一种合成手性苯基噁唑烷-2-酮的方法与流程
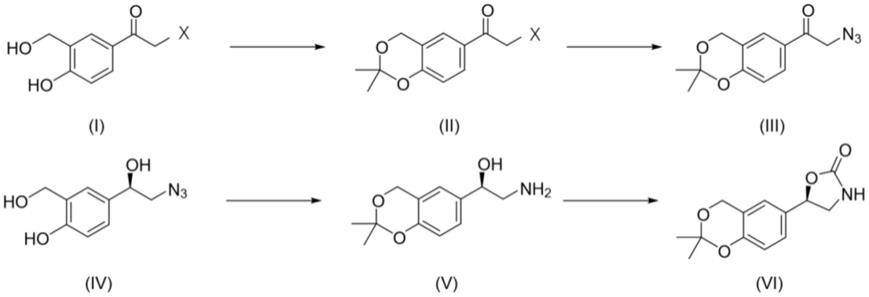
1.本发明属于有机化学合成领域,具体涉及一种合成一种合成手性苯基噁唑烷-2-酮的方法,即(r)-5-(2,2-二甲基-4h-苯并[d][1,3]二恶英-6-基)恶唑烷-2-酮的方法。
背景技术:
[0002]
如下所示,手性苯基噁唑烷-2-酮化合物(vi)是一类重要的有机合成中间体,在生物制药领域具有极为重要的作用。
[0003][0004]
目前,手性苯基噁唑烷-2-酮的合成路线以2003年由葛兰素史克药物研究中心所提出的最有代表性。该路线存在以下不足:1)在强碱条件下经buchwald反应合成化合物iii的步骤中,其收率仅有53%,在后续反应中,又以强酸的作用才可得到潜手性的β-氨基酮化合物v,反应条件苛刻;2)其关键中间体手性苯基噁唑烷-2-酮是又同样以强碱氢化钠的作用下关环生成生成,而且该步骤收率仅有70%(详见org.biomol.chem.,2003,1,1106-1111),整条路线总收率为29.7%,其收率较低,而且多次用到强酸强碱条件,后处理难度较大,为后续的合成带来众多不便。
[0005][0006]
专利cn107188813a公开了这一中间体主要合成路线,具体如下。该路线中使用到剧毒化合物溴素为溴源,不仅给反应本身操作带来困难,同时也给操作人员带来巨大的安全隐患。其次在进行关环时,所采用强碱试剂叔丁醇钾产生的碱性废物在环保处理方面也带来隐患。
[0007]
综上所述,虽然现阶段已经有多种方案可以实现手性苯基噁唑烷-2-酮的制备,但底物适用性以及操作安全性,重要的是在原子经济性上都还急需进一步提升。
技术实现要素:
[0008]
本发明针对现有的合成(r)-5-(2,2-二甲基-4h-苯并[d][1,3]二恶英-6-基)恶唑烷-2-酮路线的安全性低、收率低、难以得到高纯度产品等问题,利用催化剂(例如noyori’s催化剂),采用不对称转移氢化方法提供一种合成(r)-5-(2,2-二甲基-4h-苯并[d][1,3]二恶英-6-基)恶唑烷-2-酮的新方法。
[0009][0010]
其中,本发明由2-叠氮-1-(4-羟基-3-(羟甲基)苯基)乙烷-1-酮(化合物i)出发,经过转移氢化作用以90%收率,87%ee巧妙的构建手性羟基,之后叠氮基团经还原再次以高收率得到氨基,随后又避免强碱的使用,仅在缩合剂的作用下又以高收率得到iv,整个路线中没有使用到强碱试剂,即不会产生碱性废液,所用醇类溶剂皆可再次回收,同时给环保处理方面带来了极大的便利。
[0011]
手性苯基噁唑烷-2-酮化合物作为各类医药中间体和天然产物的合成起始物,其最具有代表性应用是葛兰素史克药物研究中心利用该中间体合成了维兰特罗,该药物在治疗慢性阻塞性肺病和哮喘等疾病中起了关键作用。本发明从2-溴代苯乙酮类化合物出发,通过保护、亲核进攻、不对称转移氢化,最后经cdi关环的方式高选择性地制备了手性苯基噁唑烷-2-酮。本发明开发了新的工艺路线,具有较好的适用性。本发明提出的合成路线及反应条件如下:
[0012][0013]
根据本发明的一种实施例方式,例如,上述路线中,x为br、cl或者i,优选为br。
[0014]
根据本发明的一种实施例方式,例如,本发明提出的一种合成(r)-5-(2,2-二甲基-4h-苯并[d][1,3]二恶英-6-基)恶唑烷-2-酮的方法包括:
[0015]
1)式i化合物在路易斯酸(例如对甲苯磺酸)的作用下,在有机溶剂中与保护试剂反应,生成双羟基保护的式ii化合物;优选的,所述有机溶剂为dcm/mtbe,采用dcm/mtbe可以方便后续处理;优选的,所述保护试剂选自2-甲氧基丙烯、2,2-二甲氧基丙烷和丙酮中的至少一种,进一步优选的,所述保护试剂为2-甲氧基丙烯。
[0016]
2)式ii化合物在叠氮试剂的亲核进攻下,在溶剂dmf中得到叠氮酮式iii化合物;优选的,所述叠氮试剂为叠氮化钠。
[0017]
3)在noyori's催化剂(例如,rhcl[(s,s)-tsdpen](p-cymene))的作用下,式iii化合物经转移氢化反应得到手性叠氮醇式iv化合物;所述转移氢化反应在转移氢化还原剂存在下进行,优选的,所述转移氢化还原剂为hco2h/et3n;优选的,该步骤的反应在有机溶剂中进行,进一步优选的,所述有机溶剂为dmf。
[0018]
4)式iv化合物催化氢化还原叠氮到氨基得到手性氨基醇v化合物;优选的,所述催化氢化还原包括:在三苯基膦作用下,以四氢呋喃为溶剂,经staudinger还原;或者,优选的,所述催化氢化还原为采用氢气还原。
[0019]
5)式v化合物在有机溶剂中,在缩合剂的作用下得到式vi化合物;优选的,所述缩合剂为n,n'-羰基二咪唑(cdi);优选的,所述有机溶剂为thf/etoh。
[0020]
根据本发明的一种实施例方式,例如,在步骤1)中,所需的保护基为2-甲氧基丙烯、2,2-二甲氧基丙烷、或者丙酮,优选用2-甲氧基丙烯进行保护,采用2-甲氧基丙烯进行保护可以得到更高的收率。采用的路易斯酸例如为氯化锌、氯化铁或者对甲苯磺酸;优选对甲苯磺酸,优选对甲苯磺酸的理由是在催化量下即可实现底物双羟基的保护。
[0021]
根据本发明的一种实施例方式,例如,在步骤2)中,所述的叠氮试剂为叠氮化钠或者三甲基叠氮硅。优选的,所述的叠氮试剂为叠氮化钠。
[0022]
根据本发明的一种实施例方式,例如,在步骤3)中,转移氢化反应所需的noyori's催化剂,可采用以下的至少一种:rucl[(s,s)-tsdpen](p-cymene)(cat.1),rucl[(s,sr)-tsdpen](mesitylene)(cat.2),rucl[(s,s)-tsdpen](benzene)(cat.3),rucl[(s,s)-fsdpen](p-cymene)(cat.4)。其中cat.1、cat.2、cat.3的制备可采用以下方法:二聚体的钌盐[rucl2(η6-arene)]2与甲苯磺酰二胺经混合后(例如,摩尔比1:1)在三乙胺的作用下,以异丙醇为溶剂与各类配体在回流条件下得到noyori's催化剂。
[0023]
四种催化剂结构如下所示:
[0024][0025]
根据本发明的一种实施例方式,例如,在步骤4)中,所述的催化氢化还原叠氮到氨基采用钯碳催化。当采用staudinger还原方法时,可采用三苯基膦作为还原试剂。
[0026]
根据本发明的一种实施例方式,例如,在步骤5)中,所述缩合试剂可采用n,n'-羰基二咪唑(cdi)进行缩合。
[0027]
采用本发明合成方法合成(r)-5-(2,2-二甲基-4h-苯并[d][1,3]二恶英-6-基)恶唑烷-2-酮,工艺路线新颖,总摩尔收率大于70%,具有路线简短、化学纯度高、易于生产的特点。
[0028]
本发明公开的合成(r)-5-(2,2-二甲基-4h-苯并[d][1,3]二恶英-6-基)恶唑烷-2-酮的方法,其中采用rhcl[(s,s)-tsdpen](p-cymene)不对称催化还原β-叠氮酮,可以快速、高效的构建高光学活性的β-叠氮醇,底物适用范围更广,对酸/碱敏感的底物都适用。整条路线中不需要强酸/碱性试剂,即无需产生酸/碱性废液,所用醇类溶剂皆可再次回收,同时给环保处理方面带来了极大的方便。同时,本发明提出的合成路线新颖,总摩尔收率大于70%,具有路线简短、化学纯度高、易于生产等优点。
附图说明
[0029]
为了更清楚地说明本发明实施例的技术方案,下面将对实施例的附图作简单地介绍,显然,下面描述中的附图仅仅涉及本发明的一些实施例,而非对本发明的限制。
[0030]
图1是实施例所得产品的核磁谱图。
具体实施方式
[0031]
下文将结合具体实施例对本发明的合成(1r)-1-(2,2-二甲基-4h-1,3-苯并二恶英-6-基)恶唑烷-2-酮的方法做更进一步的说明。应当理解,下列实施例仅为示例性地说明和解释本发明,而不应被解释为对本发明保护范围的限制。凡基于本发明上述内容所实现的技术均涵盖在本发明旨在保护的范围内。
[0032]
合成路线如下:
[0033][0034]
为解决现有路线的安全性低、收率低、难以得到高纯度产品等问题,本发明的实施例提供一种合成(r)-5-(2,2-二甲基-4h-苯并[d][1,3]二恶英-6-基)恶唑烷-2-酮的新方法。该方法包括以下反应步骤:
[0035]
1)式i化合物在对甲苯磺酸的作用下与2-甲氧基丙烯反应,生成双羟基保护的式ii化合物,其所用溶剂dcm/mtbe;
[0036]
2)式ii化合物在叠氮化钠的亲核进攻下,在溶剂dmf中得到叠氮酮式iii化合物;
[0037]
3)式iii化合物经转移氢化反应在noyori's催化剂(rhcl[(s,s)-tsdpen](p-cymene)的作用下得到手性叠氮醇式iv化合物,所需还原剂为hco2h/et3n体系;所需溶剂为dmf;
[0038]
4)式iv化合物经staudinger还原,在三苯基膦作用下,以四氢呋喃为溶剂得到手性氨基醇v化合物;
[0039]
5)式v化合物经在缩合剂cdi的作用下,以thf/etoh为溶剂得到式vi化合物;
[0040]
在另一实施例中,本发明的实施方案包括:
[0041]
步骤一:式i中x可为,氯,溴或者碘;优选x为溴。
[0042]
步骤二:所需的保护基可用为2-甲氧基丙烯,2,2-二甲氧基丙烷或者丙酮;优选用2-甲氧基丙烯进行保护。采用路易斯酸可为氯化锌,氯化铁,对甲苯磺酸;优选对甲苯磺酸,在催化量下可实现底物双羟基的保护。
[0043]
步骤三:所需的叠氮试剂为叠氮化钠,三甲基叠氮硅;优选叠氮化钠。
[0044]
步骤四:所述的转移氢化反应所需的noyori's催化剂,可采用rucl[(s,s)-tsdpen](p-cymene)(cat.1),rucl[(s,sr)-tsdpen](mesity-lene)(cat.2),rucl[(s,s)-tsdpen](benzene)(cat.3),rucl[(s,s)-fsdpen](p-cymene)(cat.4)。优选rucl[(s,s)-tsdpen](p-cymene)(cat.1)。上述四种催化剂的结构式如下所示。
[0045][0046]
步骤1):在氮气条件下,三口瓶内将化合物i(50g,0.204mol)和对甲苯磺酸(350mg,2.04mmoml)加到二氯甲烷/甲基叔丁基醚中(v/v 1:1,400ml),控制温度为-10至10℃,待反应体系温度稳定后,将2-甲氧基丙烯(36.73g,0.52mol)溶液逐滴添加到反应溶液中,控制反应温度在5-10℃,滴加完成后,维持体系温度继续搅拌1小时以完成反应,得到化合物ii 53g,收率92%。
[0047]
步骤2):将化合物ii(53g,0.186mol)溶解到dmf(230ml)中,分两批加入叠氮化钠(12.45g,0.197mol),维持体系温度在20℃,将体系继续搅拌2小时以上,直到溶液变为橘黄色,同时有白色固体析出时,反应结束。用乙酸乙酯萃取,有机相分别以饱和碳酸氢钠、盐水洗涤,合并有机相,用硫酸钠干燥,过滤,并在真空下浓缩得化合物iii为黄色固体41.4g,收率90%。
[0048]
步骤3):将化合物iii(10g,0.04mol)、rucl[(r,r)-tsdpen](p-cymene)(1.27g,2.02mmol)在氮气条件下溶解于dmf中(50ml),将9ml三乙胺以及4ml甲酸在0℃下搅拌5分钟后加入到反应体系,反应体系在室温下搅拌过夜,反应完成后,加入乙酸乙酯进行萃取,有机相用盐水洗涤,合并有机相,用硫酸钠干燥,过滤,真空浓缩得产物8.9g,收率89%,87%ee.。
[0049]
步骤4):将化合物iv(8.9g,0.036mol)溶解到thf(27ml)中,继续加入三苯基膦(11.23g,0.043mmol)和水(6.6ml,0.36mol)。反应体系在室温下混合2小时后反应完成,旋蒸除去溶剂后,将粗产品溶解到乙酸乙酯中,以hcl(1n)萃取三次后,收集水相,用固体koh将水相ph调节至12,加入二氯甲烷萃取水相,用有机相盐水洗涤,硫酸镁干燥,过滤并浓缩,得产物v 6.85g,收率86%。
[0050]
步骤5):将化合物v(6.85g,0.031mol)溶解到thf/etoh(v/v 1:1,34ml)中,加入n,n'-羰基二咪唑(5.47g,0.034mmol),将体系在室温下搅拌5小时后反应完成,旋蒸除去溶剂后,将粗产品用二氯甲烷/水进行萃取,有机相用盐水洗涤,硫酸钠干燥,过滤并浓缩,经柱层析后得产物v 6.88g,收率90%。1h nmr(600mhz,dmso-d6)δ7.65(s,1h),7.20(dd,j=8.4,2.2hz,1h),7.14(d,j=2.2hz,1h),6.83(d,j=8.4hz,1h),5.50(t,j=8.0hz,1h),
4.83(s,2h),3.82(t,j=8.8hz,1h),3.39
–
3.31(m,1h),1.47(s,6h).所得产物的核磁图谱见附图1。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1