一种N-苯磺酰甲脒化合物的制备方法及应用
一种n-苯磺酰甲脒化合物的制备方法及应用
技术领域
1.本发明涉及有机合成技术领域,具体地讲是涉及一类n-苯磺酰甲脒化合物的制备方法及应用。
背景技术:
2.n-苯磺酰甲脒是一类重要的化合物,其骨架广泛地存在于很多具有广谱药理活性和生物活性的化合物分子中。许多具有n-苯磺酰甲脒结构的生物活性化合物已被开发成具有杀菌、消炎作用的药物。例如,乙溴替丁(ebrotidine)是第一个具有胃粘膜保护作用、抗幽门螺杆菌(hp)作用及抗分泌作用的h2受体拮抗剂,1997年已在西班牙上市。
[0003][0004]
现有技术中,人们开发利用苯磺酰叠氮或苯磺酰胺的转化来实现n-苯磺酰甲脒类化合物的合成(j.am.chem.soc.,2005,127,2038;org.lett.,2011,13,6152;j.org.chem.,2020,85,2092),但是这些方法或多或少存在一定缺陷,例如:1)使用危险易爆的磺酰叠氮化合物;2)在金属催化剂的作用下,容易导致金属残留;3)反应温度较高,操作不便。
[0005]
因此,探索一种反应条件温和、收率高、操作安全简便、后处理相对容易、适宜于规模化合成n-苯磺酰甲脒化合物的制备方法,具有重要的研究意义和应用价值。此外,将开发的合成策略进一步应用到药物分子或药物关键中间体的结构后期修饰中,构建一类结构新颖的n-苯磺酰甲脒化合物,从中有望得到具有潜在杀菌活性的新化合物,具有非常广阔的研究和开发前景。
技术实现要素:
[0006]
本发明的目的在于提供一种条件温和、收率高、操作安全简便、后处理相对容易、适宜于规模化合成n-苯磺酰甲脒化合物的制备方法。
[0007]
本发明的另一个目的在于提供上述制备方法可应用于药物分子和药物中间体的结构后期修饰中,所得的化合物在杀菌方面具有潜在的应用。
[0008]
本发明的第一方面提供了制备本发明的一种n-苯磺酰甲脒化合物的方法,该方法可按照下述反应路线进行制备:
[0009][0010]
将仲胺、氰基磺酰肟醚、有机碱和溶剂加入反应器中,于一定温度下搅拌反应一段时间,反应完毕后,经洗涤、萃取、干燥、分离、旋蒸、纯化,得到目标产物n-苯磺酰甲脒化合
物。
[0011]
其中,所述的有机碱为三乙胺、1,4-二氮杂二环[2,2,2]辛烷、n,n-二甲基吡啶或1,8-二氮杂二环十一碳-7-烯。
[0012]
所述的溶剂为二甲基亚砜、n,n-二甲基甲酰胺、甲苯、二氯甲烷、四氢呋喃或乙腈。
[0013]
所述的温度为25-80℃。
[0014]
所述的仲胺、氰基磺酰肟醚、有机碱的投料物质的量之比为:仲胺:氰基磺酰肟醚:有机碱=3.0~5.0:1:1.0~2.0。
[0015]
本发明的第二个方面涉及本发明的一种n-苯磺酰甲脒化合物的制备方法可应用于药物分子和药物中间体的结构后期修饰中,所得的化合物应用于杀菌。
[0016]
本发明所述的药物分子有阿莫沙平和诺氟沙星。所述的药物中间体有3-(1-哌嗪基)-1,2-苯并异噻唑(齐拉西酮、哌罗匹隆、鲁拉西酮等药物的关键中间体)、1-(4-氯二苯甲基)哌嗪(盐酸氯环利嗪、羟嗪、西替利嗪等药物的关键中间体)和6-氟-3-(哌啶-4-基)苯并[d]异恶唑(利培酮、帕潘立酮、伊潘立酮等药物的关键中间体)。当然,本发明所述的药物分子和药物中间体不限于上述举例的范围。
[0017]
本发明的有益效果是:本发明所述的一种n-苯磺酰甲脒化合物的制备方法具有操作简便、条件温和、收率高、后处理相对容易、适宜于规模化制备、有效地避免了金属催化剂和危险试剂的使用等特点;本发明所述的一种n-苯磺酰甲脒化合物的制备方法可应用于药物分子和药物中间体的结构后期修饰中,所得的化合物在杀菌方面具有潜在的应用。
附图说明
[0018]
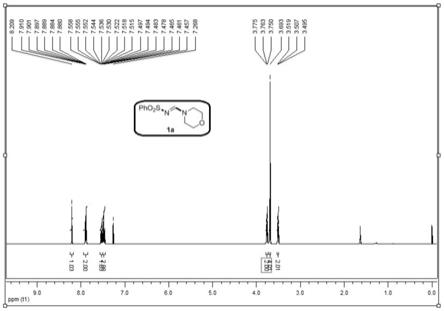
图1为本发明实施例中所制备的化合物1a的核磁共振氢谱图;
[0019]
图2为本发明实施例中所制备的化合物1a的核磁共振碳谱图;
[0020]
图3为本发明实施例中所制备的化合物1b的核磁共振氢谱图;
[0021]
图4为本发明实施例中所制备的化合物1b的核磁共振碳谱图;
[0022]
图5为本发明实施例中所制备的化合物1c的核磁共振氢谱图;
[0023]
图6为本发明实施例中所制备的化合物1c的核磁共振碳谱图;
[0024]
图7为本发明实施例中所制备的化合物1d的核磁共振氢谱图;
[0025]
图8为本发明实施例中所制备的化合物1d的核磁共振碳谱图;
[0026]
图9为本发明实施例中所制备的化合物1e的核磁共振氢谱图;
[0027]
图10为本发明实施例中所制备的化合物1e的核磁共振碳谱图;
[0028]
图11为本发明实施例中所制备的化合物1f的核磁共振氢谱图;
[0029]
图12为本发明实施例中所制备的化合物1f的核磁共振碳谱图;
[0030]
图13为本发明实施例中所制备的化合物1g的核磁共振氢谱图;
[0031]
图14为本发明实施例中所制备的化合物1g的核磁共振碳谱图;
[0032]
图15为本发明实施例中所制备的化合物1h的核磁共振氢谱图;
[0033]
图16为本发明实施例中所制备的化合物1h的核磁共振碳谱图;
[0034]
图17为本发明实施例中所制备的化合物1i的核磁共振氢谱图;
[0035]
图18为本发明实施例中所制备的化合物1i的核磁共振碳谱图;
[0036]
图19为本发明实施例中所制备的化合物2a的核磁共振氢谱图;
[0037]
图20为本发明实施例中所制备的化合物2a的核磁共振碳谱图;
[0038]
图21为本发明实施例中所制备的化合物2b的核磁共振氢谱图;
[0039]
图22为本发明实施例中所制备的化合物2b的核磁共振碳谱图;
[0040]
图23为本发明实施例中所制备的化合物2c的核磁共振氢谱图;
[0041]
图24为本发明实施例中所制备的化合物2c的核磁共振碳谱图;
[0042]
图25为本发明实施例中所制备的化合物2d的核磁共振氢谱图;
[0043]
图26为本发明实施例中所制备的化合物2d的核磁共振碳谱图;
[0044]
图27为本发明实施例中所制备的化合物2e的核磁共振氢谱图;
[0045]
图28为本发明实施例中所制备的化合物2e的核磁共振碳谱图。
具体实施方式
[0046]
下面结合实施例来进一步描述本发明,通过下述实施例有助于进一步理解本发明。这些实施例仅供叙述而并非用来限制本发明的范围或实施原则。
[0047]
制备实施例:
[0048][0049]
实施例1:n-(吗啉亚甲基)苯磺酰胺的制备
[0050][0051]
向一25ml schlenk瓶中加入氰基磺酰亚胺(150mg,0.5mmol)、吗啉(175mg,2.0mmol)、1,4-二氮杂二环[2,2,2]辛烷(85mg,0.75mmol)和2.5ml二甲基亚砜,加毕,置于60℃油浴锅搅拌反应24h。tlc检测反应完毕,加水(10ml)稀释混合物,并用乙酸乙酯(3
×
10ml)萃取,合并有机层,用饱和盐水(15ml)洗涤,无水mgso4干燥,然后在真空中浓缩,剩余物通过硅胶柱色谱法(石油醚/乙酸乙酯作为洗脱液)进行纯化,即可得到目标产物1a 100mg,白色固体,收率78%。1h nmr(400mhz,cdcl3)δ3.50(t,j=4.8hz,2h),3.69(s,4h),3.76(t,j=4.8hz,2h),7.45
–
7.49(m,2h),7.51
–
7.55(m,1h),7.88
–
7.91(m,2h),8.20(s,1h);
13
c nmr(100mhz,cdcl3)δ157.8,142.1,132.1,128.9,126.6,66.9,66.0,50.4,44.3;ir(kbr)ν2920,2850,1615,1445,1344,1147,1088,858cm-1
;hrms(esi):calcd for c
11h15
n2o3s[m+h]
+
255.0798,found 255.0523。
[0052]
实施例2:n-(硫代吗啉亚甲基)苯磺酰胺的制备
[0053][0054]
与实施例1类似的方法,硫代吗啉:氰基磺酰肟醚:1,4-二氮杂二环[2,2,2]辛烷的投料物质的量之比为:4.0:1:1.5,得到目标产物1b 99mg,白色固体,收率73%。1h nmr(400mhz,cdcl3)δ2.63(t,j=5.6hz,2h),2.70(t,j=5.2hz,2h),3.72(t,j=5.2hz,2h),3.91(t,j=5.2hz,2h),7.44
–
7.48(m,2h),7.49
–
7.53(m,1h),7.85
–
7.88(m,2h),8.18(s,1h);
13
c nmr(100mhz,cdcl3)δ158.2,142.1,132.1,128.9,126.6,53.4,46.4,28.3,26.9;ir
(kbr)ν2920,2851,1607,1446,1352,1283,1146,1089,891cm-1
;hrms(esi):calcd for c
11h15
n2o2s2[m+h]
+
271.0569,found 271.0264。
[0055]
实施例3:n-((4-甲基哌啶-1-基)亚甲基)苯磺酰胺的制备
[0056][0057]
与实施例1类似的方法,4-甲基哌啶:氰基磺酰肟醚:1,4-二氮杂二环[2,2,2]辛烷的投料物质的量之比为:4.0:1:1.5,得到目标产物1c 95mg,白色固体,收率71%。1h nmr(400mhz,cdcl3)δ0.94(d,j=6.4hz,3h),1.07
–
1.23(m,2h),1.60
–
1.70(m,2h),1.73
–
1.79(m,1h),2.78(dt,j=13.2hz,3.2hz,1h),3.25(dt,j=12.8hz,3.2hz,1h),3.54
–
3.58(m,1h),4.36
–
4.41(m,1h),7.41
–
7.50(m,3h),7.85
–
7.92(m,2h),8.11(s,1h);
13
c nmr(100mhz,cdcl3)δ157.4,142.6,131.8,128.7,126.4,51.3,44.0,34.4,32.9,30.7,21.5;ir(kbr)ν2926,2870,1614,1446,1336,1147,1088,921,872cm-1
;hrms(esi):calcd for c
13h19
n2o2s[m+h]
+
267.1162,found 267.1106。
[0058]
实施例4:n-((4-苯基哌啶-1-基)亚甲基)苯磺酰胺的制备
[0059][0060]
与实施例1类似的方法,4-苯基哌啶:氰基磺酰肟醚:1,4-二氮杂二环[2,2,2]辛烷的投料物质的量之比为:4.0:1:1.5,得到目标产物1d 118mg,白色固体,收率72%。1h nmr(400mhz,cdcl3)δ1.64
–
1.80(m,2h),1.91
–
1.94(m,1h),1.99
–
2.02(m,1h),2.75
–
2.83(m,1h),2.90(dt,j=13.2hz,3.2hz,1h),3.43(dt,j=13.2hz,2.8hz,1h),3.70
–
3.75(m,1h),4.59
–
4.64(m,1h),7.16
–
7.18(m,2h),7.21
–
7.25(m,1h),7.30
–
7.34(m,2h),7.45
–
7.54(m,3h),7.90
–
7.93(m,2h),8.20(s,1h);
13
c nmr(100mhz,cdcl3)δ157.6,144.1,142.5,131.9,128.8,126.9,126.7,126.6,51.6,44.4,42.3,33.7,32.2;ir(kbr)ν2922,2850,1614,1446,1334,1283,1146,1088,877,753cm-1
;hrms(esi):calcd for c
18h21
n2o2s[m+h]
+
329.1318,found 329.0892。
[0061]
实施例5:n-((4-苯基哌嗪-1-基)亚甲基)苯磺酰胺的制备
[0062][0063]
与实施例1类似的方法,n-苯基哌嗪:氰基磺酰肟醚:1,4-二氮杂二环[2,2,2]辛烷的投料物质的量之比为:4.0:1:1.5,得到目标产物1e 134mg,白色固体,收率81%。1h nmr(400mhz,cdcl3)δ3.18(t,j=5.2hz,2h),3.25(t,j=5.2hz,2h),3.63(t,j=5.2hz,2h),3.82(t,j=5.2hz,2h),6.90
–
6.95(m,3h),7.26
–
7.31(m,2h),7.45
–
7.56(m,3h),7.89
–
7.94(m,2h),8.24(s,1h);
13
c nmr(100mhz,cdcl3)δ157.7,150.5,142.2,132.1,129.4,128.9,126.6,121.4,117.3,50.4,50.3,49.1,43.8;ir(kbr)ν2921,2850,1614,1496,1446,1343,1287,1230,1147,1089,1016,883cm-1
;hrms(esi):calcd for c
17h20
n3o2s[m+h]
+
330.1271,found 330.1410。
[0064]
实施例6:n-((4-(4-甲氧基苯基)哌嗪-1-基)亚甲基)苯磺酰胺的制备
[0065][0066]
与实施例1类似的方法,n-对甲氧基苯基哌嗪:氰基磺酰肟醚:1,4-二氮杂二环[2,2,2]辛烷的投料物质的量之比为:4.0:1:1.5,得到目标产物1f 162mg,白色固体,收率90%。1h nmr(400mhz,cdcl3)δ3.05(t,j=4.8hz,2h),3.12(t,j=4.8hz,2h),3.62(t,j=5.2hz,2h),3.76(s,3h),3.81(t,j=5.2hz,2h),6.86(dd,j=18.4hz,9.2hz,4h),7.45
–
7.54(m,3h),7.89
–
7.92(m,2h),8.22(s,1h);
13
c nmr(100mhz,cdcl3)δ157.6,155.0,144.8,142.3,132.0,128.8,126.6,119.6,114.7,55.6,51.7,50.6,50.5,44.0;ir(kbr)ν2920,2834,1610,1512,1446,1346,1298,1247,1148,1089,1019,885cm-1
;hrms(esi):calcd for c
18h22
n3o3s[m+h]
+
360.1376,found 360.1419。
[0067]
实施例7:n-(氮杂环丙烷-1-基亚甲基)苯磺酰胺的制备
[0068][0069]
与实施例1类似的方法,环己亚胺:氰基磺酰肟醚:1,4-二氮杂二环[2,2,2]辛烷的投料物质的量之比为:4.0:1:1.5,得到目标产物1g 104mg,白色固体,收率78%。1h nmr(400mhz,cdcl3)δ1.51
–
1.57(m,4h),1.69
–
1.76(m,4h),3.47(t,j=6.0hz,2h),3.52(t,j=6.0hz,2h),7.40
–
7.49(m,3h),7.84
–
7.91(m,2h),8.17(s,1h);
13
c nmr(100mhz,cdcl3)δ159.0,142.7,131.7,128.7,126.3,52.9,47.4,29.7,27.8,26.8,25.9;ir(kbr)ν2923,2852,1604,1445,1340,1297,1145,1088,905,839,750cm-1
;hrms(esi):calcd for c
13h19
n2o2s[m+h]
+
267.1162,found 267.0795。
[0070]
实施例8:n,n-二异丙基-n
′‑
(苯磺酰基)甲酰胺的制备
[0071][0072]
与实施例1类似的方法,二异丙基胺:氰基磺酰肟醚:1,4-二氮杂二环[2,2,2]辛烷的投料物质的量之比为:4.0:1:1.5,得到目标产物1h 110mg,白色固体,收率82%。1h nmr(400mhz,cdcl3)δ1.21(d,j=6.8hz,6h),1.30(d,j=6.8hz,6h),3.63
–
3.73(m,1h),4.46
–
4.57(m,1h),7.42
–
7.50(m,3h),7.84
–
7.87(m,2h),8.24(s,1h);
13
c nmr(100mhz,cdcl3)δ156.5,142.8,131.7,128.7,126.3,48.6,48.0,23.6,19.6;ir(kbr)ν2978,2922,2850,1602,1453,1338,1282,1144,1087,891,839cm-1
;hrms(esi):calcd for c
13h21
n2o2s[m+h]
+
269.1318,found 269.1325。
[0073]
实施例9:n-甲基-n-苯基-n
′‑
(苯磺酰)甲酰胺的制备
[0074][0075]
与实施例1类似的方法,n-甲基苯胺:氰基磺酰肟醚:1,4-二氮杂二环[2,2,2]辛烷的投料物质的量之比为:4.0:1:1.5,得到目标产物1i 94mg,油状液体,收率68%。1h nmr
(400mhz,cdcl3)δ3.44(s,3h),7.19(d,j=7.6hz,2h),7.32(t,j=7.6hz,1h),7.39
–
7.44(m,2h),7.47
–
7.56(m,3h),7.92
–
7.95(m,2h),8.57(s,1h);
13
c nmr(100mhz,cdcl3)δ158.6,143.2,141.8,132.2,129.9,128.9,127.5,126.7,122.1,36.2;ir(kbr)ν2927,2852,1604,1575,1447,1338,1299,1148,1086,892,778cm-1
;hrms(esi):calcd for c
13h21
n2o2s[m+h]
+
275.0849,found 275.0484。
[0076]
应用实施例:
[0077]
实施例10:本发明的制备方法对抗抑郁药阿莫沙平的结构修饰应用
[0078][0079]
与实施例1类似的方法,阿莫沙平:氰基磺酰肟醚:1,4-二氮杂二环[2,2,2]辛烷的投料物质的量之比为:4.0:1:1.5,得到目标产物2a 183mg,白色固体,收率76%。1h nmr(400mhz,cdcl3)δ3.52
–
3.77(m,8h),7.00
–
7.05(m,1h),7.08
–
7.14(m,3h),7.19(d,j=8.4hz,1h),7.29(d,j=2.8hz,1h),7.41(dd,j=8.4hz,2.4hz,1h),7.45
–
7.54(m,3h),7.88
–
7.91(m,2h),8.27(s,1h);
13
c nmr(100mhz,cdcl3)δ159.4,158.3,157.9,151.7,142.1,139.5,133.2,132.1,130.7,128.8,128.7,127.2,126.6,126.0,125.4,124.5,123.0,120.3,49.9,47.6,46.7,43.5cm-1
;ir(kbr)ν2921,2851,1607,1559,1470,1346,1300,1284,1237,1147,1090,1009,883cm-1
;hrms(esi):calcd for c
24h22
cln4o3s[m+h]
+
481.1096,found 481.1226。
[0080]
实施例11:本发明的制备方法对抗菌药诺氟沙星的结构修饰应用
[0081][0082]
与实施例1类似的方法,诺氟沙星:氰基磺酰肟醚:1,4-二氮杂二环[2,2,2]辛烷的投料物质的量之比为:4.0:1:1.5,得到目标产物2b 127mg,白色固体,收率52%。1h nmr(400mhz,(cd3)2so)δ1.38(t,j=7.2hz,3h),2.59
–
2.62(m,2h),2.73
–
2.76(m,2h),3.32
–
3.36(m,4h),4.53(q,j=14.0hz,6.8hz,2h),5.50(s,1h),7.15(d,j=7.2hz,1h),7.42
–
7.51(m,5h),7.86(d,j=13.2hz,1h),8.89(s,1h),15.31(s,1h);
13
c nmr(100mhz,(cd3)2so)δ176.2,166.2,154.2,151.7,148.5,145.4,145.3,137.2,133.0,129.1,129.0,127.9,119.6,119.5,115.9,111.3,111.1,107.1,106.3,106.2,60.7,49.4,49.3,49.2,48.9,14.4cm-1
;ir(kbr)ν3402,2922,2851,1687,1628,1476,1275,1260,1025,999,750cm-1
;hrms(esi):calcd for c
23h24
fn4o5s[m+h]
+
487.1446,found 487.1013。
[0083]
实施例12:本发明的制备方法对抗精神病药关键中间体3-(1-哌嗪基)-1,2-苯并异噻唑的结构修饰应用
[0084][0085]
与实施例1类似的方法,3-(1-哌嗪基)-1,2-苯并异噻唑:氰基磺酰肟醚:1,4-二氮杂二环[2,2,2]辛烷的投料物质的量之比为:4.0:1:1.5,得到目标产物2c 153mg,白色固体,收率79%。1h nmr(400mhz,cdcl3)δ3.53
–
3.56(m,2h),3.61
–
3.64(m,2h),3.71
–
3.73(m,2h),3.88
–
3.90(m,2h),7.39(t,j=8.0hz,1h),7.46
–
7.55(m,4h),7.82
–
7.86(m,2h),7.89
–
7.91(m,2h),8.28(s,1h);
13
c nmr(100mhz,cdcl3)δ162.7,157.9,153.0,142.1,132.1,128.9,128.0,127.6,126.6,124.4,123.4,120.8,50.2,50.1,49.3,43.6cm-1
;ir(kbr)ν2921,2850,1610,1561,1493,1446,1345,1285,1117,1089,1012,882cm-1
;hrms(esi):calcd for c
18h19
n4o2s2[m+h]
+
387.0944,found 387.1051。
[0086]
实施例13:本发明的制备方法对抗过敏药关键中间体1-(4-氯二苯甲基)哌嗪的结构修饰应用
[0087][0088]
与实施例1类似的方法,1-(4-氯二苯甲基)哌嗪:氰基磺酰肟醚:1,4-二氮杂二环[2,2,2]辛烷的投料物质的量之比为:4.0:1:1.5,得到目标产物2d 163mg,白色固体,收率72%。1h nmr(400mhz,cdcl3)δ2.39(t,j=5.2hz,2h),2.44
–
2.47(m,2h),3.47(t,j=5.2hz,2h),3.60
–
3.70(m,2h),4.26(s,1h),7.19
–
7.35(m,9h),7.43
–
7.53(m,3h),7.85
–
7.88(m,2h),8.13(s,1h);
13
c nmr(100mhz,cdcl3)δ157.4,142.3,141.1,140.3,133.2,132.0,129.2,129.1,129.0,128.8,127.8,127.7,126.6,75.0,51.8,50.8,50.7,44.0cm-1
;ir(kbr)ν2921,2818,1614,1487,1446,1347,1284,1148,1089,999,891cm-1
;hrms(esi):calcd for c
24h25
cln3o2s[m+h]
+
454.1351,found 454.0895。
[0089]
实施例14:本发明的制备方法对抗精神病药物关键中间体6-氟-3-(哌啶-4-基)苯并[d]异恶唑的结构修饰应用
[0090][0091]
与实施例1类似的方法,6-氟-3-(哌啶-4-基)苯并[d]异恶唑:氰基磺酰肟醚:1,4-二氮杂二环[2,2,2]辛烷的投料物质的量之比为:4.0:1:1.5,得到目标产物2e 137mg,白色固体,收率71%。1h nmr(400mhz,cdcl3)δ1.92
–
2.02(m,1h),2.05
–
2.26(m,3h),3.19
–
3.26(m,1h),3.37
–
3.45(m,1h),3.48
–
3.55(m,1h),3.83(dt,j=13.6hz,3.6hz,1h),4.44(dt,j=13.6hz,3.6hz,1h),7.09(dt,j=8.8hz,2.4hz,1h),7.27(dd,j=8.4hz,2.0hz,1h),
7.45
–
7.54(m,3h),7.59
–
7.62(m,1h),7.88
–
7.91(m,2h),8.23(s,1h);
13
c nmr(100mhz,cdcl3)δ164.4(d,j
c-f
=250.1hz),164.0(d,j
c-f
=13.6hz),159.5,157.7,142.2,132.1,128.9,126.6,122.1(d,j
c-f
=11.0hz),116.9,113.0(d,j
c-f
=25.4hz),97.8(d,j
c-f
=26.5hz),50.3,43.2,33.5,30.4,29.3cm-1
;ir(kbr)ν2929,2853,1611,1495,1447,1354,1273,1147,1088,997,882cm-1
;hrms(esi):calcd for c
19h19
fn3o3s[m+h]
+
388.1126,found 388.0735。
[0092]
实施例15:本发明的制备方法所修饰的药物分子和药物中间体得到的化合物2a~2e对植物病原真菌的杀菌活性测试
[0093]
采用菌丝生长抑制法对合成的化合物进行了小麦纹枯病菌(rhizoctonia cerealis)和黄瓜炭疽病菌(colletotrichum orbiculare)的杀菌活性测试,使用浓度为100mg/l。
[0094]
准确称取一定量的目标化合物,用5ml甲醇溶解得到浓度为10mg/ml的母液。接着,准确吸取母液1ml加入到150ml psa培养基中,制成药剂浓度为100μg/ml的培养基,用此培养基进行初筛。用取样器打孔菌体,将菌饼接入上述培养基平板上,于25℃下培养3-5天,待无药对照培养基平板上的菌种长至平板的三分之二时测量其直径。每个菌种重复测试三次,计算药剂处理的生长抑制率。
[0095]
a(%)=[(b-c)/(b)]
×
100
[0096]
a为抑制率,b为空白直径,c为处理直径
[0097]
测试结果如表1所示:
[0098]
表1化合物2a~2e的初步杀菌活性数据
[0099][0100]
杀菌活性测试结果表明,所述的n-苯磺酰甲脒化合物2a~2e对小麦纹枯病菌和黄瓜炭疽病菌两种供试真菌具有一定的杀菌活性,尤其是化合物2b的杀菌活性相对较高,说明该类化合物在杀菌方面具有潜在的应用。
[0101]
上述虽然对本发明的具体实施方式进行了描述,但并非对发明保护范围的限制,所属领域技术人员应该明白,在本发明的技术方案的基础上,本领域技术人员不需要付出创造性劳动即可做出的各种修饰或变形仍在本发明的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1