环脂肽类天然产物DysoxylactamA及其类似物的制备方法
本发明涉及有机合成,尤其是涉及一种环脂肽类天然产物dysoxylactama及其类似物的制备方法。
背景技术:
1、目前,化疗是治疗恶性肿瘤的重要方法之一,旨在根除原发肿瘤和转移性恶性肿瘤。然而,在临床治疗过程中,大多数患者随着对靶向化疗产生多药耐药性(mdr),导致了抗癌药物的疗效显著降低,成为了癌症化疗失败的主要原因之一。最新研究表明,mdr有许多潜在的机制,主要的机制是由atp结合盒(abc)式转运蛋白的上调介导的药物外排增加。其中p-糖蛋白(p-gp),作为abc式转运蛋白家族的成员,负责增强化疗药物的外排,降低肿瘤细胞内化疗药物的浓度。因此,调控p-gp的药物转运功能被认为是恢复肿瘤对抗癌药物敏感性的有效途径。令人遗憾的是,迄今为止还没有一种p-gp特异性抑制剂在临床试验中取得成功。
2、天然产物是药物和先导化合物的重要来源。2019年,我们课题组从香港樫木(dysoxylum hongkongense)的树皮中分离得到一种具有逆转肿瘤细胞mdr的十七元大环脂肽类天然产物dysoxylactam a,其并未显著降低p-gp的表达,而是抑制肿瘤细胞中p-gp的转运功能。通过体外细胞实验发现,多药耐药的肿瘤细胞经过10μm的dysoxylactam a处理之后,对阿霉素(adriamycin)、长春新(vincristine)和紫杉醇(paclitaxe)的逆转系数达到28.4-1039.7倍,更重要的是,在此浓度下,dysoxylactam a并没有表现出细胞毒性。dysoxylactam a独特的化学结构和优异的逆转mdr活性,使其具有潜在的药物研发潜力。该分子一经发现后,便引起了世界许多学者的兴趣,并展开对其的全合成工作。
3、但目前的全制备方法仍然存在合成步骤繁琐,产率较低以及不便于衍生化的问题。
4、有鉴于此,特提出本发明。
技术实现思路
1、本发明的目的是提供一种天然活性产物dysoxylactam a及其类似物的通用、简便、有效的全制备方法。其中,该全制备方法包括上述化合物的消旋全合成和手性全制备方法。
2、为实现上述目的,本发明采用如下技术方案:
3、本发明提供了一种式(i)所示天然活性产物dysoxylactam a及其类似物的全制备方法。
4、
5、其中,r1选自氢、c1~c10烷氧基、c1~c10取代或未取代的烷基;取代或未取代的芳基或取代或未取代的苄基;
6、r2选自氢、c1~c10取代或未取代的烷基;取代或未取代的芳基或取代或未取代的苄基;
7、r3选自氢、c1~c10取代或未取代的脂肪酰基;取代或未取代的芳香酰基或取代或未取代的苄酰基;
8、所述天然活性产物dysoxylactam a及其类似物化合物为消旋体或r/s构型;
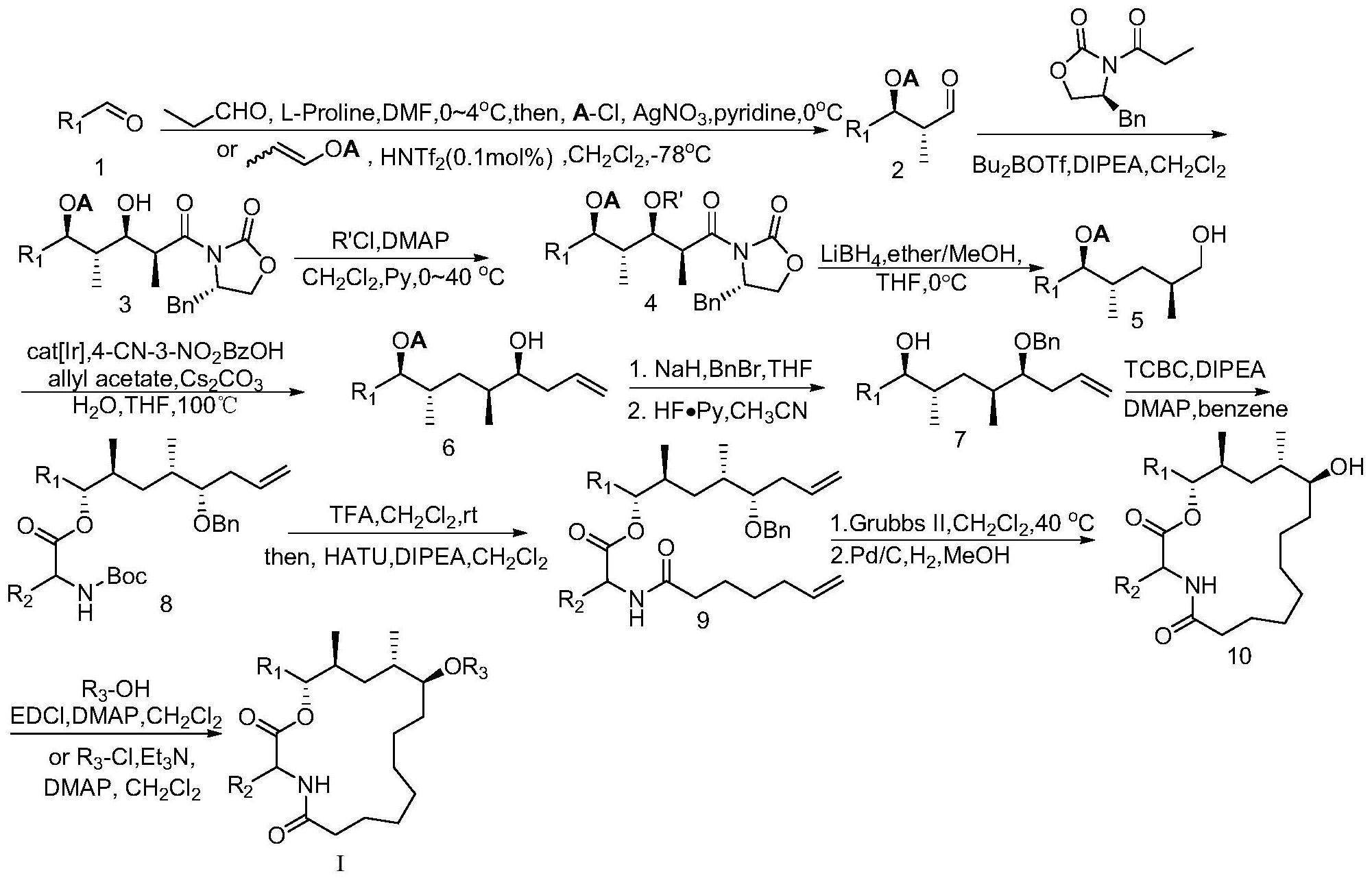
9、天然活性产物dysoxylactam a及其类似物化合物的合成包括以下步骤:
10、(a1)化合物1与丙醛在溶剂和催化剂存在下进行不对称的羟醛缩合反应,然后在碱和盐存在下加入硅醚类保护基对羟基进行保护,获得化合物2;或,
11、(a2)化合物1与被硅醚类保护的e式或z式丙烯醇在溶剂和催化剂存在下进行不对称的羟醛缩合反应,获得化合物2;
12、(b)化合物2与手性助剂在溶剂和碱存在下进行埃文斯羟醛反应,获得化合物3;
13、(c)化合物3在溶剂和碱存在下进行磺酰化反应获得化合物4;
14、(d)化合物4在溶剂存在下进行硼氢化还原反应获得化合物5;
15、(e)化合物5在溶剂和催化剂的存在下进行不对称烯丙基化反应获得化合物6;
16、(f)化合物6在碱和催化剂存在下进行羟基的苄基保护,然后在溶剂和脱保护基助剂存在下进行硅醚保护基脱除获得化合物7;
17、(g)化合物7与氨基被保护的氨基酸在溶剂、碱和反应助剂存在下进行酯化反应获得化合物8;
18、(h)化合物8在酸存在下脱除保护基,然后在溶剂、碱和缩合剂存在下进行酰胺化反应获得化合物9;
19、(i)化合物9在溶剂和催化剂存在下进行烯烃关环复分解反应和氢化还原反应脱除保护基获得化合物10;
20、(j)化合物10与羧酸或酰氯在溶剂、碱和任选的缩合剂存在下进行酯化反应获得式i化合物;
21、合成路线如下:
22、
23、式中,a选自tms、tes、tbdms、tbdps、tips、si(tms)3中的一种;
24、r’选自烷磺酰基或芳香磺酰基,优选为甲磺酰基、对甲苯磺酰基、乙基磺酰基;
25、r1、r2、r3的定义如上文所述。
26、下面,将更详细地说明本发明的新制备方法,然而说明中该方法的反应物、溶剂、碱、催化剂等所用的当量和比例、反应温度、反应所需时间等可以根据具体反应调节,均不限于下面的解释。
27、步骤(a1)
28、化合物1与丙醛在溶剂和催化剂存在下进行不对称的羟醛缩合反应,然后在碱和盐存在下加入硅醚类保护基对羟基进行保护,获得化合物2。
29、优选地,步骤(a1)的催化剂为手性配体,优选为d/l-脯氨酸;
30、优选地,步骤(a1)的溶剂为选自n,n-二甲基甲酰胺、二甲基亚砜中的一种或几种;
31、优选地,步骤(a1)的不对称的羟醛缩合反应的反应温度为-10~10℃(例如-10、-5、0、5、10℃),反应时间为12~48小时(例如12、24、36、48小时);
32、优选地,步骤(a1)的碱为有机碱,优选为选自吡啶、4-二甲氨基吡啶、n,n-二异丙基乙胺中的一种或几种;
33、优选地,步骤(a1)的盐为无机盐,优选为硝酸银;
34、优选地,步骤(a1)的羟基保护的反应温度为0~30℃(例如0、5、10、15、20、25、30℃),反应时间为2~10小时(例如2、5、6、8、10小时);
35、优选地,分步或者“一锅法”加入硅醚保护基。
36、步骤(a2)
37、化合物1与被硅醚类保护的e式或z式丙烯醇在溶剂和催化剂存在下进行不对称的羟醛缩合反应,获得化合物2。
38、优选地,步骤(a2)的催化剂为选自三氟甲烷磺酰亚胺(hntf2)、三氟化硼乙醚、四氯化钛中的一种或几种;
39、优选地,步骤(a2)的反应温度为-80~0℃,反应时间为3~12小时;
40、优选地,步骤(a2)的溶剂为选自二氯甲烷、乙腈、水中的一种或几种。
41、步骤(b)
42、化合物2与手性助剂在溶剂和碱存在下进行埃文斯羟醛反应,获得化合物3。
43、优选地,步骤(b)的手性助剂为选自(±)-4-苄基-3-丙酰基-2-噁唑烷酮、三氟甲磺酸二丁硼(bu2botf)中的一种或几种,优选为(±)-4-苄基-3-丙酰基-2-噁唑烷酮和三氟甲磺酸二丁硼;
44、优选地,步骤(b)的碱为选自三乙胺、n,n-二异丙基乙胺(dipea)中的一种或几种;
45、优选地,步骤(b)的反应温度为-78~0℃(例如-70、-60、-50、-40、-30、-20、-10、-5℃),反应时间为10-24小时;
46、优选地,步骤(b)的溶剂为选自二氯甲烷、四氢呋喃、甲苯、乙腈、乙酸乙酯中的一种或几种;
47、优选地,碱、(±)-4-苄基-3-丙酰基-2-噁唑烷酮与三氟甲磺酸二丁硼的的摩尔比为(1.3~2.0):(0.8~1.2):(1.2:1.8)。
48、步骤(c)
49、化合物3在溶剂和碱存在下进行磺酰化反应获得化合物4。
50、优选地,步骤(c)的磺酰化反应使用的磺酰化试剂为烷磺酰氯或芳香磺酰氯,优选为选自甲磺酰氯(mscl)、苯磺酰氯(tscl)中的一种或几种;
51、优选地,步骤(c)的碱为选自三乙胺、吡啶、咪唑、n,n-二异丙基乙胺中的一种或几种。
52、优选地,步骤(c)的反应温度为0~60℃(例如0、10、20、30、40、50、60℃);
53、优选地,步骤(c)的溶剂为选自二氯甲烷、四氢呋喃、甲苯、乙腈、乙酸乙酯、吡啶中的一种或几种。
54、步骤(d)
55、化合物4在溶剂存在下进行硼氢化还原反应获得化合物5。
56、优选地,步骤(d)的硼氢化还原反应使用的还原试剂为硼氢化锂;
57、优选地,步骤(d)的反应温度为0~40℃(例如0、10、20、30、40℃);
58、优选地,步骤(d)的溶剂为选自乙醚、四氢呋喃中的一种或几种;
59、优选地,还原试剂与化合物4的摩尔比为(1.0~10.0):(0.5~2.0)。
60、步骤(e)
61、化合物5在溶剂和催化剂的存在下进行不对称烯丙基化反应获得化合物6。
62、优选地,步骤(e)的催化剂为铱催化剂;
63、优选地,步骤(e)的反应原料还包括4-氯-3-硝基苯甲酸、碳酸艳和乙酸烯丙酯;
64、优选地,步骤(e)的反应温度为80~120℃(例如80、90、100、110、120℃),反应时间为24~72小时(24、36、48、56、72小时);
65、优选地,步骤(e)的溶剂为选自脱气的四氢呋喃、水、乙醚中的一种。维持惰性、密闭环境。
66、步骤(f)
67、化合物6在碱和催化剂存在下进行羟基的苄基保护,然后在溶剂和脱保护基助剂存在下进行硅醚保护基脱除获得化合物7。
68、优选地,步骤(f)的苄基保护所使用的苄基试剂为选自溴苄、氯苄中的一种或几种;
69、优选地,步骤(f)的碱为无机碱,优选为氢化钠;
70、优选地,步骤(f)的催化剂优选为四丁基碘化铵;
71、优选地,步骤(f)的脱保护基助剂为选自氟化氢吡啶、四丁基氟化铵中的一种或几种;
72、优选地,步骤(f)的溶剂为选自乙腈、四氢呋喃、二氯甲烷、n,n-二甲基甲酰胺中的一种或几种;
73、优选地,化合物6、苄基试剂、碱以及催化剂的摩尔比为1.0:(1.2~2.0):(1.2~2.0):(0~0.2)。
74、步骤(g)
75、化合物7与氨基被保护的氨基酸在溶剂、碱和反应助剂存在下进行酯化反应获得化合物8。
76、优选地,步骤(g)的氨基酸保护基为选自9-芴基甲氧基羰基保护基、叔丁氧羰基(boc)保护基中的一种;
77、优选地,步骤(g)的碱为选自三乙胺、n,n-二异丙基乙胺(dipea)、4-二甲氨基吡啶(dmap)一种或几种;
78、优选地,步骤(g)的反应助剂优选为2,4,6三氯苯甲酰氯(tcbc);
79、优选地,步骤(g)的反应温度为0~40℃(例如0、10、20、30、40℃),反应时间为8~24小时(例如8、10、12、18、24小时);
80、优选地,步骤(g)的溶剂为选自二氯甲烷、甲苯、乙腈、n,n-二甲基甲酰胺中的一种或几种。
81、步骤(h)
82、化合物8在酸存在下脱除保护基,然后在溶剂、碱和缩合剂存在下进行酰胺化反应获得化合物9。
83、优选地,步骤(h)的酸为有机酸,优选为三氟乙酸;
84、优选地,步骤(h)的碱为选自三乙胺、n,n-二异丙基乙胺(dipea)、4-二甲氨基吡啶(dmap)中的一种或几种;
85、优选地,步骤(h)的缩合剂为选自n,n,n′,n′-四甲基-o-(7-氮杂苯并三唑-1-基)六氟磷酸脲(hatu)、1h-苯并三唑-1-基氧三吡咯烷基六氟磷酸盐(pybop)中的一种或几种;
86、优选地,步骤(h)的溶剂选自二氯甲烷、甲苯、乙腈,n,n-二甲基甲酰胺中的一种或多种;
87、优选地,二氯甲烷与三氟乙酸的体积比可为(1.0~5.0):(0.5~3.0)。
88、步骤(i)
89、化合物9在溶剂和催化剂存在下进行烯烃关环复分解反应和氢化还原反应脱除保护基获得化合物10。
90、优选地,步骤(i)的烯烃关环复分解反应使用的催化剂为选自grubbs二代催化剂、二氯化钯、钯碳、氢氧化钯中的一种或几种,摩尔含量为1~10mmol%;
91、优选地,步骤(i)的烯烃关环复分解反应的反应温度为25~100℃,反应时间为15~30小时;
92、优选地,步骤(i)的氢化还原反应使用的催化剂为选自二氯化钯、钯碳、氢氧化钯碳中的一种或几种;
93、优选地,步骤(i)的溶剂为选自二氯甲烷、甲苯、甲醇、四氢呋喃中的一种或几种。
94、步骤(j)
95、化合物10与羧酸或酰氯在溶剂、碱和任选的缩合剂存在下进行酯化反应获得式i化合物。
96、若反应原料r3为羧酸时,化合物10在溶剂、缩合剂和碱的存在下进行酯化反应获得式i化合物;若反应原料r3为酰氯时,化合物10在溶剂和碱的存在下进行酯化反应获得式i化合物。
97、优选地,步骤(j)的碱为选择三乙胺、n,n-二异丙基乙胺(dipea)、4-二甲氨基吡啶(dmap)中的一种或几种;
98、优选地,步骤(j)的溶剂为选自二氯甲烷、四氢呋喃、甲苯、乙腈、乙酸乙酯中的一种或几种;
99、优选地,步骤(j)的缩合剂为选自二环己基碳二亚胺(dcc)、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edci)、偶氮二甲酸二乙酯中的一种或几种。
100、优选地,上述合成方法a~j均在惰性气体保护下进行,保护气体包括氮气、氩气、氦气中的至少一种。
101、本发明的dysoxylactam a及其类似物的制备方法以不同取代的醛以及易得的丙醛作为起始原料,通过两次羟醛缩合反应构建连续4个手性中心,再通过磺酰化反应和硼氢化还原,羟基保护基的脱除、保护以及与氨基酸的酯化反应,构建了目标分子所有的手性中心,最后再通过酰胺反应、关环复分解反应和氢化还原反应,成功获得了天然产物dysoxylactam a及其类似物。该制备方法具有较好的通适性,总收率高,步骤简洁,易衍生化,操作要求低,可大量制备,具有广阔的应用发展前景,为该类化合物后续深入的构效关系和活性研究奠定了坚实基础。
102、在上文中已经详细地描述了本发明,但是上述实施方式本质上仅是例示性,且并不欲限制本发明。此外,本文并不受前述现有技术或
技术实现要素:
或以下实施例中所描述的任何理论的限制。
- 还没有人留言评论。精彩留言会获得点赞!