一种环孢霉素A衍生物及其制备方法和应用与流程
一种环孢霉素a衍生物及其制备方法和应用
技术领域
1.本发明涉及生物技术领域,尤其涉及一种环孢霉素a衍生物及其制备方法和应用。
背景技术:
2.环孢霉素a(cyclosporin,csa)是一种内含11个氨基酸的环状多肽化合物,对细胞和体液免疫有较高选择性抑制作用,是一种淋巴细胞激活抑制剂。主要被用于各种器官移植和一些自身免疫性疾病、血液系统疾病等。但由于csa体内过程个体差异大,治疗窗窄,其血药浓度过高会引起毒副作用,尤其对肝、肾的毒性;浓度过低,则治疗效果低或无效,甚至造成器官移植排斥作用的发生。因此,监测全血中csa的血药浓度并调整其浓度在有效范围在临床合理用药中是非常必要的。
3.目前国内、外主要的检测技术包括:高效液相色谱法,放免法,荧光偏振法,化学发光法,电化学发光法,均相酶免疫法等。但这些方法在临床应用上都具有一定的缺陷性。目前市场上缺乏稳定性好、灵敏度高、特异性强的环孢霉素a监测试剂,尤其是质量好的自动化检验试剂。因此,研发生产质量达到临床要求、实用性强、性价比高,可应用于全自动生化分析仪的环孢霉素a测定试剂已成为国内外体外诊断试剂行业的热点,当前主要研究方向是合成出高效的环孢霉素a衍生物作为半抗原,通过标记与抗体发生特异性反应进行检测。
4.关于csa半抗原衍生物,目前公开的多为基于对csa中游离羟基进行修饰、对双键末端甲基碳进行碳链延长得到。其中,游离羟基由于所处区域空间位阻较大,导致羟基修饰过程收率不理想,甚至无法得到目标物;双键末端碳链延长的制备方案具有条件苛刻,技术难度较高,不利于产业化生产应用等缺陷。故开发一种符合临床要求、实用性强、性价比高,且可应用于全自动生化分析仪的环孢霉素a测定试剂盒必须克服csa半抗原衍生物制备的问题。
技术实现要素:
5.有鉴于此,本发明提供了一种环孢霉素a衍生物及其制备方法和应用。该环孢霉素a衍生物较环孢霉素a具有更强的免疫原性,作为抗原检测环孢霉素a特异性强、精密性高、准确度高、稳定性良好。
6.为了实现上述发明目的,本发明提供以下技术方案:
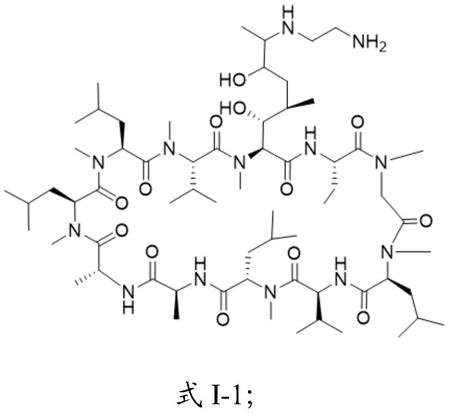
7.一种环孢霉素a衍生物,具有式i所示的结构:
[0008][0009]
r选自-nh(ch2)nnh2、其中,n=2~7,m为0或1。
[0010]
一些实施方案中,所述环孢霉素a衍生物具有式i-1~i-5所示的任意一种结构:
[0011]
[0012][0013]
本发明还提供了所述的环孢霉素a衍生物的制备方法,其特征在于,包括:
[0014]
1)、将csa溶于第一溶剂中,在氧化剂作用下进行双键环氧化反应,得到式ii结构的csa环氧乙烷化产物;
[0015]
2)、在第二溶剂中,所述csa环氧乙烷化产物与烷基二胺通过回流反应进行开环,得到环孢霉素a衍生物;
[0016][0017]
一些实施方案中,步骤1)中,将csa溶于第一溶剂中,在氧化剂作用下进行双键环
氧化反应,得到csa环氧乙烷化产物。其中,第一溶剂对csa具有良好溶解性,包括四氢呋喃、二氯甲烷、二甲基亚砜中的至少一种,包括但不限于此;所述氧化剂为过氧单磺酸钾、间氯过氧苯甲酸、叔丁基过氧化氢中的至少一种;所述csa与氧化剂的摩尔比为1:1~1:15,具体可为1:1或1:15;所述双键环氧化反应的温度为18℃~30℃,具体可为18℃或30℃。
[0018]
步骤2)中,在第二溶剂中,所述csa环氧乙烷化产物与烷基二胺通过回流反应进行开环,得到环孢霉素a衍生物。其中,所述第二溶剂对csa环氧乙烷化产物具有良好溶解性,优选沸点在60℃~100℃范围内的溶剂,具体包括四氢呋喃、1,2-二氯乙烷,1,4-二氧六环中的至少一种,包括但不限于此;所述回流反应的时间为8h~40h,具体可为8h或40h;所述烷基二胺类化合物为nh2(ch2)nnh2(n为2~7)或1,4-环己基二烷基胺,csa环氧乙烷化产物与烷基二胺类物质的摩尔比为1:2~1:50,具体可为1:2或1:50。
[0019]
一些实施方案中,所述双键环氧化反应和回流反应之后还包括分离纯化的步骤,所述分离纯化包括亚硫酸钠溶液淬灭洗涤、碳酸氢钠溶液脱酸洗涤、结晶中的至少一种。
[0020]
本发明还提供了所述环孢霉素a衍生物作为半抗原在制备检测csa的试剂盒中的应用。
[0021]
本发明还提供了所述环孢霉素a衍生物作为半抗原在制备检测csa的试剂盒中的应用。
[0022]
本发明还提供一种检测csa的试剂盒,包括酶结合物和其他试剂成分;
[0023]
所述酶结合物包括酶标记的式i结构的环孢霉素a衍生物;
[0024]
所述其他试剂成分包括包被有csa抗体的磁微粒悬浮液、csa校准品、样本稀释液、清洗液和发光底物液中的至少一种。
[0025]
一些实施方案中,所述磁微粒悬浮液中磁微粒的浓度为0.1~1.0mg/ml;
[0026]
所述csa校准品的浓度为0ng/ml~1500ng/ml;
[0027]
所述样本稀释液为含有0.2%~5%保护蛋白、50u/ml尿激酶、0.02%~0.1%防腐剂的缓冲液;所述保护蛋白为hsa;所述缓冲液为tris、bis-tris、mops、tes、hepes、tapso中的至少一种,ph值为7.0~7.8;所述防腐剂为proclin 300;
[0028]
所述清洗液为含有2%tween 20、1%proclin 300的ph7.5的2m磷酸盐缓冲液;
[0029]
所述发光底物液为鲁米诺发光底物液或amppd发光底物液。
[0030]
一些实施方案中,所述酶结合物、磁微粒悬浮液和csa校准品中还包括稀释液;
[0031]
所述稀释液包括缓冲液、0.02%~0.1%防腐剂和0.2%~5%保护蛋白;
[0032]
所述保护蛋白为hsa;
[0033]
所述缓冲液为tris、bis-tris、mops、tes、hepes、tapso中的至少一种,ph值为7.0~7.8;
[0034]
所述防腐剂为proclin 300。
[0035]
本发明提供的环孢霉素a衍生物具有式i所示的结构,免疫原性强,可满足全自动化学发光检测平台定量检测需求,与常见干扰药物间不存在交叉反应;且以该半抗原衍生物为抗原制备的csa检测试剂特异性强、精密性高、准确度高、稳定性良好。另外,本发明制备方法存在反应条件温和、操作简单、收率高、纯度高、所得目标物晶体状态好等优点,克服了以往csa半抗原衍生物制备条件苛刻、收率低、纯化难度大等缺陷。
附图说明
[0036]
图1示实施例7建立的标准曲线,以校准品浓度值为x轴,以校准品发光值取log值为y轴建立标准曲线。
具体实施方式
[0037]
本发明提供了一种环孢霉素a衍生物及其制备方法和应用。本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。本发明的方法及应用已经通过较佳实施例进行了描述,相关人员明显能在不脱离本发明内容、精神和范围内对本文的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。
[0038]
本发明采用的试材皆为普通市售品,皆可于市场购得。
[0039]
下面结合实施例,进一步阐述本发明:
[0040]
实施例1 csa环氧乙烷化产物的制备
[0041][0042]
于500ml茄形瓶中加入csa原料(5.00g,4.16mmol),用150ml二氯甲烷溶解后,加入过氧单磺酸钾(加入量与csa摩尔比1:1~1:15),室温下搅拌过夜。用200ml20%的亚硫酸钠溶液洗涤反应母液2次,200ml10%的碳酸钠水溶液进行洗涤2次。然后有机层用无水硫酸钠干燥。减压浓缩得白色固体产物4.83g,纯度96.8%,收率:92.2%。
[0043]
所得csa环氧化产物质谱及核磁氢谱数据如下:
[0044]
ms-esi(m/z):1240.73[m+na]
+
;
[0045]1h nmr(400mhz,dmso-d6)δ:7.43-7.48(m,4h,-nh-co-),5.28(s,1h,-oh),4.61-4.36(m,11h,-ch),4.05(s,2h,-ch2),3.32-2.94(m,21h,-ch3),2.73-2.46(m,4h,-ch),2.30-2.17(m,10h,-ch2),1.84-1.63(m,6h,-ch3),1.47-1.35(m,5h,-ch),1.31(m,2h,-ch2),1.24(d,3h,-ch3),0.99-0.62(m,42h,-ch3)。
[0046]
实施例2:csa胺基化衍生物1的制备
[0047][0048]
氩气保护下,向50ml无水四氢呋喃中加入csa环氧化物(500.0mg,0.41mmol),然后加入乙二胺(加入量与csa环氧化物摩尔比1:2~1:50),加热回流8h~40h。反应停止后通过旋转蒸发器减压浓缩除去溶剂,然后加入乙酸乙酯溶解剩余固体,再先后用0.01m的稀盐酸溶液洗涤1~5次,0.1m的氨水洗涤1~3次。有机相经干燥剂干燥,通过旋转蒸发器减压浓缩除去溶剂后再加入少量甲醇,然后再加入一定量纯化水(纯化水与甲醇体积比1:10~1:100)进行重结晶纯化。过滤后经干燥得到白色晶体状衍生物468.3mg,纯度99.3%,收率88.5%。
[0049]
所得csa胺基化衍生物1的质谱及核磁氢谱数据如下:
[0050]
ms-esi(m/z):1278.86[m+h]
+
;
[0051]1h nmr(400mhz,dmso-d6)δ:7.43-7.48(m,4h,-nh-co-),5.66(m,1h,-nh),5.28(s,1h,-oh),5.23(m,1h,-oh),4.61-4.36(m,12h,-ch),4.05(s,2h,-ch2),3.32-2.94(m,21h,-ch3),2.73-2.46(m,3h,-ch),2.36-2.17(m,14h,-ch2),1.84-1.63(m,6h,-ch3),1.53-1.36(m,7h,-ch,-nh2),1.31(m,2h,-ch2),1.24(d,3h,-ch3),0.99-0.62(m,42h,-ch3)。
[0052]
实施例3:csa胺基化衍生物2的制备
[0053][0054]
氩气保护下,向50ml无水四氢呋喃中加入csa环氧化物(500.0mg,0.41mmol),然后加入1,7-庚二胺(加入量与csa环氧化物摩尔比1:2~1:50),加热回流8h~40h。反应停止后通过旋转蒸发器减压浓缩除去溶剂,然后加入乙酸乙酯溶解剩余固体,再先后用0.01m的稀盐酸溶液洗涤1~5次,0.1m的氨水洗涤1~3次。有机相经干燥剂干燥,通过旋转蒸发器减压
浓缩除去溶剂后再加入少量甲醇,然后再加入一定量纯化水(纯化水与甲醇体积比1:10~1:100)进行重结晶纯化。过滤后经干燥得到白色晶体状衍生物376.9mg,纯度97.5%,收率66.4%。
[0055]
所得csa胺基化衍生物2的质谱及核磁氢谱数据如下:
[0056]
ms-esi(m/z):1349.01[m+h]
+
;
[0057]1h nmr(400mhz,dmso-d6)δ:7.43-7.48(m,4h,-nh-co-),5.66(m,1h,-nh),5.28(s,1h,-oh),5.23(m,1h,-oh),4.61-4.36(m,12h,-ch),4.05(s,2h,-ch2),3.32-2.94(m,21h,-ch3),2.73-2.46(m,3h,-ch),2.36-2.17(m,14h,-ch2),1.84-1.63(m,6h,-ch3),1.53-1.36(m,7h,-ch,-nh2),1.33-1.29(m,12h,-ch2),1.24(d,3h,-ch3),0.99-0.62(m,42h,-ch3)。
[0058]
实施例4:csa胺基化衍生物3的制备
[0059][0060]
氩气保护下,向50ml无水四氢呋喃中加入csa环氧化物(500.0mg,0.41mmol),然后加入1,4-二氨基环己烷(加入量与csa环氧化物摩尔比1:2~1:50),加热回流8h~40h。反应停止后通过旋转蒸发器减压浓缩除去溶剂,然后加入乙酸乙酯溶解剩余固体,再先后用0.01m的稀盐酸溶液洗涤1~5次,0.1m的氨水洗涤1~3次。有机相经干燥剂干燥,通过旋转蒸发器减压浓缩除去溶剂后再加入少量甲醇,然后再加入一定量纯化水(纯化水与甲醇体积比1:10~1:100)进行重结晶纯化。过滤后经干燥得到白色晶体状衍生物436.6mg,纯度97.7%,收率78.0%。
[0061]
所得csa胺基化衍生物3的质谱及核磁氢谱数据如下:
[0062]
ms-esi(m/z):1333.04[m+h]
+
;
[0063]1h nmr(400mhz,dmso-d6)δ:7.43-7.48(m,4h,-nh-co-),5.66(m,1h,-nh),5.28(s,1h,-oh),5.23(m,1h,-oh),4.61-4.36(m,12h,-ch),4.05(s,2h,-ch2),3.32-2.94(m,21h,-ch3),2.73-2.46(m,5h,-ch),2.36-2.17(m,14h,-ch2),1.84-1.63(m,6h,-ch3),1.53-1.36(m,7h,-ch,-nh2),1.33-1.29(m,6h,-ch2),1.24(d,3h,-ch3),0.99-0.62(m,42h,-ch3)。
[0064]
实施例5:csa胺基化衍生物4的制备
[0065][0066]
氩气保护下,向50ml无水四氢呋喃中加入csa环氧化物(500.0mg,0.41mmol),然后加入1,4-二氨甲基环己烷(加入量与csa环氧化物摩尔比1:2~1:50),加热回流8h~40h。反应停止后通过旋转蒸发器减压浓缩除去溶剂,然后加入乙酸乙酯溶解剩余固体,再先后用0.01m的稀盐酸溶液洗涤1~5次,0.1m的氨水洗涤1~3次。有机相经干燥剂干燥,通过旋转蒸发器减压浓缩除去溶剂后再加入少量甲醇,然后再加入一定量纯化水(纯化水与甲醇体积比1:10~1:100)进行重结晶纯化。过滤后经干燥得到白色晶体状衍生物448.6mg,纯度98.8%,收率78.17%。
[0067]
所得csa胺基化衍生物4的质谱及核磁氢谱数据如下:
[0068]
ms-esi(m/z):1360.88[m+h]
+
;
[0069]1h nmr(400mhz,dmso-d6)δ:7.43-7.48(m,4h,-nh-co-),5.66(m,1h,-nh),5.28(s,1h,-oh),5.23(m,1h,-oh),4.61-4.36(m,12h,-ch),4.05(s,2h,-ch2),3.32-2.94(m,21h,-ch3),2.73-2.46(m,5h,-ch),2.42-2.10(m,14h,-ch2),1.87-1.63(m,6h,-ch3),1.56-1.36(m,7h,-ch,-nh2),1.33-1.26(m,10h,-ch2),1.24(d,3h,-ch3),0.99-0.62(m,42h,-ch3)。
[0070]
实施例6:csa胺基化衍生物5的制备
[0071][0072]
氩气保护下,向50ml无水四氢呋喃中加入csa环氧化物(500.0mg,0.41mmol),然后加入1,4-二氨甲基环己烷(加入量与csa环氧化物摩尔比1:2~1:50),加热回流8h~40h。反
应停止后通过旋转蒸发器减压浓缩除去溶剂,然后加入乙酸乙酯溶解剩余固体,再先后用0.01m的稀盐酸溶液洗涤1~5次,0.1m的氨水洗涤1~3次。有机相经干燥剂干燥,通过旋转蒸发器减压浓缩除去溶剂后再加入少量甲醇,然后再加入一定量纯化水(纯化水与甲醇体积比1:10~1:100)进行重结晶纯化。过滤后经干燥得到白色晶体状衍生物448.6mg,纯度97.8%,收率78.77%。
[0073]
所得csa胺基化衍生物5的质谱及核磁氢谱数据如下:
[0074]
ms-esi(m/z):1354.87[m+h]
+
;
[0075]1h nmr(400mhz,dmso-d6)δ:8.66(s,2h,-nh2),7.43-7.48(m,4h,-nh),7.27-7.19(m,4h,ar-h),6.48(m,1h,-nh),5.28(s,1h,-oh),5.23(m,1h,-oh),4.61-4.36(m,12h,-ch),4.26-4.12(m,6h,-ch2),3.32-2.94(m,21h,-ch3),2.73-2.46(m,3h,-ch),2.36-2.17(m,10h,-ch2),1.84-1.63(m,6h,-ch3),1.51-1.38(m,5h,-ch),1.31(m,2h,-ch2),1.24(d,3h,-ch3),0.99-0.62(m,42h,-ch3)。
[0076]
实施例7:csa胺基化衍生物在全自动化学发光平台中的应用及检测试剂盒制备
[0077]
所制备的磁微粒化学发光法检测csa含量的试剂盒包括:csa氨基化衍生物标记的csa酶结合物,包被有csa抗体的磁微粒悬浮液,含有牛血清白蛋白的样品稀释液、含有梯度浓度csa的校准品,浓缩型清洗液和发光底物液。
[0078]
磁微粒混悬液的制备
[0079]
将粒径1~1.5μm的羧基磁微粒原液充分混匀,取30ul于1.5ml的聚苯乙烯离心管中,并置于磁力架中,用清洗液(0.1mol/l的pbs缓冲液,ph7.4)将磁微粒洗涤5次,最后一次洗涤结束后,加入100μlcouplingreagent(分别取50ul的10mg/mledc溶液和10mg/mlnhs溶液,溶于bindingbuffer中),置于旋转混匀仪中,在室温(18-25℃,下同)条件下旋转反应30分钟。活化反应结束,往离心管中加入5ul抗csa抗体和95ul的bindingbuffer,再次置于旋转混匀仪中,室温旋转反应2小时。反应结束,用封闭液封闭磁微粒三次,最后一次封闭结束后,用含有0.2%~5%hsa保护蛋白的tris-hcl缓冲液定容至3ml,于2~8℃冰箱中储存,备用。
[0080]
样品稀释液的制备
[0081]
配制tris-hcl和乙二胺四乙酸二钾的缓冲液中,调ph至7.0~7.8之间,添加0.2%~5%的hsa,50u/ml的尿激酶,0.02%~0.1%的proclin 300防腐剂混合均匀,于2~8℃冰箱中储存,备用。
[0082]
酶结合物的制备
[0083]
配制含有0.2%~5%hsa保护蛋白的tris-hcl缓冲液,ph值范围7.0~7.8之间;防腐剂:0.02%~0.1%proclin 300,按1/1500的比例加入辣根过氧化物酶标记的csa胺基化衍生物酶标抗原,混合均匀,于2~8℃冰箱中储存,备用。
[0084]
校准品的制备
[0085]
将市售csa溶于tris-hcl缓冲溶液中,制成不同浓度的校准品。以市售csa校准品为原始标准,采用其csa试剂盒对不同浓度的校准品分别检测10次,求出均值,得到csa校准品的浓度:0,40,150,400,800,1500ng/ml。
[0086]
实施例8:应用测试实验
[0087]
8.1标准曲线拟合
[0088]
试剂盒使用时,首先加入25ul校准品,100ul样品稀释液,20ul磁微粒混悬液,100ul酶结合物,37℃条件下反应17min后,用洗液洗涤,然后加入100ul发光底物,避光1分钟内测发光值,进而通过发光值拟合出csa的浓度。由于csa胺基化衍生物酶标抗原与校准品中的csa竞争结合csa抗体,所以待测样本中csa的量越多,发光值越低。
[0089]
选择适当的曲线拟合方式,以校准品浓度值为x轴,以校准品发光值取log值为y轴建立标准曲线(如表1及图1)。本方案选用四参数logistic曲线拟合方程:y=(a-d)/[1+(x/c)^b]+d,其中a=6.53832、b=0.64706、c=3772.02443、d=4.41629、r^2=0.99997。
[0090]
取待测血清或血浆样本,同法测定样本的发光值,代入校准曲线,即可计算出待测样本中csa的含量。如果血清或血浆中csa的浓度超出校准曲线范围,需对样本进行稀释后再检测以保证检测结果的准确性,结果见表1和图1,结果显示,本发明所提供试剂盒梯度良好。
[0091]
表1
[0092]
梯度浓度值(x轴)发光值(y轴)s003449632s1402716013s21502005984s34001363625s4800935761s51500608140
[0093]
8.2梯度测试
[0094]
将购于sigma公司csa溶解于无水甲醇中,配制成1mg/ml的溶液,将溶液稀释于不含csa的空白人造血清中,至浓度分别为1500,800,400,150,40,0ng/ml,将上述溶液作为样品进行elisa实验,结果如表2所示。
[0095]
表2
[0096][0097][0098]
由表2结果可知,采用本发明化合物所配制试剂盒准确性较好。
[0099]
8.3精密性测试
[0100]
将8.2中配置好的六个人造血清基质csa样品,分别用实施例7自制的csa检测试剂盒(磁微粒化学发光法)与市售的csa检测试剂盒重复测试30次,统计数据精密度(cv%),结果如表3所示。
[0101]
表3
[0102][0103]
由表3结果可知,采用本发明所提供csa试剂盒精密度明显优于某厂家市售试剂盒,精密性高。
[0104]
8.4药物干扰试验
[0105]
选取常见药物进行干扰试验,调整浓度为100ng/ml,采用实施例7自制csa检测试剂盒和8.3中市售csa检测试剂盒分别测定上述干扰物,结果如表4所示。
[0106]
表4
[0107]
[0108][0109]
由表4中结果可知,本发明所提供的csa检测试剂盒与常见干扰物无交叉反应,特异性强。
[0110]
8.5稳定性试验
[0111]
将实施例7自制csa检测试剂盒和8.3中市售csa检测试剂盒(磁微粒化学发光法)置于37℃,放置7天后进行样本评价验证,统计浓度值降幅情况,结果如表5所示。
[0112]
表5
[0113][0114]
以上仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1