一种13-甲基小檗碱类生物碱的合成方法与流程
1.本发明属于有机合成领域,具体涉及一种13-甲基小檗碱类生物碱的合成方法。
背景技术:
2.13-甲基小檗碱类生物碱主要来源是黄连的根茎提取物,它是一种c-13位甲基取代的小檗碱衍生物。小檗碱是著名传统中草药黄连中的重要成分,已有2000多年的历史,广泛分布在黄连、黄柏等植物组织中。对小檗碱的深入研究也引起了对13甲基小檗碱类生物碱的研究,近年来的研究表明其具有广泛的药理活性,如抗菌、抗炎、抗肿瘤、抗肥胖和抗高胆固醇活性,也有研究表明其可以抗动脉粥样硬化。13甲基-小檗碱类生物碱具有良好的药用前景,用黄连的根茎提取来获得此类生物碱已经无法满足实际需求,而目前此类化合物的合成方法在成本、安全性和产率等方面还存在诸多不足,影响了此类生物碱的实际应用和推广。
3.目前应用化学合成方法制备13甲基-小檗碱类生物碱的方法主要有以下几种方式。
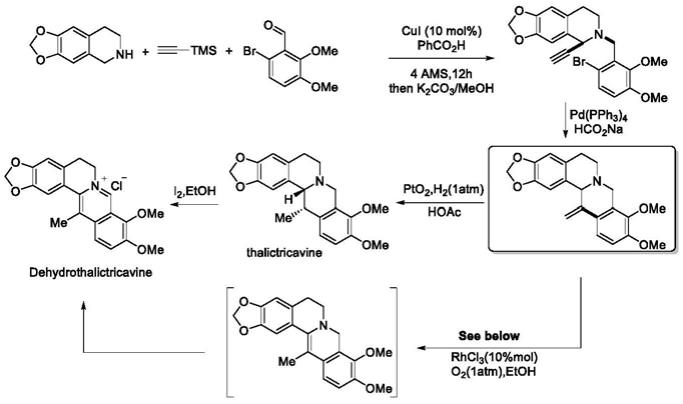
4.其一以四氢异喹啉、tms保护的乙炔及溴代苯甲醛为起始原料,依次经铜催化的redox-a3反应、钯催化还原外碳环化、通过铑催化的异构化和伴随氧化制备得13-甲基小檗碱,或者在关环后经氢化还原再氧化得到13-甲基小檗碱(zhou,s.;tong,r.chem.eur.j.2016,22,7084-7089)
[0005][0006]
其二以3,4-二甲氧基为起始原料,经过硼氢化还原、羟基保护、区域选择性的friedel
–
crafts酰化反应得到芳基甲基酮,与乙二醇保护的溴代芳醛在钯催化下发生偶联反应得1,5-二羰基中间体,然后芳构化环化,最后脱保护后氯代亲和加成三步制得13-甲基巴马汀碱(gatland,pilgrim.angew.chem.int.ed.2014,53,14555
–
14558)。
[0007][0008]
其三以小檗碱为起始原料,用定量的nabh4还原为二氢小檗碱,然后在乙酸和乙醇的混合溶剂中,与37%的甲醛溶液反应,再酸化得到13-甲基小檗碱(yan zhang,chaoran wang,current biology 24,117-123,january 20,2014)。
[0009][0010]
上述方法存在以下缺陷:1、合成步骤相对繁琐,操作复杂;2、为了更好的提高转化率,制备需要价格昂贵的多种金属催化剂及过量的酸催化,因此产能低,价格昂贵。因此,开发一种工艺简单、成本低廉、环境友好的13-甲基小檗碱类生物碱的制备方法具有重要的意义。
技术实现要素:
[0011]
鉴于此,有必要针对上述问题提供一种13-甲基小檗碱类生物碱的合成方法,该方法克服现有工艺的缺点,具有工艺简单,生产成本低,产品收率高纯度高的特点,实验条件简单。
[0012]
本发明是通过以下技术方案实现的:
[0013]
本发明所述的13-甲基小檗碱类生物碱,结构如下:
[0014][0015]
本发明的13-甲基小檗碱类生物碱的合成路径如下:
[0016][0017]
其中:
[0018]
x为单取代或二取代位于其所在苯环上的任何可取代的位置上,并且x为氢、直链
或支链c1~c5烷基、直链或支链c1~c5烷氧基或卤素;或者连接在一起形成环;
[0019]
y为单取代或二取代位于其所在苯环上的任何可取代的位置上,并且y为氢、直链或支链c1~c5烷基、直链或支链c1~c5烷氧基或卤素;或者连接在一起形成环。
[0020]
本发明的13-甲基小檗碱类生物碱的合成方法,包括:
[0021]
反应a:以芳基乙胺(ⅱ)和芳基甲醛(ⅲ)为主要起始原料,在其他辅料参与下,反应得到仲胺盐酸盐(ⅳ);
[0022]
反应b:以仲胺盐酸盐(ⅳ)和丙酮醛(
ⅴ
)为主要原料,在其他辅料参与下,反应得到13-甲基小檗碱类生物碱(i)。
[0023]
进一步的,所述反应a的具体操作包括:
[0024]
a1:在氩气环境中加入4ams粉末,再将芳基乙胺(ⅱ)和芳基甲醛(ⅲ)加入其中,用溶剂分散均匀后置室温搅拌12小时,反应完全后,将反应液过分子筛,旋干溶液得西佛碱粗品。
[0025]
4ams粉末的用量为化合物(ii)和化合物(iii)的总量,化合物(ii)和化合物(iii)的重量比为1:1。此处溶剂为干燥的二氯甲烷,体积用量为每1g混合物用10ml溶剂分散;
[0026]
a2:在氩气保护下将a1所得的西弗碱粗品用溶剂分散后预冷,再向其中加入硼氢化钠,反应完全后,将反应液在冰浴下用饱和氯化铵溶液淬灭,去除有机溶剂,再萃取得仲胺;
[0027]
西弗碱粗品作为此步骤的原料,溶剂为二氯甲烷与甲醇按照1:1体积比混合,溶剂用量为每1g西弗碱粗品原料用10ml溶剂溶解;硼氢化钠为1.5个当量。此处加入硼氢化钠固体需要缓慢加入是为了防止反应液剧烈产气导致反应液喷出圆底烧瓶,因此需要少量多次缓慢加入。
[0028]
a3:将a2所得的仲胺用有机溶剂分散均匀后,放入-20℃预冷半小时,向其中缓慢加入盐酸乙醇,使其成盐酸盐,待成盐充分后滤出干燥得仲胺盐酸盐。
[0029]
仲胺为原料,有机溶剂为乙醇,用量为每1g原料用10ml溶剂。盐酸乙醇的含量为3mol/l,用量为1.5个当量,缓慢加入是为了成盐更为均匀充分,加入过快会形成不均匀块状固体,滴加速度尽量为一滴每秒。
[0030]
进一步的,反应a中,所述芳基乙胺包括3,4-亚甲二氧基苯乙胺、3,4-二甲氧基苯乙胺等。
[0031]
进一步的,反应a中,所述芳基甲醛包括2,3-二甲氧基苯甲醛、2,3-亚甲二氧基苯甲醛或3,4-二甲氧基苯甲醛等。
[0032]
进一步的,在反应b中,仲胺盐酸盐(ⅳ)和丙酮醛(
ⅴ
)在酸溶液中,在除水剂和氧化剂作用下,发生皮克特-施彭格勒反应/傅-克羟基烷基化/脱水反应/氧化反应得到13-甲基小檗碱类生物碱(i)。
[0033]
进一步的,在反应b完成后,还包括操作:反应液经淬灭,阴离子转化,萃取浓缩,打浆纯化后获得化合物(i)。
[0034]
进一步的,反应b中,反应中酸为无水甲酸、冰乙酸、草酸、对甲苯磺酸、甲基磺酸、苯甲酸、丁二酸、柠檬酸、酒石酸、樟脑磺酸及硼酸中的任意一种或多种,更优选为无水甲酸。
[0035]
进一步的,反应b中,反应的除水剂为无水硫酸钠、无水硫酸镁、无水硫酸钙、无水
氯化钙、无水碳酸钾中的任意一种或多种,更优选为无水硫酸镁。
[0036]
进一步的,反应b中,反应的氧化剂为过氧化氢、过氧乙酸、硝酸、过硫酸铵、次氯酸钠、过碳酸铵、过硼酸钠、过硼酸钾、氯化铁、氯化铜、碘单质中的任意一种或多种,更有选为碘单质。
[0037]
进一步的,反应b中,反应的温度在-20℃~100℃,更优选为100℃。
[0038]
进一步的,所述13-甲基小檗碱类生物碱包括13-甲基小檗碱、13-甲基巴马汀碱、13-甲基黄连碱、13-甲基伪小檗碱或13-甲基伪黄连碱等。
[0039]
本发明有益效果:
[0040]
本发明能够以大于80%的收率制备13-甲基小檗碱类生物碱。
[0041]
本发明提供了一种合成13-甲基小檗碱类生物碱的方法。以芳基乙胺和芳基甲醛为原料合成仲胺盐酸盐,再与丙酮醛在无水甲酸溶液中,以除水剂、氧化剂辅助反应,发生皮克特-施彭格勒反应/傅里德-克拉夫茨羟基烷基化反应/脱水反应/氧化反应得13-甲基小檗碱类生物碱,合成步骤简化,易操作;不需要价格昂贵的多种金属催化剂及过量的酸催化,产能高,成本低。
具体实施方式
[0042]
为了更好的说明本发明技术方案所要解决的问题、采用的技术方案和达到的有益效果,现结合具体实施方式进一步阐述。值得说明的是,本发明技术方案包含但不限于以下实施方式。
[0043]
本发明实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购等途径获得的常规产品。
[0044]
实施例1
[0045]
13-甲基小檗碱的制备:
[0046][0047]
所述步骤(a)具体操作:
[0048]
一、将双颈瓶烘干后,再氩气保护下加入4ams粉末,再将3,4-亚甲二氧基苯乙胺和2,3-二甲氧基苯甲醛加入其中,用干燥后的溶剂将其分散均匀后置室温300-400r/min搅拌12小时,待tlc检测反应完全后,将反应液用硅藻土滤除分子筛,旋干溶液得西佛碱粗品。
[0049]
二、取双颈瓶,烘干后在氩气保护下将西弗碱粗品用溶剂分散后置冰浴中预冷,再向其中缓慢加入硼氢化钠,tlc检测反应。将反应液在冰浴下用饱和氯化铵溶液淬灭,旋除有机溶剂,再萃取得仲胺。
[0050]
三、将仲胺用有机溶剂分散均匀后,放入-20℃预冷半小时,向其中缓慢加入盐酸乙醇,使其成盐酸盐,待成盐充分后滤出干燥得仲胺盐酸盐。
[0051]
所述步骤(b)具体操作:
[0052]
在封管中加入无水硫酸镁,烘干后加入甲酸分散均匀,再加入丙酮醛水溶液密封,
于60℃搅拌活化30分钟。活化后依次加入仲胺盐酸盐和碘,密封后升温至100℃反应过夜,tlc检测反应完全后,将反应液用饱和亚硫酸钠溶液淬灭,旋干溶剂,固体残渣用dcm复溶后,加入少量碱水转化其阴离子,用dcm萃取,收集有机层减压浓缩。浓缩液加入少量盐酸水溶液,将阴离子转化为氯离子,用dcm萃取水层,收集有机层,无水硫酸钠干燥除水后减压旋除溶剂,得到粗品。粗品用甲苯/乙醇混合比例溶剂打浆纯化得13-甲基小檗碱(黄色固体,87%收率)。
[0053]
熔点:232.4-233.5℃,1h nmr(400mhz,dmso-d6)δ9.89(s,1h),8.22
–
8.16(m,2h),7.47(s,1h),7.15(s,1h),6.18(s,2h),4.83
–
4.81(m,2h),4.10(s,3h),4.09(s,3h),3.12
–
3.10(m,2h),2.93(s,3h).
13
c nmr(101mhz,dmso-d6)δ150.32,148.95,146.38,144.06,143.97,135.89,133.74,132.93,130.02,125.89,121.37,120.73,120.36,110.62,108.16,102.02,62.03,57.02,56.66,27.25,17.68.
[0054]
实施例2
[0055]
13-甲基巴马汀碱的制备:
[0056][0057]
所述步骤(a)中操作与实施例1相同,将芳基乙胺换成3,4-二甲氧基苯乙胺。
[0058]
所示步骤(b)中操作与实施例1相同,得到黄色固体,收率91%。
[0059]
熔点:169.3-171.4℃,1h nmr(400mhz,dmso-d6)δ9.91(s,1h),8.21(s,1h),8.19(d,j=9.5hz,1h),7.39(s,1h),7.18(s,1h),4.89
–
4.82(m,2h),4.11(s,3h),4.09(s,3h),3.89(s,3h),3.86(s,3h),3.14(d,j=5.9hz,2h),2.99(s,3h).
13
c nmr(151mhz,dmso-d6)δ150.63,150.18,147.19,144.03,143.98,136.06,133.08,131.81,129.73,125.91,121.29,120.70,119.12,114.34,111.01,62.04,57.02,56.83,56.18,55.86,26.82,17.72
[0060]
实施例3
[0061]
13-甲基黄连碱的制备:
[0062][0063]
所述步骤(a)中操作与实施例1相同,芳基乙胺为3,4-亚甲二氧基苯乙胺,芳醛换成2,3-亚甲二氧基苯甲醛。
[0064]
所示步骤(b)中操作与实施例1相同,得到黄色固体,收率87%。
[0065]
熔点:231-232.7℃,1h nmr(400mhz,dmso-d6)δ9.97(s,1h),8.05(d,j=8.9hz,1h),7.98(d,j=8.9hz,1h),7.46(s,1h),7.14(s,1h),6.56(s,2h),6.18(s,2h),4.78(t,j=5.9hz,2h),3.15
–
3.06(m,2h),2.91(s,3h).
13
c nmr(101mhz,dmso-d6)δ148.93,147.09,146.36,144.75,143.09,135.42,133.71,132.24,130.74,120.39,120.12,119.51,110.84,110.73,108.16,104.71,102.02,56.51,27.19,18.14.
[0066]
实施例4
[0067]
13-甲基伪小檗碱的制备:
[0068][0069]
所述步骤(a)中操作与实施例1相同,将芳基乙胺为3,4-亚甲二氧基苯乙胺,芳醛换成3,4-二甲氧基苯甲醛。
[0070]
所示步骤(b)中操作与实施例1相同,得到浅黄色固体,收率90%。
[0071]
熔点:255-257℃,1h nmr(400mhz,dmso-d6)δ9.57(s,1h),7.76(s,1h),7.56(s,1h),7.47(s,1h),7.16(s,1h),6.18(s,2h),5.76(s,1h),4.73
–
4.62(m,2h),4.14(s,3h),4.01(s,3h),3.14
–
3.06(m,2h),2.91(s,3h).
13
c nmr(101mhz,dmso-d6)δ157.29,151.92,148.94,146.32,144.29,136.77,136.42,133.65,127.98,121.19,120.55,110.62,108.21,107.37,103.71,102.00,56.89,56.38,55.99,54.93,27.44,17.82.
[0072]
实施例5
[0073]
13-甲基伪黄连碱的制备:
[0074][0075]
所述步骤(a)中操作与实施例1相同,将芳基乙胺为3,4-亚甲二氧基苯乙胺,芳醛换成3,4-亚甲二氧基苯甲醛。
[0076]
所示步骤(b)中操作与实施例1相同,得到浅黄色固体,收率90%。
[0077]
熔点:285-286.3℃,1h nmr(400mhz,dmso-d6)δ9.56(s,1h),7.81(s,1h),7.75(s,1h),7.47(s,1h),7.15(s,1h),6.45(s,2h),6.18(s,2h),4.67
–
4.64(m,2h),3.12
–
3.09(m,2h),2.84(s,3h).
13
c nmr(151mhz,dmso-d6)δ156.23,150.44,149.05,146.33,144.57,138.78,137.27,133.70,128.59,122.65,120.36,110.60,108.18,104.33,103.98,102.02,101.48,55.89,27.34,18.10.
[0078]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1