一类新型Smad3蛋白降解剂及其应用的制作方法
本发明属于药物化学领域。具体而言,涉及一类新型的靶向smad3蛋白的protac分子,其制备方法,以及包含该类化合物的药物组合物。
背景技术:
1、泛素-蛋白酶体途径是较普遍的一种内源蛋白降解方式,需要降解的蛋白先被泛素化修饰,然后被蛋白酶体分解为较小的多肽、氨基酸以及可以重复使用的泛素。protac(proteolysis targeting chimeras),即蛋白降解靶向嵌合体,是近年来新兴的热门研究领域[1]。protac分子一般可分为三个部分,一端为与特定靶蛋白结合的小分子片段(warhead),另一端为具有泛素化功能的e3连接酶的配体(e3 ligase ligand),以及将两者连接起来的连接体(linker)。 protac分子利用了细胞的蛋白泛素化降解途径,可选择性的降解目标靶蛋白。具体而言,由于protac分子的两端分别为靶蛋白与e3连接酶的配体片段,所以protac分子可同时与靶蛋白和e3连接酶结合,促进了靶蛋白的泛素化,进而被蛋白酶体识别并加以降解。传统的小分子抑制剂,不仅需要与靶蛋白具有高的结合力,同时需要与靶蛋白结合后,能够降低靶蛋白的功能活性。与传统小分子抑制剂不同,protac分子中与靶蛋白结合的小分子片段,并不一定需要对靶蛋白的活性有影响,只要具有较好的结合力即可。这一特点,使得protac分子可以靶向一些传统意义上的“不可成药”靶点,比如转录因子,骨架蛋白等等[2]。这类蛋白往往缺少明显的活性位点,从而很难利用传统的小分子抑制剂发挥药效作用。因此,protac具有非常广阔的应用前景。目前报道的protac分子,不仅应用于肿瘤领域中常见的一些激酶靶点,如egfr[3],alk[4],cdk[5]等,还可应用于表观遗传领域的 brd4[2,6],hdac[7],以及核受体ar[8],er[9]等等。
2、smad家族的蛋白根据其分子结构和不同的生物学功能,可分为r-smad(receptor-regulated smad)、co-smad(common-mediated smad)和i-smad (inhibitorysmad)三个亚群。它们做为转化生长因子-β(transforming growth factor beta,tgf-β)信号通路中的转运蛋白,参与介导细胞外的tgf-β信号到细胞核内调控相关靶基因的表达。tgf-β与细胞膜上的ii型受体结合后,招募并激活i型受体(alk5),进而磷酸化细胞内的r-smad;磷酸化的 r-smad与co-smad及其他转录因子形成复合物,进入细胞核内调控下游基因的转录[10]。smad3属于r-smad的一员,介导tgf-β信号通路参与的一系列生物学反应,包括促细胞上皮-间质转化、促组织纤维化、促血管生成、促肿瘤的免疫逃逸等[11]。
3、目前已知tgf-β是促进肾脏的纤维化进程中的一个关键因子,smad3 被tgf-β激活后形成的转录因子复合物可以直接结合到一系列胶原形成基因启动子区域,促进基质层的形成[12]。将小鼠体内smad3基因敲除后可以抑制多种肾病中的纤维化[13-16],在肾病小鼠模型中过表达smad7以抑制 smad3活性也可以有效延缓肾脏损伤进程[17]。一个特异性的smad3小分子抑制剂,sis3,可以有效地抑制糖尿病肾病和梗阻性肾病小鼠模型中的肾脏纤维化进程[18,19]。此外,也有多项研究提示smad3在多种肿瘤进展中起到重要作用[20],在llc肺癌和b16f10黑色素瘤细胞cdx小鼠模型中,smad3 基因敲除和药理学抑制对于癌症生长,侵入和转移都产生了显著的抑制效果[21]。这些结果提示smad3是一个极具开发前景的组织纤维化和肿瘤治疗药物靶点。
4、中山大学附属第一医院的王欣等报道了靶向smad3蛋白的protac分子,其降解作用通过招募von hippel-lindau(vhl)e3连接酶实现[22]。本专利申请中,设计并合成了一系列招募cereblon e3连接酶的smad3 protac 分子,并且其对smad3蛋白的降解作用优于文献化合物。
技术实现思路
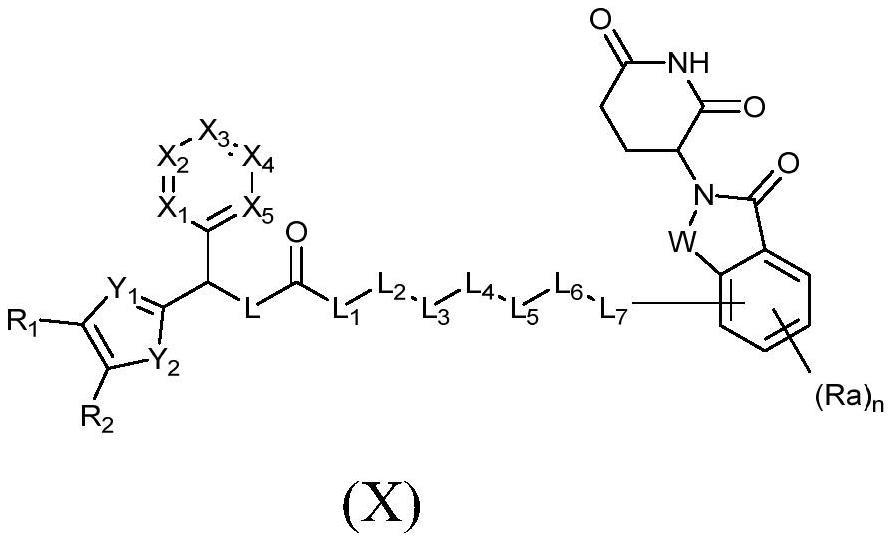
1、本发明的目的在于提供一种能够降解smad3蛋白的protac分子。具体而言,在一方面中,本发明提供了式(i)化合物,或其互变异构体、立体异构体、前药、晶型、药学上可接受的盐、水合物或溶剂合物:
2、
3、其中,
4、w为crr’或c=o;
5、其中r和r’独立地选自h、d、卤素、c1-6烷基或c1-6卤代烷基;
6、l为nr”;
7、其中r”独立地选自h、c1-6烷基或c1-6卤代烷基;
8、x1为crx1或n;
9、x2为crx2或n;
10、x3为crx3或n;
11、x4为crx4或n;
12、x5为crx5或n;
13、其中rx1、rx2、rx3、rx4和rx5独立地选自h、d、卤素、c1-6烷基或c1-6卤代烷基;
14、y1为cry1或n;
15、y2为o、s或nry2;
16、其中ry1独立地选自h、d、卤素、c1-6烷基或c1-6卤代烷基;
17、ry2选自h、c1-6烷基或c1-6卤代烷基;
18、r1和r2独立地选自h、d、卤素、c1-6烷基或c1-6卤代烷基;
19、或者r1和r2相连,并且与它们连接的原子一起形成
20、其中z1为crz1或n;
21、z2为crz2或n;
22、z3为crz3或n;
23、z4为crz4或n;
24、其中rz1、rz2、rz3和rz4独立地选自h、d、卤素、c1-6烷基或c1-6卤代烷基;
25、l1为cr1r1’;
26、l2为o、s、nr2”或cr2r2’;
27、l3为o、s、nr3”或cr3r3’;
28、l4为o、s、nr4”或cr4r4’;
29、l5为o、s、nr5”或cr5r5’;
30、l6为o、s、nr6”或cr6r6’;
31、l7为o、s、nr7”或cr7r7’;
32、或者l1、l5和l6分别独立地不存在;
33、或者-l6-l7-结合形成-ch=ch-或-c≡c-;
34、或者l2和l5的取代基相连,并且与l2、l3、l4和l5一起形成c5-7亚环烷基、5-7元亚杂环基、c6-10亚芳基或5-7元亚杂芳基;
35、或者l2和l4的取代基相连,并且与l2、l3和l4一起形成c5-7亚环烷基、5-7元亚杂环基、c6-10亚芳基或5-7元亚杂芳基;
36、或者l3和l5的取代基相连,并且与l3、l4和l5一起形成c5-7亚环烷基、5-7元亚杂环基、c6-10亚芳基或5-7元亚杂芳基;
37、或者-l2-l3-l4-表示c5-7亚环烷基、5-7元亚杂环基、c6-10亚芳基或5-7 元亚杂芳基;
38、或者-l3-l4-l5-表示c5-7亚环烷基、5-7元亚杂环基、c6-10亚芳基或5-7 元亚杂芳基;
39、其中r1、r1’、r2、r2’、r3、r3’、r4、r4’、r5、r5’、r6、r6’、 r7和r7’独立地选自h、d、卤素、c1-6烷基或c1-6卤代烷基;
40、或者r2和r2’、r3和r3’、r4和r4’、r5和r5’、r6和r6’、r7和 r7’结合形成=o;
41、r2”选自h、c1-6烷基或c1-6卤代烷基;
42、r3”选自h、c1-6烷基或c1-6卤代烷基;
43、r4”选自h、c1-6烷基或c1-6卤代烷基;
44、r5”选自h、c1-6烷基或c1-6卤代烷基;
45、r6”选自h、c1-6烷基或c1-6卤代烷基;
46、r7”选自h、c1-6烷基或c1-6卤代烷基;
47、条件是,相邻的两个原子不能同时为杂原子。
48、在另一方面,本发明提供了本发明化合物的制备方法。
49、在另一方面,本发明提供了一种药物组合物,其含有本发明化合物和药学上可接受的赋形剂。在具体实施方案中,本发明化合物以治疗有效量提供。在具体实施方案中,本发明化合物以预防有效量提供。
50、在另一方面,本发明提供了本发明化合物或本发明的药物组合物在制备用于治疗和/或预防smad3蛋白介导的疾病的药物中的用途。
51、在另一方面,本发明提供了一种在受试者中治疗和/或预防smad3蛋白介导的疾病的方法,包括向所述受试者给药本发明化合物或其药物组合物。
52、在另一方面,本发明提供了本发明化合物或其药物组合物,其用于治疗和/或预防smad3蛋白介导的疾病。
53、在另一方面,上文提及的smad3蛋白介导的疾病选自自身免疫疾病及炎症,组织纤维化和肿瘤等。
54、由随后的具体实施方式、实施例和权利要求,本发明的其他目的和优点将对于本领域技术人员显而易见。
- 还没有人留言评论。精彩留言会获得点赞!