一种通过基因编辑改良水稻稃尖和叶鞘颜色的方法及其专用sgRNA和载体与流程
3’;a1u6cr:5
’ꢀ
aaacggtcctcggactcgaagtc 3’。
8.进一步的,本发明将所述接头引物与pylgrna-u6c质粒连接,得到sgrna 连接产物;以sgrna连接产物为模板,经两轮pcr扩增得到sgrna表达盒。
9.作为优选的,本发明第一轮pcr扩增引物为u-f:5
’ꢀ
ctccgttttacctgtggaatcg 3’;grna-r:5
’ꢀ
cggaggaaaattccatccac 3’;第二轮pcr扩增引物为b1’:5
’ꢀ
ttcagaggtctctctcgcactggaatcggcagcaaagg 3’;bl:5
’ꢀ
agcgtgggtctcgaccgggtccatccactccaagctc 3’。
10.优选的,本发明所述crispr/cas9编辑系统为pylcrispr/cas9-mh。
11.进一步优选的,本发明所述sgrna表达盒与pylcrispr/cas9-mh连接获得打靶载体pylcrispr/cas9-a1。
12.进一步的,本发明将所述载体pylcrispr/cas9-a1转化水稻愈伤,用潮霉素引物对t0代转基因植株进行pcr扩增,筛选转化成功的阳性株系,用靶标基因检测引物筛选纯合转化植株。
13.进一步的,所述靶标基因检测引物为 a1df:5’cctgtgagctctctcatcgt 3’,a1dr:5
’ꢀ
cgttgttcctaacccacgac 3’。
14.作为优选的,本发明所述方法还包括在所述纯合转化植株中筛选无转基因成分的纯合突变株系:采用潮霉素引物和核酸酶cas9引物对转化植株的基因组进行扩增,均没有检测到扩增条带的为无转基因成分的纯合突变株系。
15.进一步的,所述潮霉素引物为hygf:5’tattgcatctcccgccgtgc 3’; hygr:5’agcgcgtctgctgctccata 3’;所述核酸酶cas9引物为cas9f:5
’ꢀ
acgacgacagcctgaccttt 3’,cas9r:5’tatcccgcccattctgcagg 3’。
16.本发明另一方面还提供一种改良水稻稃尖和叶鞘颜色的sgrna,其靶标序列为5’gacttcgagtccgaggacc 3’。
17.本发明再一方面还提供一种改良水稻稃尖和叶鞘颜色的载体,所述的载体为插入编码上述sgrna编码序列的pylcrispr/cas9-mh载体。
18.本发明与现有技术相比,具有以下优点和效果:
19.本发明提供了一种通过pylcrispr/cas9-mh基因编辑系统定向改良水稻稃尖和叶鞘颜色的方法。通过对水稻花青素合成途径关键基因a1进行编辑,抑制色素原基因c的表达,从而快速高效创制稃尖和叶鞘着色改变且主要农艺性状没有改变的新种质资源,在水稻育种中有重要的应用价值。
20.利用基因组编辑技术可以精准编辑花青素合成途径中的c、a或p基因,一般t0代就能筛选到目标基因被定点编辑的纯合转化植株,t1代可以获得剔除潮霉素和cas9外源基因的新材料,育种周期短,效率高。
附图说明
21.图1:a1基因的结构示意图。
22.图2:打靶载体pylcrispr/cas9-a1的结构示意图;其中,rb:载体右边界;u6c:水稻u6c启动子;sgrna:向导rna;nos:nos终止子;cas9:核酸酶蛋白;ubi:ubi启动子;35s:hygro:35s启动子-潮霉素基因;lb:载体左边界。
23.图3:pcr扩增检测含有pylcrispr/cas9-a1质粒的阳性克隆。
24.图4:pcr鉴定t1代转基因株系中无转基因成分的突变体;其中,m:dnamarker;1:阳性对照;2-7:转基因株系a1-1的t1代单株;8-13:转基因株系a1-14的t1代单株;14:野生型(中早39)。
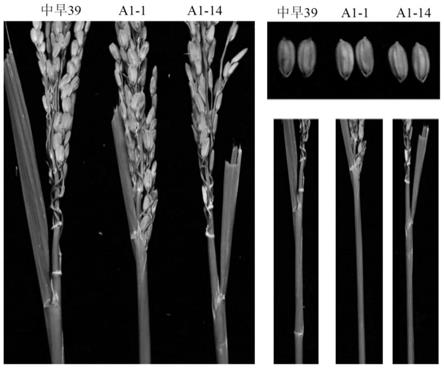
25.图5:t1代纯合转基因株系a1-1、a1-14和野生型(中早39)稃尖和叶鞘的颜色。
具体实施方式
26.本发明采用crispr/cas9系统对稃尖和叶鞘着色关键基因a1进行编辑,构建转基因敲除载体,利用农杆菌介导法进行遗传转化,获得t0代转基因株系;紧接着通过pcr检测t0代转基因株系中基因的编辑效率,编辑的类型;对t1 代纯合的且没有转基因成分的株系进行表型鉴定,筛选出稃尖和叶鞘颜色为常规稻草白且其他农艺性状(株高、全生育期、千粒重、有效穗数等)没有改变的新种质。
27.本发明编辑的靶标基因为花青素合成途径中的激活基因a1,在 http://rice.uga.edu/登录号loc_os01g44260,基因编码序列如seq id no.1所示,编辑的靶位点序列为5’gacttcgagtccgaggacc 3’,位于第一个外显子 (图1)。
28.本发明采用的编辑系统为pylcrispr/cas9-mh,可以采用商业购买,本发明所用编辑系统来自于华南农业大学刘耀光老师实验室。
29.作为一种实施方式,本发明载体的构建方法如下:根据选定的激活基因 a1的靶位点序列,设计靶位点接头引物,两轮pcr扩增sgrna表达盒,sgrna 表达盒与pylcrispr/cas9-mh连接获得打靶载体pylcrispr/cas9-a1,转化感受态dh10b,菌落pcr筛选阳性克隆。
30.优选的,在所述的载体构建方法中,所用的靶位点接头引物为:a1u6cf: 5’tcaggacttcgagtccgaggacc 3’;a1u6cr: 5’aaacggtcctcggactcgaagtc 3’。
31.所述sgrna表达盒第一轮pcr扩增引物为u-f: 5’ctccgttttacctgtggaatcg 3’;grna-r: 5’cggaggaaaattccatccac 3’;第二轮pcr扩增引物为b1’: 5’ttcagaggtctctctcgcactggaatcggcagcaaagg 3’;bl:5
’ꢀ
agcgtgggtctcgaccgggtccatccactccaagctc 3’。
32.所述菌落pcr引物为:sp1:5’cccgacatagatgcaataa 3’;a1u6cr: 5’aaacggtcctcggactcgaagtc 3’。
33.本发明将构建得到的载体进行遗传转化。作为一种实施方式,本发明利用农杆菌eh105a介导,将构建的目标载体pylcrispr/cas9-a1转化水稻愈伤,用潮霉素引物对t0代转基因植株进行pcr扩增,筛选转化成功的阳性株系。优选的,所述潮霉素引物为hygf:5’tattgcatctcccgccgtgc 3’; hygr:5’agcgcgtctgctgctccata 3’。
34.本发明将a1被成功编辑的转基因株系进行鉴定,筛选纯合的突变株系,同时考察t0代转基因株系的表型。作为一种实施方式,本发明对转化成功的 t0代转基因植株,采用靶标基因检测引物进行pcr扩增,扩增产物进行sanger 测序,测序结果与靶位点序列进行比对,确定突变的类型。优选所述靶标基因检测引物为a1df:5’cctgtgagctctctcatcgt 3’,a1dr:5
’ꢀ
cgttgttcctaacccacgac 3’。
35.本发明在纯合转化植株中筛选无转基因成分的纯合转基因株系,同时考察t1代转基因株系的表型。作为一种实施方式,本发明将上述筛选鉴定的纯合突变株系,自交繁殖后获得a1基因定点突变的t1代植株,采用上述潮霉素引物hygf/hygr和核酸酶cas9引物cas9f:5
’ꢀ
acgacgacagcctgaccttt 3’,cas9r:5’tatcccgcccattctgcagg 3’对其进行pcr扩
增,均没有检测到潮霉素基因hygro和核酸酶基因cas9 的株系,即为剔除转基因成分的纯合突变体。
36.本发明将筛选鉴定的无转基因成分纯合突变株系,利用靶标基因检测引物a1df/a1dr再次检测突变的类型,获得基因突变类型能够稳定遗传且稃尖和叶鞘颜色改变但其他农艺性状(株高、全生育期、千粒重、有效穗数等) 没有改变的新种质,得到稃尖和叶鞘颜色被改变的新种质。
37.下面结合具体实施例对本发明作进一步说明,但本发明并不限于以下实施例。实施例中所使用的实验方法如无特殊说明,均为常规方法,所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
38.以下实施例中选取的背景材料为中早39,该品种为超级早籼稻,具有高产稳产、稻瘟病抗性强,且持久、结实率高、种植方式轻简多样化、耐寒、广适性、加工品质优等特点。但是中早39的稃尖和叶鞘为紫色,使得在大田种植时稻穗暗沉,外观品相不受青睐。本实施例通过crispr/cas9基因编辑技术对中早39 的a1基因进行定点编辑,获得稃尖和叶鞘颜色为稻草白的且其他农艺性状没有改变的中早39,实现中早39的定向改良。
39.实施例1
40.打靶载体pylcrispr/cas9-a1的构建
41.1、中早39的a1基因的靶位点设计
42.采用北京全式金生物科技有限公司的植物基因组提取试剂盒,提取中早 39的基因组dna。以早39的基因组dna为模板,用引物a1zz39f:5’43.tagctatcatatattctgcg 3’,a1zz39r:
[0044]5’
taaagtcaaaggtatatcttg 3’,扩增a1基因,pcr产物在擎科生物科技(杭州)有限公司进行测序。测序结果进行blast比对分析,发现中早 39的a1编码区5’端序列与水稻日本晴相同。
[0045]
根据中早39的a1基因的序列,在该基因编码区5’端的第一个外显子上选取5’gacttcgagtccgaggacc 3’作为基因编辑的靶位点(图1)。
[0046]
2、crispr/cas9基因编辑载体的构建
[0047]
基因编辑载体构建的过程,按以下步骤进行:
[0048]
(1)靶位点接头制备
[0049]
将接头引物(a1u6cf:tcaggacttcgagtccgaggacc;a1u6cr: aaacggtcctcggactcgaagtc)用1x te(ph8.0)溶解成100μm母液,各取1μl加入到98μl 0.5x te混合稀释到1μm。约90℃30s,移至室温冷却完成退火,即获得靶位点接头。
[0050]
(2)sgrna表达盒制备
[0051]
首先配制bsai酶切-连接反应液(10μl):加入约10ng pylgrna-u6c质粒,1.0μl接头,5u bsai(neb公司),35u t4 dna ligase(takara),1.0μl 10x dna ligase buffer(takara);然后在pcr仪上进行反应,反应程序为37℃ 5min,20℃5min,5个循环。反应产物即为sgrna连接产物。
[0052]
接着以sgrna连接产物为模板,经两轮pcr扩增sgrna表达盒,第一轮 pcr的反应体系(15μl)如下:
[0053][0054][0055]
pcr反应中的引物分别为u-f和grna-r序列:
[0056]
u-f:5’ctccgttttacctgtggaatcg3’;
[0057]
grna-r序列5’cggaggaaaattccatccac3’;
[0058]
其中,kod fx高保真dna聚合酶为toyoto公司产品。
[0059]
反应程序:95℃1min;95℃10s,60℃15s,68℃20s,10个循环;95℃10s, 60℃15s,68℃30s,22个循环。
[0060]
第二轮pcr:取第一轮pcr产物1μl用h2o稀释20倍,取2μl为模板,按照如下体系(50μl)进行:
[0061][0062]
引物b1’和bl的序列分别为:
[0063]
b1’:5’ttcagaggtctctctcgcactggaatcggcagcaaagg3’;
[0064]
bl:5’agcgtgggtctcgaccgggtccatccactccaagctc3’。
[0065]
pcr反应程序:95℃10s,60℃15s,68℃20s,25个循环。获得的pcr产物即为sgrna表达盒。
[0066]
(3)sgrna表达盒连接pylcrispr/cas9-mh载体
[0067]
将上述获得的50μl sgrna表达盒用pcr产物纯化试剂盒纯化(全式金),将纯化后的sgrna按照下面的变温循环酶切链接反应体系连接到 pylcrispr/cas9-mh载体。
[0068]
反应体系及过程:取约20-70ng纯化好的sgrna表达盒产物,加入约60-80ng 未切割的pylcrispr/cas9-mh,10u bsai 37℃酶切10min,加入1.5μl 10x dna
ꢀꢀ
ligase buffer和35u ligase,ddh2o补齐至15μl。反应程序:37℃5min;10℃5min, 20℃5min,15个循环。即获得打靶载体pylcrispr/cas9-a1的连接产物。
[0069]
pylsgrna-u6c是中间载体,为sgrna表达盒提供启动子和引导序列骨架; pylcrispr/cas9p35s-h载体为植物双元表达载体。两个载体均由华南农业大学刘耀光教授团队开发提供。
[0070]
(4)转化大肠杆菌dh10b及验证
[0071]
将pylcrispr/cas9-a1的连接产物用42℃热激90s转化dh10b感受态, sob培养基复苏45min后,取400菌液涂布于含有50mg/l卡那霉素的lb平板上,培养约12-16h。挑取平板上长出的单菌落,37度200rpm在lb液体培养基中培养12h,取1μl菌液为模板,按照以下pcr反应体系进行阳性克隆鉴定。
[0072][0073]
所使用的检测引物序列分别为:
[0074]
sp1:5’cccgacatagatgcaataa3’[0075]
u6cr:5’aaacggtcctcggactcgaagtc3’[0076]
pcr反应程序:95℃5min;98℃15s,55℃30s,68℃30s,30个循环;68℃7min。
[0077]
扩增产物经1%琼脂糖凝胶电泳分离,能检测到大小约为924bp特异性条带的即为阳性克隆(图3)。最后将阳性克隆的菌液用质粒提取试剂盒(全式金) 提取质粒,质粒经测序验证正确后,即获得打靶载体pylcrispr/cas9-a1(图2)。
[0078]
实施例2
[0079]
稃尖和叶鞘颜色改变的新种质的获得
[0080]
1、打靶载体pylcrispr/cas9-a1的遗传转化
[0081]
首先将构建好的pylcrispr/cas9-a1质粒按照常规热激法转入农杆菌 eha105。然后经过水稻愈伤诱导,农杆菌侵染、抗性愈伤选择、生根诱导等常规农杆菌介导法的步骤将pylcrispr/cas9-a1转入水稻品种中早39。
[0082]
2、t0代阳性植株的获得及靶位点检测
[0083]
经过3-4个月的遗传转化,获得了16株t0代阳性转基因苗,经潮霉素引物 hygf/hygr扩增均能获得特异的478bp大小的条带。
[0084]
为了鉴定阳性转基因植株a1基因靶位点序列的编辑情况,我们提取了16株 t0代阳性苗的基因组dna(全式金植物基因组提取试剂盒),设计了检测a1df: 5’cctgtgagctctctcatcgt 3’,a1dr:5’cgttgttcctaacccacgac 3’对16株阳性苗进行pcr扩增,扩增的反应具体如下:
[0085][0086]
pcr反应程序:95℃2min;98℃15s,55℃30s,68℃30s,30-35个循环;68℃7min。扩增产物经1.5%琼脂糖凝胶电泳分离,将能检测到大小为480bp的 pcr产物测序,测序结果表明在16株阳性苗中,有3株(a1-6、a1-10和a1-12) 没有发生突变;13株发生了突变,总的突变率为81.25%;在13株发生突变的阳性苗中,4株为纯合突变(a1-1、a1-4、a1-5和a1-14),纯合突变率为25%; 6株为双等位突变(a1-3、a1-7、a1-8、a1-9、a1-11和a1-15),双等位突变率为37.5%;3株为杂合突变(a1-2、a1-13和a1-13),杂合突变率为18.75%;突变的类型有碱基的插入,缺失和替代,具体的变异情况见表1。
[0087]
表1t0代转基因株系的突变位点、突变类型和突变频率
[0088]
[0089][0090]
其中,加粗部分为a1的靶位点序列;单下划线部分为插入的碱基;双下划线部分为替代的碱基;-为缺失的碱基。
[0091]
3、无转基因成分的纯合转基因株系的获得
[0092]
为获得无转基因成分的基因编辑株系,将两个t0代获得的纯合突变株系 a1-1和a1-14自交繁殖下一代构成t1代突变群体,其中a1-1为插入碱基a造成移码突变,a1-14为缺失11个碱基造成移码和缺失突变(表1)。提取a1-1 和a1-14的t1代植株的基因组dna,用潮霉素和核酸酶cas9的特异引物扩增基因组序列。
[0093]
潮霉素引物hygf/hygr:
[0094]
hygf:5’tattgcatctcccgccgtgc 3’;
[0095]
hygr:5’agcgcgtctgctgctccata。
[0096]
核酸酶cas9引物cas9f/cas9r:
[0097]
cas9f:5’acgacgacagcctgaccttt 3’,
[0098]
cas9r:5’tatcccgcccattctgcagg 3’。
[0099]
两对引物不能同时扩增出478bp和372bp的dna片段的植株即不含转基因成分的突变株(图4为部分t1植株的鉴定)。
[0100]
进一步,利用靶位点检测引物a1df/a1dr分别扩增上述鉴定的不含转基因成分的a1-1和a1-14的单株,测序表明这些单株靶位点的突变情况与t0代是一致的,表明基因编辑的这种突变在剔除cas9载体骨架后是能够稳定遗传的。至此,我们就获得无转基因成分的a1基因定点突变的纯合转基因株系。
[0101]
4、纯合突变株系稃尖和叶鞘颜色及其他农艺性状的考察
[0102]
获得了纯合突变株系a1-1和a1-14 t1代无转基因成分的单株,我们对单株的稃尖和叶鞘颜色及其他农艺性状进行了考察,结果表明其所有谷粒的稃尖以及叶鞘的颜色变为稻草白,而野生型中早39为紫色(图5);且纯合突变株系的株高、全生育期、剑叶长宽、千粒重,有效穗数、每穗实粒数同野生型中早39相比均没有显著变化(表2)。至此,我们将获得的纯合突变株系t1代无转基因成分的单株,继续自交繁殖就获得了由a1基因的突变造成的稃尖叶鞘颜色改变的新种质资源,实现了对超级早籼稻中早39的定向改良。
[0103]
表2转基因株系a1-1、a1-14和中早39的主要农艺性状
[0104][0105]
上述实施例为本发明最佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
[0106]
[0107]
[0108]
[0109]
[0110]
[0111]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1