以有机芳香酸为反应介质一步法制备聚酰亚胺的方法与流程
1.本发明属于聚合物合成技术领域,具体涉及一种以有机芳香酸为介质的一步法合成聚酰亚胺的制备方法。
背景技术:
2.聚酰亚胺(pi)是主链上含有酰亚胺环的一类聚合物,具有优异的热稳定性、机械性能和介电性能等,作为特种涂料、薄膜、纤维和复合材料广泛应用在电子电器、航空航天、工业分离、光电显示和机械部件等领域([1]丁孟贤.聚酰亚胺:单体合成聚合方法及材料制备[m].科学出版社,国际标准书号isbn:9787030310804,2021。[2]宋晓峰.聚酰亚胺的研究与进展[j].纤维复合材料,2007,3:33。)。
[0003]
聚酰亚胺又可分为均苯型pi,可溶性pi,聚酰胺-酰亚胺(pai)和聚醚酰亚胺(pei)四类。根据合成与加工方法,聚酰亚胺又可分为热塑性聚酰亚胺和热固性聚酰亚胺。热塑性聚酰亚胺具有良好的热塑加工性能,它不仅可采用热固性聚酰亚胺的所有加工方式成型,还可采用适合于热塑性塑料的挤出、注塑、模压、吹塑等方法成型,因此特别适于一次成型结构复杂的制品,无需二次加工,解决了传统热固性聚酰亚胺成型加工困难、产品形式单一等问题。典型的热塑性聚酰亚胺是聚醚酰亚胺,如沙比特的牌号ultem(pei-1)和extem(pei-2)。ultem(pet-1)具有非卤化阻燃(osu/faa可用)、强度极高、热阻能力出色(玻璃化温度tg为217~230℃)、金属粘附性极佳等性能,其型号ultem1010的特性粘度(间甲酚为溶剂,30℃)为0.5dl/g;extem(pei-2)的韧性好且异常耐热(tg为247~300℃),并具有出色的金属附着性,其粘度约为0.3~0.4dl/g。这两个热塑性聚醚亚胺常用于复合材料、涂料和粘合剂添加剂,热塑性复合材料和纤维等。
[0004]
pei-1和pei-2的化学结构如下,其合成方法之一是由2,2
’‑
双[4-(3,4-二羧苯氧基)苯基]丙烷四酸二酐(bpada)分别与间苯二胺(mpda)和4,4
’‑
二氨基二苯砜(dds)缩聚得到。
[0005][0006]
聚酰亚胺主要由二元酐和二元胺合成,二酐、二胺品种繁多,不同的组合就可以获得不同性能的聚酰亚胺。合成方法主要分为一步法和二步法。
[0007]
二步法通常适用于制备有机溶剂不溶的聚酰亚胺,第一步是将二酐和二胺在极性溶剂,如dmf,dmac,nmp或the/甲醇混合溶剂中先进行低温缩聚,获得可溶的聚酰胺酸;第二
步是成膜或纺丝后加热至300℃左右脱水实现亚酰胺化,得到聚酰亚胺。对于可溶的聚酰亚胺,可以向聚酰胺酸中加入乙酐和叔胺类催化剂,进行亚酰胺化,得到聚酰亚胺溶液和粉末。
[0008]
一步法是将二胺和二酐还可以在高沸点溶剂中直接加热缩聚,获得聚酰亚胺。反应过程也是经过聚酰氨胺酸中间体的形成,然后经过亚酰胺化生成聚酰亚胺。根据反应机理,一步法聚合的关键一步仍然是亚酰胺化。大量研究表明,酸性催化剂可使亚酰胺化反应加速。一步法的基本前提条件是聚酰亚胺能溶解在聚合所用的溶剂中。由于聚酰亚胺含有刚性的杂环结构,通常难溶于常用的有机溶剂中。所以,可用于一步法合成的溶剂较少,常用的溶剂为间甲基苯酚和对氯苯酚。这两个溶剂都是酚类化合物,室温下为粘稠液体,沸点超过200℃,聚合反应一般在此高温下进行,确保溶解聚酰亚胺,促进亚酰胺化反应。已知,有机酸或有机碱,如苯磺酸和异喹啉,常作为催化剂加入反应溶液中,被认为能促进亚酰胺化反应,但并没有在反应机理和发应动力学方面有较深入的研究(alexandera.kuznetsov,et al.,macromolecular symposia 128(1):203
–
219,1998.)。酚类化合物的酸性较弱,难以有效地催化亚酰胺化反应。根据二酐和二胺的化学结构,在间甲基苯酚和对氯苯酚中的聚合发应通常在180~200℃下进行,几小时到十几个小时完成亚酰胺化,得到聚酰亚胺。
[0009]
因此,行业内普遍认为以间甲基苯酚或对氯苯酚的酚类溶剂为介质的一步法制备聚酰亚胺的合成工艺技术具有以下有待改进的不足及缺陷:1)常温下,溶剂的气味大、毒性较大;2)聚合反应温度高、时间长,否则亚酰胺化不完全,得到聚酰胺酸-聚酰亚胺的共聚物;在高温长时间下反应,一些聚酰亚胺或聚酰胺酸中间体容易发生分解,聚合物的分子量或溶液粘度较低;3)需要加入有机酸或有机碱催化剂,促使亚酰胺化反应完全;4)分离和纯化聚合物需要用到溶剂量4~5倍以上的甲醇为沉淀剂,聚合物需要反复洗涤才能除去粘性较高的溶剂。溶剂与甲醇的分离与回收会造成对环境一定的污染和制备成本的增加。
技术实现要素:
[0010]
针对现有技术存在的问题,本发明提供了一种以有机芳香酸为反应介质一步法制备聚酰亚胺的方法,例如,该方法可用于制备聚醚酰亚胺pei-1和pei-2。
[0011]
为了实现以上目的,本发明采用以下技术方案:
[0012]
一种以有机芳香酸为反应介质一步法制备聚酰亚胺的方法,所述方法包括以下步骤:
[0013]
步骤(1):将等当量的芳香二酐和芳香二胺单体与作为反应介质的有机芳香酸混合均匀;
[0014]
步骤(2):在恒定温度下进行缩聚反应,得到缩聚反应液,经后处理得到聚酰亚胺;其中,
[0015]
所述有机芳香酸选自苯甲酸、水杨酸和5-氯水杨酸中的一种或两种。
[0016]
上述方法中,作为一种优选实施方式,芳香二酐和芳香二胺单体在所述反应介质中的总固含量为30%~45%(例如,32%、35%、40%、43%)。
[0017]
本发明中的单体的固含量,是指两种单体在反应介质中的总的质量百分比含量,以100g反应介质为例,100g反应介质中加入30~45g两种单体。
[0018]
上述方法中,作为一种优选实施方式,芳香二胺选自间苯二胺(mpda)、4,4
’‑
二氨
基二苯砜(dds)中的至少一种。
[0019]
上述方法中,作为一种优选实施方式,芳香二酐选自2,2
’‑
双[4-(3,4-二羧苯氧基)苯基]丙烷四酸二酐(bpada)。
[0020]
上述方法中,作为一种优选实施方式,当芳香二胺选自间苯二胺和4,4
’‑
二氨基二苯砜中的至少一种,芳香二酐选自2,2
’‑
双[4-(3,4-二羧苯氧基)苯基]丙烷四酸二酐时,制备得到的聚酰亚胺为聚醚酰亚胺。
[0021]
上述方法中,作为一种优选实施方式,当有机芳香酸为二元混合有机芳香酸(即,两种有机芳香酸的混合物)时,缩聚反应的温度为110~140℃(例如,115℃、120℃、125℃、130℃、135℃),反应时间为0.8~1.5h(例如,0.9h、1h、1.2h、1.3h、1.4h),优选为1h。
[0022]
本发明中,二元混合有机芳香酸中两种有机芳香酸可以以任意比例混合,优选两种有机芳香酸的质量混合比为1:4~4:1(即1:4~0.25),再优选为1:2~2:1(即1:2~0.5)。
[0023]
本发明中,两种有机芳香酸的混合比例不同,则,二元混合有机芳香酸的熔点不同,也意味着聚合反应温度不同,即,聚合反应温度与两种有机芳香酸的混合比例有关。
[0024]
上述方法中,作为一种优选实施方式,当有机芳香酸为二元混合有机芳香酸时,制备得到的聚醚酰亚胺的粘度超过0.3dl/g。
[0025]
上述方法中,作为一种优选实施方式,当有机芳香酸为单一有机芳香酸时,缩聚反应的温度为170~200℃(例如,175℃、180℃、185℃、190℃、195℃),反应时间为1~2h(例如,1.2h、1.5h、1.7h、1.8h、1.9h),优选为1.5h。
[0026]
上述方法中,作为一种优选实施方式,当有机芳香酸为单一有机芳香酸混合物时,制备得到的聚醚酰亚胺的粘度超过0.3dl/g。
[0027]
上述方法中,作为一种优选实施方式,所述步骤(1)中,所述混合为搅拌混合,混合温度为室温,这里室温是指20~25℃;优选地,所述混合在氮气保护氛围下进行。
[0028]
上述方法中,作为一种优选实施方式,所述后处理包括:趁热将聚酰亚胺反应液倒在不锈钢板或铝箔纸上,冷却至室温后用粉碎机碾碎,得到所述聚醚酰亚胺/有机芳香酸混合物粉末,除去有机芳香酸后,可得到聚醚酰亚胺粉末。
[0029]
上述方法中,作为一种优选实施方式,所述方法还包括:
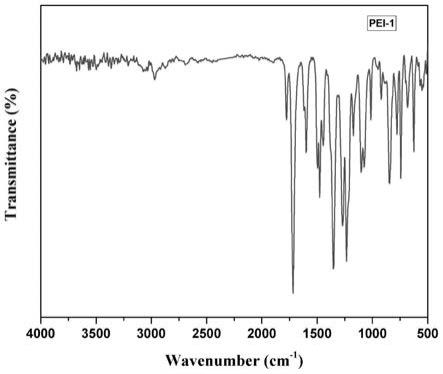
[0030]
步骤(3):用热水或甲醇洗涤所述聚醚酰亚胺/有机芳香酸混合物粉末;优选地,将聚酰亚胺/有机芳香酸混合物粉末在沸水或甲醇中搅拌洗涤纯化,趁热过滤,烘干;更优选地,洗涤时间为1~4h(例如,1.5h、2.0h、2.5h、3.0h、3.5h)。
[0031]
上述方法中,作为一种优选实施方式,所述方法还包括:
[0032]
步骤(4):有机芳香酸的回收:将水中结晶析出有机芳香酸,回收再用;或将甲醇中回收的有机芳香酸升华纯化,回收再用。
[0033]
本发明所述方法还适用于制备其他的在所述有机芳香酸或二元混合有机芳香酸介质中的聚合过程中可溶解的聚酰亚胺。
[0034]
本发明中,在相互不冲突的情况下,上述技术特征可以自由组合形成新的技术方案。
[0035]
基于本发明,与传统的酚类溶剂介质相比,
[0036]
1)分别以苯甲酸,水杨酸和5-氯水杨酸为反应介质,可以在较低反应温度下和较短时间内得到分子量或粘度较高的、完全亚酰胺化的聚酰亚胺,例如,pei-1和pei-2聚醚酰
亚胺;
[0037]
2)以苯甲酸,水杨酸和5-氯水杨酸的二元混合物为介质,在不同的混合比例范围内,可以在更低反应温度下和更短的时间内即可得到分子量或粘度较高的pei-1和pei-2聚醚酰亚胺;
[0038]
3)本发明中的这些有机芳香酸既可以作为反应介质,又可以作为亚酰胺化的催化剂,能加速聚合反应、使聚酰胺酸中间体全部亚酰胺化。
[0039]
以所述有机芳香酸为介质能在较低温度下、很短的反应时间内得到高粘度的聚酰亚胺的反应原理可归结于以下独特的几点:
[0040]
1)所述有机芳香酸的酸性比常用的间甲基苯酚酸性要强,或pka值较低(见表1)。
[0041]
2)在一定混合比例时,所述二元混合有机芳香酸的熔点低于单独每个有机芳香酸(参见表1),可使反应物在较低温度下熔化,进行反应。
[0042]
3)有机芳香酸或二元混合有机芳香酸酸对聚酰亚胺,如pei-1和pei-2,具有较好的溶解性,利于聚合反应进行。
[0043]
表1聚合反应介质的物性和适合聚合反应的参考温度
[0044]
溶剂熔点℃沸点℃pka反应温度℃间甲基苯酚1020310.09200水杨酸1592102.98≥1605-氯水杨酸1723202.00≥170苯甲酸1222494.20≥120水杨酸/5-氯水杨酸(重量比:1:1)140
‑‑‑‑‑‑
≥140水杨酸/苯甲酸(重量比:1:2)120
‑‑‑‑‑‑
≥1205-氯水杨酸/苯甲酸(重量比:1:2)110
‑‑‑‑‑‑
≥110
[0045]
与现有技术相比,本发明具有以下有益效果:
[0046]
(1)与以酚类化合物为反应介质、常用的一步法工艺技术相比,本发明以苯甲酸、水杨酸和5-氯水杨酸的二元混合物为介质,在不同的质量混合比例范围内,在较低反应温度下(110~140℃),反应时间大大缩短(比如缩短到1.5h),得到分子量较高或粘度较高的、完全亚酰胺化的聚酰亚胺,例如,pei-1和pei-2聚醚酰亚胺;
[0047]
(2)分别以苯甲酸、水杨酸和5-氯水杨酸为介质,在170~200℃反应温度下,聚合反应时间大大缩短(比如缩短到1.5h),得到分子量较高或粘度较高的、完全亚酰胺化的聚酰亚胺,例如,pei-1和pei-2聚醚酰亚胺;
[0048]
(3)有机芳香酸的酸性大于酚类(如,间甲基苯酚)溶剂,能更有效地催化亚酰胺化环合反应,使聚酰胺酸中间体全部亚酰胺化;
[0049]
(4)通过用热水洗涤,可分离和纯化聚酰亚胺产物;有机芳香酸可从冷水中析出,进一步升华纯化,反复使用;
[0050]
(5)有机芳香酸安全无毒绿色环保,可回收再用,适合工业化生产。
附图说明
[0051]
图1为本发明实施例1所得pei-1红外图谱。
[0052]
图2为本发明实施例5所得pei-2的红外图谱。
[0053]
图3为本发明对比例2中d2-3所得pei-2产物的红外图谱。
具体实施方式
[0054]
下面将结合说明书附图对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员所获得的所有其他实施例,都属于本发明保护的范围。
[0055]
本发明中,聚酰亚胺的特性粘度测试方法为:在30℃下,以间甲基苯酚为溶剂,使用乌氏粘度计,采用毛细管法测试。
[0056]
下面实施例中所有反应加热介质为油浴,反应温度为反应液体的温度。
[0057]
聚合物的分离可采用如下两种方法:
[0058]
方法1:将聚合反应液倒入甲醇中,搅拌,过滤,再用甲醇洗涤,过滤,烘干。
[0059]
方法2:适用于有机芳香酸为反应介质。将热反应液倒在不锈钢板或铝箔纸上,冷却至室温后用粉碎机碾碎成粉末;再将聚酰亚胺粉末在甲醇或沸水中搅拌洗涤纯化,趁热过滤,真空烘干。
[0060]
有机芳香酸的回收利用:
[0061]
将水中结晶析出有机芳香酸,回收再用;或将甲醇中回收的有机芳香酸升华纯化,回收再用。
[0062]
以bpada和mpda制备pei-1聚醚酰亚胺
[0063]
对比例1
[0064]
本实施例以间甲基苯酚为反应介质,以bpada和mpda制备pei-1聚醚酰亚胺,具体地,
[0065]
在室温(20~25℃)和氮气氛围下,将7g的间甲基苯酚(bp 202.3℃)加入到50ml的反应管中,再加入升华后的mpda(0.541g,5mmol),磁力搅拌至完全溶解。溶液澄清透明后,加入bpada(2.602g,5mmol),配置成单体固含量为30%重量百分比的反应液。将含液体的反应管部分完全浸没在恒温加热油浴中,搅拌下由室温升温至200℃反应14h后停止加热。保持搅拌让其自行冷却至室温,后将聚合物溶液缓慢倒入250ml快速搅拌下的甲醇中,得到白色絮状固体。将过滤收集的产物用甲醇(50ml)洗涤3次,然后在索氏提取器中用乙醇洗涤24h,将产物置于真空烘箱中干燥(60℃/12h,70℃/1h,80℃/1h,90℃/1h,100℃/6h)。pei-1产物的收率为87.7%(2.60g),特性粘度为0.98dl/g。
[0066]
表2在不同反应条件下得到的pei-1产物的收率及粘度
[0067][0068]
*聚合物含有未亚酰胺化环合的部分,即为聚酰胺酸-聚酰胺的共聚物。
[0069]
以间甲基苯酚反应介质,改变固含量、反应温度和反应时间,重复上述步骤,得到如表2所示的pei-1产物的收率及粘度值。
[0070]
该实施例表明,以间甲基苯酚为溶剂,降低反应温度或缩短反应时间,不能得到完全亚酰胺化的pei-1产物。
[0071]
实施例1
[0072]
本实施例以水杨酸为反应介质,以bpada和mpda制备pei-1聚醚酰亚胺。具体地,
[0073]
在室温(20~25℃)和氮气氛围下,将mpda(0.541g,5mmol)加入到50ml反应管中,再加入水杨酸(3.5g)后摇动混合。然后再加入bpada(2.602g,5mmol)和水杨酸(3.5g),配置成单体固含量为30%重量百分比的反应混合物。将含有物料的反应管部分完全浸没在恒温加热油浴中,搅拌下由室温(20~25℃)升温至165~170℃时打开搅拌,待水杨酸固体完全熔化为液体时开始计时。在170℃下恒温搅拌1.5h后,拿出反应管降温到140~160℃,趁热将反应液倒到不锈钢板或铝箔纸上,充分冷却至室温后用粉碎机碾碎成粉末。将固体粉末倒入250ml甲醇中洗涤3次,后过滤,在真空烘箱中程序升温下干燥(60℃/12h,70℃/1h,80℃/1h,90℃/1h,100℃/6h),得到2.67g白色固体粉末产物(90.1%收率)。产物pei-1的tg为225℃,特性粘度为1.18dl/g,红外特征峰为1771cm-1
、1704cm-1
、1380cm-1
、720cm-1
,如图1所示。
[0074]
以水杨酸为反应介质,改变固含量、反应温度和反应时间,重复上述步骤,得到如表3所示的pei-1产物的粘度值。
[0075]
表3实施例1中在不同反应条件下得到的pei-1产物的收率及粘度
[0076][0077]
该实施例表明,以水杨酸为反应介质,在170℃温度下反应1.5小时即可得到高分子量的pei-1产物。在相同反应条件下,增加单体固体含量,产物的粘度增加,分子量更高;且产物完全亚酰胺化,不含酰胺酸基团。
[0078]
实施例2
[0079]
本实施例以5-氯水杨酸为反应介质,以bpada和mpda制备pei-1聚醚酰亚胺。具体地,
[0080]
在室温(20~25℃)和氮气氛围下,将mpda(0.541g,5mmol)加入到50ml反应管中,再加入5-氯水杨酸(3.5g)后摇动混合。然后再加入bpada(2.602g,5mmol)和5-氯水杨酸(3.5g),配置成单体固含量为30%重量百分比的反应液。将含有物料的反应管部分完全浸没在恒温加热油浴中,搅拌下由室温(20~25℃)升温至180~185℃时打开搅拌,待5-氯水杨酸固体完全熔化为液体时开始计时。在185℃下恒温搅拌1.5h后,拿出反应管降温到140~160℃,趁热将反应液倒到不锈钢板或平铺铝箔纸上,充分冷却至室温后用粉碎机碾碎成粉末。固体粉末倒入250ml甲醇中洗涤3次,后过滤,在真空烘箱中程序升温下干燥(60℃/12h,70℃/1h,80℃/1h,90℃/1h,100℃/6h)得到白色固体粉末2.72g(91.8%收率),特性测试粘度为0.81dl/g。红外光谱检测正式产物完全亚酰胺化。
[0081]
改变固含量为45%,重复上述步骤,得到pei-1产物的收率为92%,特性粘度为0.97dl/g。
[0082]
该实施例表明,以5-氯水杨酸为反应介质,在185℃温度下反应1.5小时即可得到高分子量、完全亚酰胺化的聚醚酰亚胺pei-1。在相同反应条件下,提高单体固体含量,产物的粘度增加,分子量更高。
[0083]
实施例3
[0084]
本实施例以苯甲酸为反应介质,以bpada和mpda制备pei-1聚醚酰亚胺。具体地,
[0085]
在室温(20~25℃)和氮气氛围下,将mpda(0.541g,5mmol)加入到50ml反应管中,再加入苯甲酸(3.5g)后摇动混合。然后再加入bpada(2.602g,5mmol)和苯甲酸(3.5g),配置成单体固含量为30%重量百分比的反应混合物。将含有物料的反应管部分浸没在恒温加热油浴中,搅拌下由室温(20~25℃)升温至200℃时打开搅拌,待苯甲酸固体熔化为液体时开始计时。在200℃下恒温搅拌2h后,拿出反应管降温到140~160℃,趁热将反应液倒到不锈钢板或铝箔纸上,充分冷却至室温后用粉碎机碾碎成粉末。固体粉末倒入250ml甲醇中洗涤3次,后过滤,在真空烘箱中程序升温下干燥(60℃/12h,70℃/1h,80℃/1h,90℃/1h,100℃/6h)得到白色固体粉末2.75g(92.8%收率)。产物pei-1的tg为225℃,特性粘度为0.86dl/g。
[0086]
改变固含量和反应温度,重复上述步骤,得到如表4所示的pei-1的粘度值。
[0087]
表4实施例3中在不同反应条件下得到的pei-1产物的收率及粘度
[0088][0089]
*聚合物含有未能亚酰胺化环合的部分,即为聚酰胺酸-聚酰胺的共聚物。
[0090]
该实施例表明,以苯甲酸为反应介质,在较高(200℃)温度下反应2小时即可得到高分子量或高粘度的、完全亚酰胺化的聚醚酰亚胺pei-1。在相同反应条件下,增加单体固体含量,产物的粘度增加,分子量更高。
[0091]
实施例4
[0092]
本实施例以水杨酸/5-氯水杨酸/苯甲酸的二元混合物为反应介质,以bpada和mpda制备pei-1聚醚酰亚胺。具体地,
[0093]
在室温(20~25℃)和氮气氛围下,将mpda(0.541g,5mmol)加入到50ml反应管中,再加入3.5g混合均匀的水杨酸(1.75g)和5-氯水杨酸(1.75g)后摇动混合。然后再加入bpada(2.602g,5mmol)和3.5g混合均匀的水杨酸(1.75g)和5-氯水杨酸(1.75g),配置成单体固含量为30%重量百分比的反应混合物。将含液体的反应管部分完全浸没在恒温加热油浴中,搅拌下由室温(20~25℃)升温至140℃时打开搅拌,待有机芳香酸熔化时开始计时。在140℃下恒温搅拌1h后,拿出反应管,趁热将反应液倒到不锈钢板或平铺铝箔纸上,充分冷却至室温后用粉碎机碾碎成粉末。固体粉末倒入250ml甲醇中洗涤3次,后过滤,在真空烘箱中程序升温下干燥(60℃/12h,70℃/1h,80℃/1h,90℃/1h,100℃/6h)得到白色固体粉末
2.79g(94.1%收率),特性粘度为0.86dl/g。
[0094]
改变介质、固含量和反应温度,重复上述步骤,得到pei-1的特性粘度如下表5所示。
[0095]
该实施例表明,以二元混合酸为反应介质,与单酸(水杨酸或5-氯水杨酸)相比,可在较低温度(如110℃)下反应1小时得到高粘度的、完全亚酰胺化的聚醚酰亚胺pei-1。在同一反应介质中,提高单体固体含量或升高反应温度,均能提高产物的粘度。介质的选择和比例对聚合物的粘度有影响。
[0096]
表5实施例4中在不同反应条件下得到的pei-1产物的收率及粘度
[0097][0098]
以bpada和dds制备聚醚酰亚胺pei-2
[0099]
对比例2
[0100]
本实施例以间甲基苯酚为反应介质,以bpada和dds制备pei-1聚醚酰亚胺。具体地,
[0101]
在室温(20~25℃)和氮气氛围中,将8.5g间甲基苯酚加入到50ml反应管中,再加入dds(1.241g,5mmol),磁力搅拌至完全溶解,溶液澄清透明后再加入bpada(2.602g,5mmol),配置成单体固含量为30%重量百分比的反应液。将含液体的反应管部分浸没在恒温加热油浴中,搅拌下由室温(20~25℃)升温至200℃反应14h后停止加热,保持搅拌让其自行冷却至室温。然后将聚合物溶液缓慢倒入250ml剧烈搅拌下的甲醇中,得到白色纤维状固体。将过滤收集的产物用甲醇(50ml)洗涤3次,然后在索氏提取器中用乙醇洗涤24h,将产物置于真空烘箱中干燥(60℃/12h,70℃/1h,80℃/1h,90℃/1h,100℃/6h)。pei-2产物的收率为87.7%(2.60g),特性粘度为0.6dl/g。
[0102]
改变固含量、反应温度和反应时间,重复上述步骤,得到如下表6所示的pei-2粘度值。
[0103]
表6对比例2中在不同反应条件下得到的pei-2产物的收率及粘度
[0104][0105]
*聚合物含有未能亚酰胺化环合的部分,即为聚酰胺酸-聚酰胺的共聚物。
[0106]
该实例结果表明以间甲基苯酚为反应介质,降低反应温度或缩短反应时间,不能得到完全亚酰胺化的pei-2产物(参见图3所示的对比例d2-3中的pei-2产物的红外图谱,图3中3500cm-1
附近(如图3中的箭头所指方向)和1400cm-1
附近(图3中圆圈标记处)的吸收峰归属于酰胺酸基团,证明亚酰胺化不完全,即,证明有聚酰胺酸-聚酰胺的共聚物存在)。
[0107]
实施例5
[0108]
本实施例以水杨酸为反应介质,以bpada和dds制备pei-1聚醚酰亚胺。具体地,
[0109]
在室温(20~25℃)和氮气氛围中,将dds(1.241g,5mmol)加入到50ml反应管中,再加入4.5g水杨酸后摇动混合。然后再加入bpada(2.602g,5mmol)和4.5g水杨酸,配置成单体固含量为30%重量百分比的反应混合物。将含液体的反应管部分浸没在恒温加热油浴中,搅拌下由室温(20~25℃)升至165~170℃后打开搅拌,待水杨酸固体熔化为液体时开始计时。在170℃下恒温搅拌1.5h后,拿出反应管降温到140~160℃,趁热将反应液倒到不锈钢板或铝箔纸上,充分冷却至室温后用粉碎机碾碎成粉末。固体粉末倒入250ml甲醇中洗涤3次,后过滤,在真空烘箱中程序升温下干燥(60℃/12h,70℃/1h,80℃/1h,90℃/1h,100℃/6h)得到白色固体粉末3.26g(89.0%收率)。产物pei-2的tg为251℃,特性粘度为0.47dl/g,红外特征峰为1780cm-1
、1704cm-1
、1380cm-1
、720cm-1
(参见图2所示的pei-2的红外图谱)。
[0110]
改变单体固含量为45%,重复上述步骤,得到pei-2的收率为91.0%,特性粘度为0.83dl/g。
[0111]
该实施例结果表明,以水杨酸为反应介质,在170℃温度下反应1.5小时即可得到高分子量或高粘度的聚醚酰亚胺pei-2。在相同反应条件下,提高单体固体含量,产物的粘度升高,分子量更高。所得pei-2完全亚酰胺化,不含酰胺酸基团。
[0112]
实施例6
[0113]
本实施例以5-氯水杨酸为反应介质,以bpada和dds制备pei-1聚醚酰亚胺。具体地,
[0114]
在室温(20~25℃)和氮气氛围中,将dds(1.242g,5mmol)加入到50ml反应管中,再加入4.5g的5-氯水杨酸后摇动混合。然后再加入bpada(2.602g,5mmol)和4.5g的5-氯水杨酸,配置成单体固含量为30%重量百分比的反应混合物。将含液体的反应管部分完全浸没在恒温加热油浴中,搅拌下由室温(20~25℃)升温至180~185℃时打开搅拌,待所有固体完全熔化为液体时开始计时。在185℃下恒温搅拌1.5h后,拿出反应管降温到140~160℃,趁热将反应液倒到不锈钢板或平铺的铝箔纸上,充分冷却至室温后用粉碎机碾碎成粉末。固体粉末倒入250ml甲醇中洗涤3次,后过滤,在真空烘箱中程序升温下干燥(60℃/12h,70
℃/1h,80℃/1h,90℃/1h,100℃/6h)得到白色固体粉末3.20g(87.4%收率),特性粘度为0.39dl/g。
[0115]
改变单体固含量为45%,重复上述步骤,得到完全亚酰胺化的pei-2的特性粘度为0.90dl/g。
[0116]
该实例结果表明以水杨酸为反应介质,在185℃温度下反应1.5小时即可得到高分子量或高粘度的聚醚酰亚胺pei-2。在相同反应条件下,增加单体固体含量,产物的粘度增加,分子量更高。产物完全亚酰胺化,不含酰胺酸基团。
[0117]
实施例7
[0118]
本实施例以苯甲酸为反应介质,以bpada和dds制备pei-1聚醚酰亚胺。具体地,
[0119]
在室温(20~25℃)和氮气氛围中,将dds(1.242g,5mmol)加入到50ml反应管中,再加入4.5g的苯甲酸后摇动混合。然后再加入bpada(2.603g,5mmol)和4.5g的苯甲酸,配置成单体固含量为30%重量百分比的反应混合物。将含液体的反应管部分完全浸没在恒温加热油浴中,搅拌下由室温(20~25℃)升温至200℃时打开搅拌,待苯甲酸固体熔化为液体时开始计时。在185℃下恒温搅拌2h后,拿出反应管降温到140~160℃,趁热将反应液倒到不锈钢板或平铺的铝箔纸上,充分冷却至室温后用粉碎机碾碎成粉末。固体粉末倒入250ml甲醇中洗涤3次,后过滤,在真空烘箱中程序升温下干燥(60℃/12h,70℃/1h,80℃/1h,90℃/1h,100℃/6h)得到白色固体粉末3.20g(87.4%收率),特性粘度为0.36dl/g。
[0120]
改变固含量和反应温度,重复上述步骤,得到如表7的pei-2的收率及粘度值。
[0121]
表7实施例7中在不同反应条件下得到的pei-2产物的收率及粘度
[0122][0123]
*聚合物含有未能亚酰胺化环合的部分,即为聚酰胺酸-聚酰胺的共聚物。
[0124]
该实例结果表明,以苯甲酸为反应介质,在200℃温度下反应2小时即可得到高分子量或高粘度的聚醚酰亚胺pei-2。在相同反应条件下,增加单体固体含量,产物的粘度增加,分子量更高。产物完全亚酰胺化,不含酰胺酸基团。
[0125]
实施例8
[0126]
本实施例以水杨酸/5-氯水杨酸/苯甲酸的二元混合物为反应介质,以bpada和dds制备pei-1聚醚酰亚胺。具体地,
[0127]
在室温(20~25℃)和氮气氛围中,将dds(1.242g,5mmol)加入到50ml反应管中,再加入4.5g混合研磨均匀的水杨酸(2.25g)和5-氯水杨酸(2.25g),摇动混合。然后再加入bpada(2.602g,5mmol)和4.5g混合研磨均匀的水杨酸(2.25g)和5-氯水杨酸(2.25g),配置成单体固含量为30%重量百分比的反应混合物。将含液体的反应管部分浸没在恒温加热油浴中,搅拌下由室温(20~25℃)升温至140℃时打开搅拌,待有机芳香酸熔化时开始计时。在140℃下恒温搅拌1h后,拿出反应管,趁热将反应液倒到平铺的铝箔纸上,充分冷却至室
温后用粉碎机碾碎成粉末。将固体粉末倒入250ml甲醇中洗涤3次,后过滤,在真空烘箱中程序升温下干燥(60℃/12h,70℃/1h,80℃/1h,90℃/1h,100℃/6h)得到白色固体粉末3.35g(91.4%收率),特性粘度为0.59dl/g。
[0128]
改变介质、固含量和反应温度,重复上述步骤,得到pei-2的特性粘度如下表8所示。
[0129]
表8实施例8中在不同反应条件下得到的pei-2产物的收率及粘度
[0130][0131]
该实例表明以混合酸为反应介质,与单酸(苯甲酸或5-氯水杨酸)相比,可在较低温度(如110℃)下反应1小时得到高分子量或高粘度的、完全亚酰胺化的聚醚酰亚胺pei-2。在同一反应介质中,提高单体固体含量或升高反应温度,能增加产物的粘度。介质的选择和比例对聚合物的粘度有影响。
[0132]
反应介质的回收与纯化
[0133]
实施例9
[0134]
本实施例水杨酸的回收与纯化,以bpada和dds制备pei-1聚醚酰亚胺。具体地,
[0135]
第一种回收方法:将实施例1或实施例5中所得的聚合物/水杨酸混合物粉末,未经甲醇洗涤,加入到500ml的沸水中,搅拌洗涤1~4h,趁热过滤,聚合物粉末再用300ml沸水洗涤1~3h,趁热过滤得到pei-1聚醚酰亚胺粉末。将所有滤液合并,冷却至室温,静置2h,过滤收集析出的晶体,在空气对流烘箱60℃干燥24h,即得水杨酸。
[0136]
第二种回收方法:将实施例1或实施例5中所得的聚合物/水杨酸混合粉末经甲醇清洗好后,收集所得甲醇洗液,浓缩抽干后,得到水杨酸固体,待升华纯化。
[0137]
纯化:收集合并多次聚合后得到的水杨酸,然后取25g水杨酸平铺在100ml升华器底部,保持0.1~2.0mmhg的压力和90℃的温度,1~2h完成升华,得到24.2g白色晶体水杨酸。
[0138]
以上所述仅为本发明的部分实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均在本发明的权利要求保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1