(500mhz,dmso)δ8.02(d,j=4.2hz,1h),7.41(dd,j=23.5,8.4hz,4h),7.13(d,j=4.2hz,1h),4.02-3.95(m,2h),3.86(dt,j=16.4,4.3hz,1h),3.83-3.77(m,1h),3.67(dd,j=13.8,2.9hz,1h),3.62
–
3.56(m,1h),3.46-3.39(m,1h),3.18(dd,j=12.2,5.9hz,2h),2.99-2.91(m,1h),2.39(d,j=6.9hz,2h),1.97-1.88(m,1h),1.56-1.45(m,1h),1.16(ddd,j=18.6,10.7,5.7hz,4h).[a]
d20
=+11.2(c=0.1334g/100ml,ch3oh)。
[0119]
实施例5:式ia所示的硝基噻吩甲胺类光学异构体的合成
[0120]
步骤1)(s)-3-((4-氯苄基)氨基)甲基)吡咯烷-1-羧酸叔丁酯(10a)的合成
[0121][0122]
于50ml圆底烧瓶中加入400.6mg(2mmol)(s)-1-boc-3-氨甲基吡咯烷(7a),加入10ml 1,2-二氯乙烷溶解,并将281.1mg(2mmol)对氯苯甲醛(9)溶解于5ml的1,2-二氯乙烷中,滴入上述溶液,加入240mg冰醋酸,室温搅拌2h;将三乙酰氧基硼氢化钠分五批次加入反应液中,每次169.5mg(0.8mmol),时间间隔1h。tlc检测反应完全。将反应液倒入20ml水中,用二氯甲烷萃取(15ml
×
4),合并有机相,无水硫酸钠干燥,过滤滤除干燥剂,滤液浓缩,得透明油状物粗品10a。
[0123]
步骤2)(s)-3-((((4-氯苄基)((5-硝基噻吩-2-基)甲基)氨基)甲基)吡咯烷-1-羧酸叔丁酯(5a)的合成
[0124][0125]
于50ml圆底烧瓶中加入649.7mg(2mmol)化合物10a、314.2mg(2mmol)5-硝基噻吩-2-甲醛、240mg(4mmol)冰醋酸,再加入15ml1,2-二氯甲烷,室温反应1h;将三乙酰氧基硼氢化钠分5批加入反应液中,每次169.5mg(0.8mmol),时间间隔1h。tlc检测反应完全。将反应液倒入20ml水中,用二氯甲烷(15ml
×
4)萃取,合并有机相,无水硫酸钠干燥,过滤滤除干燥剂,滤液减压蒸馏除去有机溶剂,得黄色油状物。将残留物通过柱层析分离,以石油醚:乙酸乙酯=5:1至4:1进行梯度洗脱,收集所需组分,减压蒸干得609mg黄色油状物(5a),收率65.3%。[a]
d20
=-13.8(c=0.1050g/100ml,ch3oh)。
[0126]
步骤3)(r)-n-(4-氯苄基)-1-(5-硝基噻吩-2-基)-n-(吡咯烷-3-基甲基)甲胺盐酸盐(6a)的合成
[0127][0128]
在圆底烧瓶(100ml)中加入盐酸/乙酸乙酯(2.3ml,9.0mmol,4m),加入乙酸乙酯(15ml),将化合物5a(1398mg,3.0mmol)溶于15ml乙酸乙酯,在低温下(≤5℃),逐滴缓慢滴入盐酸/乙酸乙酯中,得到类白色不溶固体。tlc检测无原料点。减压蒸馏除去有机溶剂得到1.30g类白色固体(6a),收率98.7%。
[0129]
步骤4)光学异构体(s)-3-((((4-氯苄基)((5-硝基噻吩-2-基)甲基)氨基)甲基)吡咯烷-1-甲酸乙酯(ia)的合成
[0130]
反应式:
[0131][0132]
于50ml圆底烧瓶中加入778mg(1.77mmol)化合物6a,加入5ml无水二氯甲烷,置于-5℃冰盐浴中,滴加537.4mg(5.31mmol)三乙胺,反应20min,将384.1mg(3.54mmol)氯甲酸乙酯溶于3ml二氯甲烷中,缓慢滴入上述反应液。tlc检测反应完全。反应液倒入15ml水中,用二氯甲烷(10ml
×
4)萃取,合并有机相,无水硫酸钠干燥,过滤滤除干燥剂,滤液减压蒸馏浓
缩,残留物通过柱层析分离,以石油醚:乙酸乙酯=5:1至3:1梯度洗脱,收集所需组分,减压蒸干得0.667g黄色油状物(ia),收率86.1%。[a]
d20
=-10.9(c=0.1581g/100ml,ch3oh)。
[0133]
实施例6:式ib所示的硝基噻吩甲胺类光学异构体的合成
[0134]
步骤1)(r)-3-((4-氯苄基)氨基)甲基)吡咯烷-1-羧酸叔丁酯(10b)的合成
[0135]
反应式:
[0136][0137]
于50ml圆底烧瓶中加入400.6mg(2mmol)(r)-1-boc-3-氨甲基吡咯烷(7b),加入10ml 1,2-二氯乙烷溶解,并将281.1mg(2mmol)对氯苯甲醛(9)溶解于5ml的1,2-二氯乙烷中,滴入上述溶液,加入240mg冰醋酸,室温搅拌2h;将三乙酰氧基硼氢化钠分五批次加入反应液中,每次169.5mg(0.8mmol),时间间隔1h。tlc检测反应完全。将反应液倒入20ml水中,用二氯甲烷萃取(15ml
×
4),合并有机相,无水硫酸钠干燥,过滤滤除干燥剂,滤液浓缩,得透明油状物粗品10b。
[0138]
步骤2)(r)-3-((((4-氯苄基)((5-硝基噻吩-2-基)甲基)氨基)甲基)吡咯烷-1-羧酸叔丁酯(5b)的合成
[0139][0140]
于50ml圆底烧瓶中加入649.7mg(2mmol)化合物10b、314.2mg(2mmol)5-硝基噻吩-2-甲醛、240mg(4mmol)冰醋酸,再加入15ml1,2-二氯甲烷,室温反应1h;将三乙酰氧基硼氢化钠分5批加入反应液中,每次169.5mg(0.8mmol),时间间隔1h。tlc检测反应完全。将反应液倒入20ml水中,用二氯甲烷(15ml
×
4)萃取,合并有机相,无水硫酸钠干燥,过滤滤除干燥剂,滤液减压蒸馏除去有机溶剂,得黄色油状物。将残留物通过柱层析分离,以石油醚:乙酸乙酯=5:1至4:1进行梯度洗脱,收集所需组分,减压蒸干得624mg黄色油状物(5b),收率66.9%。[a]
d20
=+13.2(c=0.1062g/100ml,ch3oh)。
[0141]
步骤3)(r)-n-(4-氯苄基)-1-(5-硝基噻吩-2-基)-n-(吡咯烷-3-基甲基)甲胺盐酸盐(6a)的合成
[0142]
[0143]
在圆底烧瓶(100ml)中加入盐酸/乙酸乙酯(2.0ml,7.8mmol,4m),加入乙酸乙酯(15ml),将化合物5a(1215mg,2.6mmol)溶于15ml乙酸乙酯,在低温下(≤5℃),逐滴缓慢滴入盐酸/乙酸乙酯中,得到类白色不溶固体。tlc检测无原料点。减压蒸馏除去有机溶剂得到1.13g类白色固体(6a),收率98.6%。
[0144]
步骤4)光学异构体(r)-3-((((4-氯苄基)((5-硝基噻吩-2-基)甲基)氨基)甲基)吡咯烷-1-甲酸乙酯(ib)的合成
[0145][0146]
于50ml圆底烧瓶中加入1097mg(2.5mmol)化合物6b,加入10ml无水二氯甲烷,置于-5℃冰盐浴中,滴加759mg(7.5mmol)三乙胺,反应10min,将542.5mg(5mmol)氯甲酸乙酯溶于3ml二氯甲烷中,缓慢滴入上述反应液。tlc检测反应完全。反应液倒入20ml水中,用二氯甲烷(15ml
×
4)萃取,合并有机相,无水硫酸钠干燥,过滤滤除干燥剂,滤液减压蒸馏浓缩,残留物通过柱层析分离,以石油醚:乙酸乙酯=5:1至3:1梯度洗脱,收集所需组分,减压蒸干得0.909g黄色油状物光学异构体(ib),收率83.0%,[a]
d20
=+11.2(c=0.1334g/100ml,ch3oh)。ms(esi,m/z):438.11(m+h)
+
,460.09(m+na)
+
,1h-nmr(500mhz,dmso)δ8.02(d,j=4.2hz,1h),7.41(dd,j=23.5,8.4hz,4h),7.13(d,j=4.2hz,1h),4.02
–
3.95(m,2h),3.86(dt,j=16.4,4.3hz,1h),3.83-3.77(m,1h),3.67(dd,j=13.8,2.9hz,1h),3.62-3.56(m,1h),3.46-3.39(m,1h),3.18(dd,j=12.2,5.9hz,2h),2.99-2.91(m,1h),2.39(d,j=6.9hz,2h),1.97-1.88(m,1h),1.56
–
1.45(m,1h),1.16(ddd,j=18.6,10.7,5.7hz,4h)。
[0147]
测试实施例1:mdck细胞毒性活性测试
[0148]
药品用dmso配成母液,再用培养液作稀释,各8个稀释度,其中sr9009测试浓度分别为0.15625um、0.3125um、0.625um、1.25um、2.5um、5.0um、10um、20um;ia和ib测试浓度分别为0.625um、1.25um、2.5um、5.0um、10um、20um、40um、80um。mdck细胞接种96孔培养板(5
×
104个细胞),置5%co2,37℃的培养箱培养。加入药液,于测试浓度下孵育48小时,加入2-(2-甲氧基-4-硝基苯基)-3-(4-硝基苯基)-5-(2,4-二磺酸苯)-2h-四唑单钠盐(cck8)(10ul),再孵育4小时,测定450nm的光密度值(od值),计算细胞活力。结果如图1所示,sr9009在5um的浓度下即有一定的细胞毒性,10um下细胞活力约为10%,显示较大的细胞毒性;而光学异构体ia和ib有毒浓度为20um;80um的浓度下细胞活力依然接近70%,毒性比较小;由此光学异构体ia和ib可见细胞毒性明显弱于sr9009,说明光学异构体ia和ib具有较好的安全性。sr9009是ia和ib的混合物,但是它的毒性比ia和ib都要大很多,这提示光学异构体具有sr9009不具备的安全性。文献报道sr9009有不依赖于rev erbα的药理活性(pnas 2019,116(25):12147
–
12152),这提示sr9009具有多靶效应。进一步,手性对映体ia和ib也具有多靶效应,二者的多靶效应在毒性方面具有叠加效应,造成了sr9009具有较大的细胞毒性。
[0149]
测试实施例2:抗病毒活性测试
[0150]
采用定量qpcr法检测sr9009,ia和ib对流感病毒rna的影响。在无毒剂量下测试化合物的抗病毒活性。根据测试实施例1结果,在无毒浓度或低毒浓度下,测试化合物的抗病毒活性,sr9009测试浓度为0.156-5.0um;ia和ib的测试浓度为0.1-20um。将2
×
105cells/ml mdck细胞均匀铺于12孔板中,1ml/孔。约20h后用pbs洗2遍,将pr8原液稀释9000倍后,感染mdck细胞2h,感染结束后,用pbs洗1遍,加入不同化合物处理(用含有0.1%bsa稀释药物),同时设置病毒对照组(细胞只加病毒处理不加药物)和细胞空白对照组(细胞正常培养,不加病毒也不加药物),在感染20h后提取细胞总rna,测定rna浓度,再使用逆转录试剂盒对500ng总rna进行逆转录反应,最后,根据标准曲线法,计算出待测样品中pr8病毒np蛋白的拷贝数,跟模型组进行比较,计算抑制率。结果如图1显示,sr9009低剂量下较弱的抗病毒活性,抑制率≤40%;而高浓度≥5um浓度下,细胞毒全部死亡,无法提取mrna,毒性效应大于抗病毒药效,不具备药用潜力;ia活性较弱,ib而具有较好的抗病毒活性,半数有效浓度低于5um。由于sr9009存在多靶效应,而本发明显示ia和ib抗病毒活性的差异,提示ia和ib具有不同的作用靶标,具有不同的药物用途。
[0151]
测试实施例3:光学异构体的小鼠减肥药效测试
[0152]
4-5w周龄的c57bl/6n雄性小鼠140只,给与高饲料造模,然后选择造模成功的肥胖小鼠100只,随机分组,每组25只,分别给于sr9009、ia和ib,剂量为100mg/kg体重,每天2次;设置溶媒对照组,给药期间给药组和溶媒对照组均给予高脂饲料喂养。同时,设置正常小鼠,给与普通饲料作为对照。记录小鼠体重,以给药的第一天体重为100%,其它时间体重与第一天的比值为体重增长率,得到体重增长率,结果如下图3。结果显示sr9009在给药第一周具有比较显著的降低体重的作用,与文献(nature,2012,485:123-127)一致。但是,随着时间延长,小鼠体重呈上升趋势,减肥效应不能持续。而ia未显示减肥作用,ib显示持续的减肥作用。
[0153]
测试实施例4:抗衰老药效实验
[0154]
首先,进行秀丽隐杆线虫的培养与同期化。线虫(品系n2,中国科学院张宏实验室惠赠)的培养参照brenner所用方法,将l4线虫挑于含e.coli op50的ngm板中,将培养皿置于20℃生化培养箱中培养,每3天转一次皿。线虫的同步化采用次氯酸钠裂解法,用已灭菌的m9溶液将正在产卵期的线虫从ngm皿上冲洗下来,3000rpm离心3min,重复此步骤2~3次。向管中加入5.5ml裂解液(250μl naclo、250μl 10m naoh和5ml高压水,现用现配,注意避光),用涡旋震荡仪震荡8~10min,直至虫体全部消失。再往管中加5ml m9溶液,终止裂解,3000rpm离心3min,弃上清收集沉淀(虫卵沉淀呈白色)。沉淀用m9溶液反复清洗3次,再转到m9溶液中20℃培养,16-20h后转l1线虫至含e.coli op50的ngm板中,约48h线虫发育至l4时期,完成同期化。然后,进行秀丽隐杆线虫生命周期实验,参照jiang y等的方法,将实验分为对照组和实验组,其中实验组包含1个浓度(在大肠埃希菌op50(中国科学院张宏实验室惠赠)菌液中的摩尔浓度为1mm)的lxc051或lxc0350,对照组用等体积的高压水代替。每个浓度设置3个平行,每个平行挑取120只l4时期的线虫到各组ngm板中,置于20℃培养,此时计为线虫寿命的第0天。在线虫生命的早期(第0~10天)使用含5μm fudr的ngm板来抑制线虫产卵,第11天转至普通的ngm板中。为了保证药物的浓度、充足的食物和防止污染,每3天将线虫转至新的皿中。同时每天记录线虫的存活、死亡、丢失等数量,直至所有的线虫均死亡。无移动及吞咽动作,用铂金丝轻触虫体无任何反应的线虫判定为死亡;钻进培养基中、
丢失、爬至培养皿壁上干死、阴部炸裂和形成虫袋的线虫应被剔除。数据统计分析,采用graphpad prism 8软件进行统计学分析,所得结果采用x
±
s表示,组间比较采用单因素方差分析,两两比较采用t检验,以p<0.05为差异有统计学意义。结果显示,如图4所示,ia组线虫生存曲线明显长于空白溶媒(2%dmso)组,显著延长线虫的寿命,中位数寿命显示,ia组寿命约为27天,长于溶媒对照组的24天。如图5所示,ib组线虫生存曲线明显长于空白溶媒(2%dmso)组,显著延长线虫的寿命,中位数寿命显示,ib组寿命约为26天,长于溶媒对照组的24天。ia的中位数生存期长于ib。
[0155]
测试实施例5:抗早幼粒白血病细胞实验
[0156]
采用mtt比色法检测细胞增殖,其检测原理为活细胞中线粒体内琥珀酸脱氢酶在mtt作用下还原成不溶于水的蓝紫色结晶甲瓒并在细胞内沉积,而死细胞并没有这种特性。二甲基亚砜(dmso)能溶解细胞内沉积的甲瓒,在酶联免疫检测仪的492nm波长处检测光吸收值(od值)。在一定细胞数内mtt结晶与细胞数成正比,通过计算od值可反映活细胞数。
[0157]
实验步骤包括:
[0158]
1.制备hl细胞(军事医学研究院放射医学研究所余祖胤课题组惠赠)悬液:取对数增殖期细胞,调整细胞密度为1.1
×
105个/ml。
[0159]
2.接种铺板:96孔板,每孔接种90ul(1
×
104个细胞)。
[0160]
3.实验分组:设置调零孔(仅含完全培养基)和空白对照孔(加入不含药物的细胞悬液),8个给药孔(终浓度分别为100、50、25、12.5、6.25、3.125、1.56、0.78μm),每个浓度设5个复孔,每孔分别含10ul相应浓度ia所示的光学异构体药物,同样地设置ib所示的光学异构体药物的给药孔。
[0161]
4.37℃5%co2孵育48h。孵育48h之后加入配制好的mtt溶液(5mg/ml),孵育4h后离心(2000r/min
×
10min),小心吸出上清,加入100ul dmso,低速振荡10min,使结晶物充分溶解。在酶标仪492nm测量各孔od值,并用origin检测半数抑制浓度(ic50)值。结果如图6所示,抗白血病活性ia》sr9009》ib。
[0162]
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应所述以权利要求的保护范围为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1