一种ARV-471中间体的制备方法与流程
一种arv
‑
471中间体的制备方法
技术领域
1.本发明涉及一种医药中间体的制备,具体涉及一种arv
‑
471中间体(4
‑
(3
‑
(溴甲基)
‑4‑
(甲氧羰基)苯基)哌嗪
‑1‑
羧酸叔丁酯)的制备方法。
背景技术:
2.目前临床使用的大部分小分子药物其治疗策略都是靶向性,并利用“占据驱动”作用模式抑制靶蛋白的功能,而发挥治疗疾病的作用,但由于受体突变等原因靶向药物易产生耐药性。不同于传统的小分子抑制剂和拮抗剂,蛋白降解技术药物由于其能够直接诱导靶蛋白的降解,因而具有更好临床效果如可以初步解决耐药性问题,蛋白降解药物近年来发展迅速。其优点是:(1)作用范围更广、活性更高、可靶向“不可成药”靶点;(2)提高选择性、活性和安全性;(3)克服药物的耐药性。
3.arvinas公司所开发的arv
‑
471是新型雌激素受体(er)降解剂类抗乳腺癌药物。在临床研究中发现,arv
‑
471可诱导多个er
+
bc细胞株中的er大幅度降解,表现出令人鼓舞的临床疗效和耐受性特征。arv
‑
471和palbociclib联合给药对于er
+
型乳腺癌可达到临床完全清除效果,获得重大进展。
4.arv
‑
471目前处于期临床。2021年上半年辉瑞公司同arvinas公司达成了20.5亿美元的arv
‑
471开发协议,其中预付款为6.5亿美元。
[0005]4‑
(3
‑
(溴甲基)
‑4‑
(甲氧羰基)苯基)哌嗪
‑1‑
羧酸叔丁酯(式i所示化合物)是制备arv
‑
471的重要中间体,其结构为:。
[0006]
专利cn201780085150.9和cn201880020007.6公开了4
‑
(3
‑
(溴甲基)
‑4‑
(甲氧羰基)苯基)哌嗪
‑1‑
羧酸叔丁酯(式i所示化合物)的制备方法:5
‑
溴苯并呋喃
‑
1(3h)
‑
酮和哌嗪
‑1‑
羧酸叔丁酯在加热条件下经过buchwald反应得到化合物2;化合物2经过naoh碱性条
件开环得到化合物3;再经过三甲基硅烷重氮化甲烷得到化合物4;化合物4在三苯基膦和四溴化碳的条件下得到式i所示化合物。
[0007][0007][0007]
但现有技术存在些欠缺,尤其是第一步buchwald反应中,如1)贵金属催化剂如二亚苄基丙酮二钯pd2(dba)3的使用,导致成本过高;2)反应过程中需要隔绝空气,操作条件严苛;3)重金属pd的引入,导致原料药制备中需引入额外的除重金属钯工艺;4)需特别指出的是,本团队在重复此方法时发现化合物2的收率偏低,只有25%左右,特别是在百克级以上放大量反应中,发现体系中有大量副产物4
‑
溴
‑2‑
(羟甲基)苯甲酸存在,且此副产物收率超过50%;5)此外在最后一步中,目标产物纯化过程困难,易残留杂质三苯氧磷。
[0008]
因此,对现有4
‑
((3
‑
溴甲基)
‑4‑
(甲氧羰基)苯基)哌嗪
‑1‑
羧酸叔丁酯的工艺改进,具有重要的实际价值。
技术实现要素:
[0009]
发明要解决的问题为了解决现有技术中存在的上述问题,本发明提供了一种操作简便、经济性好、适宜工业化生产的arv
‑
471中间体(4
‑
(3
‑
(溴甲基)
‑4‑
(甲氧羰基)苯基)哌嗪
‑1‑
羧酸叔丁酯)的制备方法。
[0010]
用于解决问题的方案一种制备如式i所示的arv
‑
471中间体的方法,所述方法包括如下步骤s3:化合物5的质子酸盐与nbs在引发剂存在下发生反应得到如式i所示的arv
‑
471中间体;
,。
[0011]
进一步地,如上所述的制备方法,所述引发剂选自过氧化二苯甲酰(bpo)、间氯过氧化苯甲酸、偶氮二异丁腈、偶氮二异庚腈和偶氮二异丁酸二甲酯中的一种或多种。
[0012]
进一步地,如上所述的制备方法,所述质子酸选自有机酸和无机酸。
[0013]
优选地,所述有机酸选自甲酸、乙酸、丙酸、苯甲酸、柠檬酸、酒石酸、马来酸、富马酸、琥珀酸和苹果酸。
[0014]
优选地,所述无机酸选自盐酸、氢溴酸、硫酸、硝酸、磷酸,优选为盐酸。
[0015]
进一步地,如上所述的制备方法,所述反应的温度为50℃~130℃。
[0016]
进一步地,如上所述的制备方法,所述化合物5的质子酸盐与nbs的摩尔比为1:1~10。
[0017]
进一步地,如上所述的制备方法,该反应是在溶剂中进行。
[0018]
优选地,所述溶剂选自有机溶剂。
[0019]
优选地,所述有机溶剂选自四氯化碳、二氯甲烷、氯仿和1,2
‑
二氯乙烷中的一种或多种。
[0020]
进一步地,如上所述的制备方法,所述方法还包括如下步骤s2:化合物5在质子酸存在下反应得到化合物5的质子酸盐。
[0021]
进一步地,如上所述的制备方法,所述步骤s2是在溶剂中进行。
[0022]
优选地,所述溶剂选自乙醚、甲基叔丁基醚、四氢呋喃、2
‑
甲基四氢呋喃、乙酸乙酯、乙酸异丙酯和水中的一种或多种。
[0023]
进一步地,如上所述的制备方法,所述步骤s2的反应温度为0℃~100℃,优选为室温。
[0024]
进一步地,如上所述的制备方法,所述方法还包括如下步骤s1:;步骤s1:化合物4和哌嗪
‑1‑
羧酸叔丁酯在碱存在下反应得到化合物5。
[0025]
进一步地,如上所述的制备方法,所述步骤s1中所述碱选自有机碱和无机碱中的一种或多种。
[0026]
优选地,所述有机碱选自三乙胺、吡啶、n,n
‑
二异丙基乙胺、正丁基锂、二异丙基氨基锂、醋酸钠、醋酸钾、叔丁醇钠、叔丁醇钾和1,8
‑
二氮杂二环十一碳
‑7‑
烯中的一种或多
种。
[0027]
优选地,所述无机碱选自氢化钠、磷酸钾、碳酸钠、碳酸钾、碳酸铯、氢氧化钠、氢氧化锂和氢氧化钾中的一种或多种。
[0028]
进一步地,如上所述的制备方法,所述步骤s1反应温度为50℃~150℃;优选为80℃~130℃。
[0029]
优选地,所述步骤s1中化合物4与哌嗪
‑1‑
羧酸叔丁酯的摩尔比为1:0.5~5。
[0030]
进一步地,如上所述的制备方法,所述步骤s1是在溶剂中进行。
[0031]
优选地,所述溶剂选自有机溶剂。
[0032]
优选地,所述有机溶剂选自二甲亚砜、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺、n
‑
甲基吡咯烷酮中的一种或多种。
[0033]
发明的效果(1)本公开创造性的发现,4
‑
(4
‑
(甲氧羰基)
‑3‑
甲基苯基)哌嗪
‑1‑
羧酸叔丁酯的盐如盐酸盐进行溴化时,可以较高收率得到目标苄溴衍生物4
‑
((3
‑
溴甲基)
‑4‑
(甲氧羰基)苯基)哌嗪
‑1‑
羧酸叔丁酯(式i所示化合物)(如实施例3)。但常规的典型策略,如游离态的4
‑
(4
‑
(甲氧羰基)
‑3‑
甲基苯基)哌嗪
‑1‑
羧酸叔丁酯直接溴化时,并不能得到目标苄溴产物,而仅得到苯环溴代化合物4
‑
(2
‑
溴
‑4‑
(甲氧羰基)
‑3‑
甲基苯基)哌嗪
‑1‑
羧酸叔丁酯和4
‑
(2
‑
溴
‑4‑
(甲氧羰基)
‑5‑
甲基苯基)哌嗪
‑1‑
羧酸叔丁酯(如比较例1),从而有力的表明4
‑
(4
‑
(甲氧羰基)
‑3‑
甲基苯基)哌嗪
‑1‑
羧酸叔丁酯成盐使用的极端重要性。
[0034]
(2)本公开所述方法避免重金属物质的使用和产生、环境友好、简单、高效、反应条件温和、操作容易控制、安全可靠、成本低、经济效益好。同时本公开的制备方法反应收率高,所获得的产物纯度高,适宜工业化生产。
附图说明
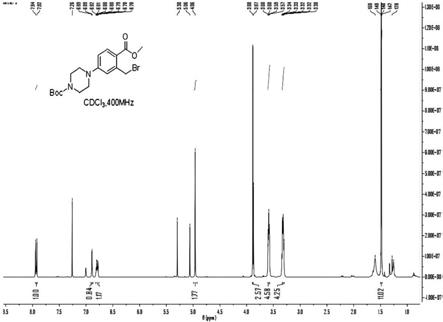
[0035]
图1:实施例3制备得到的4
‑
((3
‑
溴甲基)
‑4‑
(甲氧羰基)苯基)哌嗪
‑1‑
羧酸叔丁酯(式i所示化合物)的1h nmr图谱。
具体实施方式
[0036]
为使本公开的技术方案和有益效果能够更加明显易懂,下面通过列举具体实施例的方式进行详细说明。其中,附图不一定是按比例绘制的,局部特征可以被放大或缩小,以更加清楚的显示局部特征的细节;除非另有定义,本文所使用的技术和科学术语与本技术所属的技术领域中的技术和科学术语的含义相同。
[0037]
本公开提供了一种制备如式i所示的arv
‑
471中间体的方法,所述方法包括如下步骤s3:化合物5的质子酸盐与nbs在引发剂存在下发生反应得到如式i所示的arv
‑
471中间体;
,。
[0038]
在某些实施方案中,所述引发剂选自过氧化二苯甲酰(bpo)、间氯过氧化苯甲酸、偶氮二异丁腈、偶氮二异庚腈和偶氮二异丁酸二甲酯等中的一种或多种。
[0039]
在某些实施方案中,所述质子酸选自有机酸和无机酸。
[0040]
在某些实施方案中,所述有机酸选自甲酸、乙酸、丙酸、苯甲酸、柠檬酸、酒石酸、马来酸、富马酸、琥珀酸和苹果酸等。
[0041]
在某些实施方案中,所述无机酸选自盐酸、氢溴酸、硫酸、硝酸、磷酸等。
[0042]
在某些实施方案中,所述无机酸选自盐酸。
[0043]
在某些实施方案中,所述反应的温度为50℃~130℃。
[0044]
在某些实施方案中,所述反应的温度为60℃~110℃。
[0045]
在某些实施方案中,所述反应的温度为70℃~90℃。
[0046]
在某些实施方案中,所述化合物5的质子酸盐与nbs的摩尔比为1:1~10。
[0047]
在某些实施方案中,所述化合物5的质子酸盐与nbs的摩尔比为1:1~5。
[0048]
在某些实施方案中,所述化合物5的质子酸盐与nbs的摩尔比为1:1~3。
[0049]
在某些实施方案中,该反应是在溶剂中进行。
[0050]
在某些实施方案中,所述溶剂选自有机溶剂。
[0051]
在某些实施方案中,所述有机溶剂选自四氯化碳、二氯甲烷、氯仿和1,2
‑
二氯乙烷等中的一种或多种。
[0052]
在某些实施方案中,所述方法还包括如下步骤s2:化合物5在质子酸存在下反应得到化合物5的质子酸盐。
[0053]
在某些实施方案中,所述步骤s2是在溶剂中进行。
[0054]
在某些实施方案中,所述溶剂选自乙醚、甲基叔丁基醚、四氢呋喃、2
‑
甲基四氢呋喃、乙酸乙酯、乙酸异丙酯和水等中的一种或多种。
[0055]
在某些实施方案中,所述步骤s2的反应温度为0℃~100℃。
[0056]
在某些实施方案中,所述步骤s2的反应温度为10℃~50℃。
[0057]
在某些实施方案中,所述步骤s2的反应温度为20℃~40℃。
[0058]
在某些实施方案中,所述步骤s2的反应温度为室温。
[0059]
在某些实施方案中,所述方法还包括如下步骤s1:;
步骤s1:化合物4和哌嗪
‑1‑
羧酸叔丁酯在碱存在下反应得到化合物5。
[0060]
在某些实施方案中,所述步骤s1中所述碱选自有机碱和无机碱中的一种或多种。
[0061]
在某些实施方案中,所述有机碱选自三乙胺、吡啶、n,n
‑
二异丙基乙胺、正丁基锂、二异丙基氨基锂、醋酸钠、醋酸钾、叔丁醇钠、叔丁醇钾和1,8
‑
二氮杂二环十一碳
‑7‑
烯等中的一种或多种。
[0062]
在某些实施方案中,所述无机碱选自氢化钠、磷酸钾、碳酸钠、碳酸钾、碳酸铯、氢氧化钠、氢氧化锂和氢氧化钾等中的一种或多种。
[0063]
在某些实施方案中,所述步骤s1反应温度为50℃~150℃。
[0064]
在某些实施方案中,所述步骤s1反应温度为60℃~140℃。
[0065]
在某些实施方案中,所述步骤s1反应温度为80℃~130℃。
[0066]
在某些实施方案中,所述步骤s1中化合物4与哌嗪
‑1‑
羧酸叔丁酯的摩尔比为1:0.5~5。
[0067]
在某些实施方案中,所述步骤s1中化合物4与哌嗪
‑1‑
羧酸叔丁酯的摩尔比为1:1~3。
[0068]
在某些实施方案中,所述步骤s1中化合物4与哌嗪
‑1‑
羧酸叔丁酯的摩尔比为1:1~2。
[0069]
在某些实施方案中,所述步骤s1是在溶剂中进行。
[0070]
在某些实施方案中,所述溶剂选自有机溶剂。
[0071]
在某些实施方案中,所述有机溶剂选自二甲亚砜、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺、n
‑
甲基吡咯烷酮等中的一种或多种。
[0072]
除非有相反陈述,在说明书和权利要求书中使用的英文缩写具有下述含义:nbs:n
‑
溴代琥珀酰亚胺。
[0073]
bpo:过氧化二苯甲酰。
[0074]
下面通过具体实施例对本发明的方法进行说明,应理解,这些实施例是用于说明本发明的基本原理、主要特征和优点,而本发明不受以下实施例的范围限制;实施例中采用的实施条件可以根据具体要求做进一步调整,未注明的实施条件通常为常规实验中的条件。
[0075]
下述实施例中1h nmr图谱是用bruker仪器(400mhz)测定而得,化学位移用ppm表示。使用四甲基硅烷内标准(0.00ppm)。1h nmr的表示方法:s=单峰,d=双重峰,t=三重峰,q=四重峰,m=多重峰,br=变宽的,dd=双重峰的双重峰,dt=三重峰的双重峰。若提供偶合常数时,其单位为hz。
[0076]
质谱是用quattro microtm api三重四极杆质谱仪测定得到,离子化方式为esi。
[0077]
tlc:薄层色谱法。薄层层析硅胶板使用烟台黄海hsgf254或青岛gf254硅胶板,薄层色谱法(tlc)使用的硅胶板采用的规格是0.2mm
‑
0.3mm,薄层层析分离纯化产品采用的规格是0.4mm
‑
0.5mm。
[0078]
柱层析一般使用烟台黄海硅胶200
‑
300目硅胶为载体。
[0079]
在下列实例中,除非另有指明,所有温度为摄氏温度,除非另有指明,各种起始原料和试剂来自市售或者是根据已知的方法合成,市售原料和试剂均不经进一步纯化直接使用,除非另有指明,市售厂家包括但不限于国药集团,百灵威科技有限公司,梯希爱(上海)
化成工业发展有限公司,上海毕得医药科技有限公司和上海迈瑞尔化学科技有限公司等。
[0080]
实施例中无特殊说明,反应中的溶液是指水溶液。
[0081]
实施例中无特殊说明,反应的温度为室温,为20℃~30℃。
[0082]
实施例中的反应进程的监测采用薄层色谱法(tlc),反应所使用的展开剂,纯化化合物采用的柱层析的洗脱剂的体系或薄层色谱法的展开剂体系包括:a:石油醚和乙酸乙酯体系;b:二氯甲烷和甲醇体系;c:正己烷:乙酸乙酯;其中溶剂的体积比根据化合物的极性不同而不同,也可以加入少量的酸性或碱性试剂进行调节,如醋酸或三乙胺等。
[0083]
实施例14
‑
(4
‑
(甲氧羰基)
‑3‑
甲基苯基)哌嗪
‑1‑
羧酸叔丁酯将化合物4
‑
氟
‑2‑
甲基苯甲酸甲酯(100g,595mmol)和哌嗪
‑1‑
羧酸叔丁酯(120g,655mmol)溶于dmso(1.5l)中,加入碳酸钾(247g,1.785mol),120℃反应过夜。次日tlc监测反应完全。将反应体系冷却至室温过滤,滤液加入乙酸乙酯和水分液,有机相浓缩使用快速柱层析法得产物110g,收率为78%。
[0084]1hnmr(400mhz,cdcl3):δ7.89(d,j=8.0hz,1h),6.71
‑
6.67(m,2h),3.84(s,3h),3.58
‑
3.55(m,4h),3.29
‑
3.26(m,4h),2.59(s,3h),1.48(s,9h)。
[0085]
ms
‑
esi:333.8[m
‑
h]
+
。
[0086]
实施例24
‑
(4
‑
(甲氧羰基)
‑3‑
甲基苯基)哌嗪
‑1‑
羧酸叔丁酯盐酸盐将化合物4
‑
(4
‑
(甲氧羰基)
‑3‑
甲基苯基)哌嗪
‑1‑
羧酸叔丁酯(67.0g,200mmol)溶于甲基叔丁基醚(600ml)中,加入盐酸/1,4
‑
二氧六环溶液(4.0m,150ml),室温搅拌1小时后,反应体系中有大量固体析出,反应体系过滤,滤饼烘干至恒重得白色固体59.6g,收率为80%。
[0087]1hnmr(400mhz,cdcl3):δ8.00(d,j=8.0hz,1h),7.54
‑
7.49(m,2h),4.05(s,4h),3.90(s,3h),3.42(s,4h),2.64(s,3h),1.49(s,9h)。
[0088]
实施例34
‑
(3
‑
(溴甲基)
‑4‑
(甲氧羰基)苯基)哌嗪
‑1‑
羧酸叔丁酯将4
‑
(3
‑
(溴甲基)
‑4‑
(甲氧羰基)苯基)哌嗪
‑1‑
羧酸叔丁酯盐酸盐(10.0g,
27mmol)溶于四氯化碳溶液(100ml)中,再加入过氧化二苯甲酰 (70.0mg,0.3 mmol)和nbs(4.8g,27.0mmol)。加料完毕,回流反应。tlc监测反应基本完全,减压蒸去溶剂,加入饱和碳酸氢钠水溶液和乙酸乙酯分液,减压蒸去溶剂,残留物用快速柱层析分离。得产物6.0g,收率为54%。
[0089]1h nmr (400 mhz, cdcl3) :δ 7.93 (d, j = 8.0 hz ,1h), 6.89 (s, 1h), 6.80 (d, j = 4.0 hz, 1h), 4.96 (s, 2h), 3.88 (s, 3h), 3.60
‑
3.57 (m, 4h), 3.34
‑
3.30 (m, 4h), 1.48 (s, 9h)。
[0090]
ms
‑
esi: 413.3 [m+h]
+
。
[0091]
比较例1
ꢀꢀ4‑
(2
‑
溴
‑4‑
(甲氧羰基)
‑3‑
甲基苯基)哌嗪
‑1‑
羧酸叔丁酯和4
‑
(2
‑
溴
‑4‑
(甲氧羰基)
‑5‑
甲基苯基)哌嗪
‑1‑
羧酸叔丁酯将化合物4
‑
(4
‑
(甲氧羰基)
‑3‑
甲基苯基)哌嗪
‑1‑
羧酸叔丁酯(366mg,1.1mmol)溶于四氯化碳溶液(10ml)中,加入过氧化二苯甲酰(2.4mg,0.01mmol),nbs(196mg,1.1mmol),反应体系搅拌加热回流。tlc监测反应基本完全,反应体系冷却至室温并浓缩,使用快速柱层析法得化合物4
‑
(2
‑
溴
‑4‑
(甲氧羰基)
‑5‑
甲基苯基)哌嗪
‑1‑
羧酸叔丁酯(250mg)和4
‑
(2
‑
溴
‑4‑
(甲氧羰基)
‑3‑
甲基苯基)哌嗪
‑1‑
羧酸叔丁酯(74mg)。
[0092]4‑
(2
‑
溴
‑4‑
(甲氧羰基)
‑5‑
甲基苯基)哌嗪
‑1‑
羧酸叔丁酯。1h nmr (400 mhz, cdcl3) :δ 8.15 (s, 1h), 6.80 (s, 1h), 3.87 (s, 3h), 3.62
‑
3.60 (m, 4h), 3.06
‑
3.03 (m, 4h), 2.55 (s, 3h), 1.49 (s, 9h)。
[0093]4‑
(2
‑
溴
‑4‑
(甲氧羰基)
‑3‑
甲基苯基)哌嗪
‑1‑
羧酸叔丁酯。1h nmr (400 mhz, cdcl3) :δ 7.75 (d, j = 8.0 hz, 1h), 6.88 (d, j = 8.0hz, 1h), 3.88 (s, 3h), 3.62
‑
3.60 (m, 4h), 3.02
‑
2.99 (m, 4h), 2.69 (s, 3h), 1.48 (s, 9h)。
[0094]
应当理解,以上实施例均为示例性的,不用于包含权利要求所包含的所有可能的实施方式。在不脱离本公开的范围的情况下,还可以在以上实施例的基础上做出各种变形和改变。同样的,也可以对以上实施例的各个技术特征进行任意组合,以形成可能没有被明确描述的本发明的另外的实施例。因此,上述实施例仅表达了本发明的几种实施方式,不对本发明专利的保护范围进行限制。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1