一种测序接头及其测序分析系统的制作方法
1.本发明涉及分子生物学技术领域,尤其是核酸测序,具体涉及一种测序接头及其测序分析系统。
背景技术:
2.自2014年新英格兰医学杂志发表宏基因组二代测序(mngs)确诊钩体病的首例临床应用案例以来,mngs在新发病原体鉴定、罕见重要病原体诊断等方面取得诸多进展,临床上也认可了mngs在急危重症感染领域的应用。病原mngs是指把疑似感染部位的样本,提取样本中的核酸,将核酸片段接上和测序芯片可以杂交的dna接头,接头上含有可以区分不同样本的标签序列(index),通过高通量测序仪测序,把测得的序列和含有各种病原体的数据库进行比对,可以快速锁定病原体。同时通过区分标签序列index,就可以实现在一次运行中同时并行测序多个样本,充分利用测序通量,并降低成本。
3.常规的truseq测序接头如附图1a,接头末端有一个t碱基垂悬,用来和加入到目标片段中的样本中的末端a碱基垂悬互补进行t-a连接。read1测序引物最后一位含有t,因此测序时会直接先测到插入片段,而不会测到t碱基。read 1测序完成后,换标签序列测序引物来获得标签序列。一般用来做病原体测序的时候,read 1测序部分约需要500分钟,完成index 1标签测序大概需要50分钟。也就是整体测序完大概需要550分钟(~9小时),测序仪才可以获得全部的序列,并可以区分是哪个具体样本。
4.综上所述,加上文库制备的时间(4小时)和测序时间(9 ~ 10小时),整体上从开始准备样本到最终可以开始分析每一个样本需要14个小时。如果是illumina nextseq类似通量的测序仪,每次产生约20g数据,需要分析一个小时左右。 因此从最初样本到产生结果至少要15个小时,大致流程耗时如附图1b所示。检测时效性差,亟需改进。
技术实现要素:
5.本发明的目的是提供一种测序接头,可以在保证较高的测序质量和通量下,实现多个样本的边测序边分析,极大地缩短了周转时间(tat),提高了检测的时效。
6.通过对现有测序接头检测过程的分析,为了提高检测的时效性,有两个关键的时间点需要去解决,1.测序时间长,占了整体tat时间的50%;2. 分析需要一个小时,并且只有等待全部测序结束后,也就是需要进行至少14个小时以后,获得每一个样本的标签序列index后才能拆分数据再开始进行序列比对分析。
7.为了达到上述目的,本发明采用如下的技术方案:一种测序接头(如附图2所示),呈部分互补配对的y字形结构,其中一条链从5’至3’依次包括:内部index序列、index1测序引物结合区域序列、index1序列、与芯片探针结合的p7序列;另一条链上从5’至3’依次包括:与芯片探针结合的p5序列、read1测序引物结合区域序列、内部index序列和t碱基垂悬。这两条链的内部标签序列区域完全互补配对,测序引物结合区域序列部分互补配对。在p5序列与read1测序引物结合区域序列之间还可以增
加index2序列。
8.新的接头由于在read1测序引物结合区域下游添加了内部index序列,会出现在测序的过程中,到固定的位置(t-a连接处),测序结果都是t碱基。如附图3所示,单独采用长度为8 bp的内部index序列接头测序时各循环数下碱基比例会在第九个循环时出现极高比例的t碱基,这样会造成单一碱基荧光强度过强,而其它碱基一律没有信号,a/t/c/g四个碱基之间的平衡比例被打破,增加了测序仪分析具体碱基的难度,会被分析软件判断为该位置碱基的测序质量存在问题,导致较大比例的测序序列无法通过质控,大大减少了有效数据产出。对于二代测序仪来说,测序刚开始的几个碱基尤为重要,起到定位簇位置的作用,因此前十个循环内,出现整个测序芯片在某一个循环都是相同的碱基会大大降低测序质量和数量。
9.为了解决此问题,本发明进一步优化设计用于区分不同样品的内部index序列,设计具有两种至四种以上长度的内部index序列,且相邻长短的内部index序列之间的长度差可以是一个碱基、两个碱基或者多个碱基,但为了节约测序成本和减少花在测序内部index上的时间,长度差优选为一个碱基。在使用的时候必须要有不同长度的内部index序列接头组合使用,避免t-a连接的t碱基出现在同一个测序循环中。使用的所有内部index序列在组合后应达到内部index序列在各位置轮测序循环中的碱基比例基本平衡,index这样尽可能提高前10个碱基测序质量。
10.附图4为采用6 bp、7 bp和8 bp三种长短的内部index序列接头时的各轮循环下的碱基比例结果,通过混合不同的index长度的接头,从而错开了碱基t出现的循环,从附图4可以看到有三个循环出现稍高比例的t,并不是都集中出现在同一个循环,这样优化后可以得到高质量的测序结果。
11.推荐至少两种至四种以上的内部index序列组合完成多样本的标记和测序。且各种内部index长度的接头的实际使用比例要达到均衡。为了节约测序成本和减少花在测序内部index上的时间,最优使用为6bp、7bp和8bp的三种内部index序列长度组合,且每种内部index长度的接头要占接头总量的三分之一左右;或最优使用为6bp、7bp、8bp和9bp的四种内部index序列长度组合,且每种内部index长度的接头要占接头总量的四分之一左右。比如两种长短的内部index序列组合的时候,一种内部index序列长度为6个碱基,一种内部index序列长度为7个碱基,两类样本各50%混合。这样会出现,第七个碱基测序结果为50%的序列为t(6碱基内部index序列的t-a连接处),剩下的50%的序列为7碱基长度的内部index序列的第七个碱基(且不允许设计为t)。这个组合在测序到第八个碱基就会出现50%的信号又是t的现象(7碱基内部index序列的t-a连接处)index。从第九个碱基开始,所有的序列都是插入片段中的序列。如果有三至四种不同长度的内部index序列组合,能更好地在各个循环把碱基比例均匀分配。如三种不同长短的内部index序列组合,一种内部index序列长度为6个碱基,一种内部index序列长度为7个碱基,一种内部index序列长度为8个碱基,各占1/3混合。或者是四种不同长短的内部index序列组合,一种内部index序列长度为6个碱基,一种内部index序列长度为7个碱基,一种内部index序列长度为8个碱基,还有一种内部index序列长度为9个碱基,各占1/4混合。
12.相邻长短的内部index序列之间的长度差可以是一个碱基、两个碱基或者多个碱基,但index优选为一个碱基,如6个碱基,7个碱基和8个碱基的组合。
13.本发明进一步优选,使用的所有内部index序列在组合后达到内部index序列在各轮测序循环中的碱基比例基本平衡。。一般而言,在一次测序中文库数(或使用的index数)大于等于4个时,内部index序列在各轮测序循环中的atcg四种碱基的比例分别各自控制在8% ~ 50%为合适,比例控制在12.5% ~ 37.5%为最优。
14.除上述要求外,使用的所有内部index序列还应满足:(1) 任意两个内部index序列的最小汉明距离为3;(2) 排除含有三个以上相同连续碱基的index序列;(3) 内部index的前两个碱基不应该是“gg”。一般而言,index序列的长度越长,atcg四种碱基能组合创造出的index种类越多。为了设计出足够多的index用于多样本测序,且任意两个index序列之间的最小汉明距离大于等于3,内部index的序列长度为6个碱基以上为宜。
15.由于改变了测序序列的产生方式,测序开始后,几个循环后就可以测得内部index序列来区分每一个样本,因此可以不用等待全部测序(9~10个小时)完成以后才开始分析具体样本的序列。另外由于测序循环数越多,测得的序列会越来越长,本发明随着测序的进展,可实现实时分析得到不同长度序列的比对和分析结果。
16.本发明的另一目的是针对上述新的接头结构,提供新的测序分析系统(见附图2b),进行边测序边分析,实时分析得到序列比对和分析结果。本系统具有实时循环分析、分析时间短、准确性高的优点。
17.本发明的测序分析系统包括:1.测序监控模块:用于实时监控测序进度并触发分析任务。
18.测序监控模块会定时扫描测序目录,监测测序进度。当测序进行到足够长度(最短长度为22bp)时,由监控程序发出信号触发后续分析步骤,并随着测序进行对延伸后的序列持续进行实时分析,可在完成上一轮分析后马上启动下一轮分析。
19.2.数据生成模块:用于将测序生成的bcl文件转换成fastq文件,并过滤低质量序列;同时针对特殊设计的接头使用特异性分析程序将序列数据拆分至对应的样本中。
20.数据生成模块将测序生成的bcl文件转换成fastq文件,并对测序数据进行质控,去除低质量数据和含接头序列,保证进入后续分析流程的数据质量可靠。同时,在测序时使用特殊设计的接头使其既可用于区分不同的样本,也适用于极速分析过程,并使用特异性分析程序将序列数据拆分至对应的样本中。
21.3.数据过滤模块:用于去除通过质控的序列中的人源序列。
22.数据过滤模块将通过质控的序列用快速比对软件与人源基因组数据库进行比对,去除比对上的人源序列。输出未比对上的序列,得到去除了人源序列的非人源数据。
23.4. 数据分析模块:用于将非人源序列比对到病原微生物基因组数据库中;数据分析模块将非人源数据与病原微生物基因组数据库进行比对,得到微生物序列比对结果。对于有多条比对结果的序列,系统会选取比对得分在得分区间[l, u]内的比对结果,计算这些参考序列所属分类单元(taxon)的最近共同祖先(lca),作为该序列的最终比对结果。得分区间的确定方式为:,,其中代表理论上比对最高得分,代表理论上比对最低得分,代表该序列的比对结果的最高得分,代表得分区间范围参数,默认值为20。在分析比对结果时,同时记录每
条序列所比对上的物种是否唯一,是否是完全比对等信息。
[0024]
5. 报告生成模块:用于统计分析比对结果,输出分析报告。
[0025]
报告生成模块根据序列的比对结果,统计每个分类单元测到的序列数目,对于含有更小节点的taxon,既统计本taxon上的序列数目,也统计该taxon及其所有子节点的序列数目,并统计每个taxon的唯一比对和完全比对的序列数目。
[0026]
通过实施上述技术方案,相比于现有技术的核酸测序,本发明具有如下的优点:1.内部index位于测序引物和插入片段之间,极速分析时候,首先测得index,这样就可以在测序早期将来自于不同样本的序列分开,不用等待测序全部完成。
[0027]
2.index至少采用两种以上不同长短的长度(优选3种长度,分别是6/7/8 bp)。不同长短的index序列,避免了常规方法会出现在同一个循环处,都是t的结果,从而降低了测序质量。
[0028]
3.index每一个位置的碱基要求碱基比例分布均匀。
[0029]
4.分析软件在测序仪获得22个序列以后,就开始分析病原体信息,每个循环持续跟进分析,达到ngs实时分析的目的。
[0030]
5.结合不同长短的index接头和实时分析的方法,把原本上机后至少需要11个小时才能知道结果,缩短到测序开始后5个小时左右就可以第一时间知道样本中微生物的基本情况,达到ngs极速分析的目的。
附图说明
[0031]
附图1a为现有技术中常规测序接头结构示意图和测序流程示意图;附图1b为现有技术中常规测序接头系统各流程耗时分布图;附图2为本发明所述的测序结构示意图和测序流程示意图;附图2b为使用本发明的测序接头的测序分析系统流程示意图;附图3为单独采用长度为8 bp的内部index序列接头测序时各循环数下碱基比例;附图4为采用长度为6 bp、7 bp和8 bp三种长度的内部index序列接头测序时各循环数下碱基比例;附图5 为实施例1中所使用的测序接头详细序列结构;附图6为实施例1中使用本发明测序接头和传统的illumina truseq接头测序时各循环数下碱基比例对比图;附图7为实施例1中使用本发明测序接头和传统的illumina truseq接头测序时测序质量和最终文库数据量对比图;附图8为实施例2中嗜肺军团菌在各分析循环测得的序列数;附图9为实施例2中克氏柠檬酸杆菌在各分析循环测得的序列数。
具体实施方式
[0032]
需要说明的是,以下实施案例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施案例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施案例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施案例技术方案的范围。
[0033]
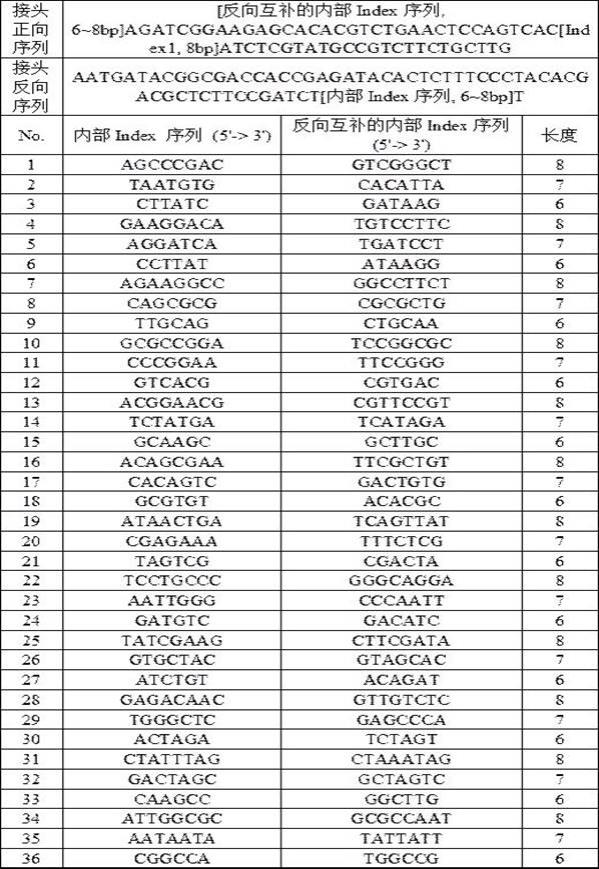
实施例1本实施例的一种测序接头,呈部分互补配对的y字形结构。其中一条链从5’至3’依次包括:内部index序列、index1测序引物结合区域序列、index1序列、与芯片探针结合的p7序列。另一条链上从5’至3’依次包括:与芯片探针结合的p5序列、read1测序引物结合区域序列、内部index序列和t碱基垂悬。这两条链的内部标签序列区域完全互补配对,测序引物结合区域序列部分互补配对。其结构如附图5所示,采用三种长短的内部index序列,长度分别为6 bp、7 bp和8 bp。
[0034]
本实施例共设计了48个内部index序列。 他们被分为16组,每组都有6 bp、7 bp和8 bp长短的内部index序列。
[0035]
内部index序列满足以下要求:(1) 任意两个内部index序列的最小汉明距离为3 (2) 排除含有三个以上相同连续碱基的index序列。 (3) 内部index的前两个碱基不应该是“gg”。 (4) 7bp index的第7个碱基和8bp index的第7个碱基不应为t,8bp index的第8个碱基不应为t。(5)组合内的index各个测序位置的碱基比例都是人工调整以达到相对平衡。
[0036]
具体序列和设计如下:
使用上述内部index接头和传统的illumina truseq接头各建了153个文库,然后分别分批上机测序:内部index接头文库分成8次上机测序,每次上机约18~20个文库,且每种内部index长度的接头要占该轮测序中使用的接头总量的三分之一左右;truseq接头文库分成5次上机测序,每次上机约30-31个文库。对比两种接头的测序质量,结果如附图6(两种接头测序时各循环数下碱基比例对比)和附图7所示(两种接头测序时测序质量和最终文
库数据量对比)。
[0037]
如图6所示,使用优化后的内部index接头能够提供较平衡的碱基比例,仅仅是第9个循环测到的t碱基比例稍高(相对truseq接头而言),但对测序质量没有影响。
[0038]
如图7所示,使用优化后的内部index接头能够保证较高的合格簇百分比和q30分数,这些测序质量指标和truseq接头的数据对比时并无显著差异。使用优化后的内部index接头拆分数据时既可以单独使用内部index拆分,也可以使用内部index + index1 做双index拆分,并且最终得到的文库数据量和使用truseq时也无显著差异。
[0039]
实施例2为了评估本系统的分析性能,我们用本发明的测序分析系统对两个临床阳性样本进行了分析。其中样本1的临床结果为嗜肺军团菌感染,样本2的临床结果为克氏柠檬酸杆菌感染。两个样本的分析时间及检测结果如下表1所示。嗜肺军团菌在各分析循环测得的序列数见附图8,克氏柠檬酸杆菌在各分析循环测得的序列数见附图9。
[0040]
表1. 临床样本检测分析时间统计分析结果表明,在测序读长为22bp的极速分析第一份报告中,本系统已经能够敏感地检测到阳性病原菌;随着测序进行,检测到的病原菌序列数缓慢升高,在几个循环后趋于稳定。因此,对于病原体感染阳性样本,本系统能够在极早期检出阳性病原体,并给出可靠的分析结果。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1