1-苄基-2,4-二芳基咪唑类化合物、合成方法及其在抗肿瘤上的应用
1.本发明属于生物技术领域,涉及一种1-苄基-2,4-二芳基咪唑类化合物的化学制备方法及其抗肿瘤应用。
背景技术:
2.乳腺癌是导致女性死亡的第二大肿瘤疾病。乳腺癌的治疗方法主要包括手术治疗、放疗、辅助化疗、内分泌治疗和靶向治疗。尽管目前已有多种针对乳腺癌的药物上市,但仍存在如不良反应严重,易耐药等问题,因此需要更多具有较高有效性和安全性的新药。
3.hsp90是研究治疗乳腺癌药物的重要药物靶标(acta pharmaceutica sinica b,2021,11(6),1446-1468;)。hsp90抑制剂能够下调包括akt,her2和erk等多种hsp90客户蛋白的表达,从而起到杀死肿瘤细胞的作用。17-aag(17-烯丙胺基-17-去甲氧基格尔德霉素)是第一个进入临床试验的hsp90抑制剂。然而后续实验却显示它的溶解性较差、口服利用度低。其它hsp90抑制剂也因为成药性,耐药性或毒性等问题陆续在临床试验中进展缓慢或失败(current drug targets,2020,21(3),302-317.),因此急需更新颖的hsp90抑制剂的开发。
4.咪唑是构成药物分子的重要化学片段(g.e.schiltz,use of carbonyl derivatives for heterocyclic synthesis,editor(s):paul knochel,comprehensive organic synthesis(second edition),elsevier,2014,pages 555-572)。2012年,cai等人发表了由三氟化硼乙醚和cui共同催化苯乙酮和苄胺合成1-苄基-2,4-二芳基咪唑类化合物的方法(organic letters,2012,14(23),6068-6071.)。2013年,huang等人发表了由碘单质为催化剂将苯乙酮和苄胺转化成1-苄基-2,4-二芳基咪唑类化合物的方法(advanced synthesis&catalysis,2013,355(1),170-180.)。2015年,xiang等发现了以碘单质为催化剂,苄胺和乙烯基叠氮化合物为原料合成1-苄基-2,4-二芳基咪唑类化合物的方法(chemical communications,2015,51(30),6598-6600)。2016年,cao等人以烯胺和苄胺为原料,溴化铜为催化剂合成1-苄基-2,4-二芳基咪唑类化合物(rsc advances,2016,6(62),57232-57235)。2017年,liu等发现亚硝酸钠能够促进苯乙酮和苄胺转化成1-苄基-2,4-二芳基咪唑类化合物(organic chemistry frontiers,2017,4(8),1508-1512.)。2020年,yang等人采用电化学的方法由苯乙酮和苄胺合成1-苄基-2,4-二芳基咪唑类化合物(journal of organic chemistry,2020,85(9),5952-5958.)。上述方法均能简单高效地由简单原料合成1-苄基-2,4-二芳基咪唑类化合物,但难以实现1-位苄基苯环和2-位苯环取代基不同的目标化合物的合成。2010年,hirao等人公开了一种由2,5-二芳基咪唑和氯苄反应合成1-苄基-2,4-二芳基咪唑类化合物,并将其应用于金属防蚀(jp2010156043a)。1994年,wang等人发现了由2-溴咪唑类化合物和三甲基苯基锡反应得到1-苄基-2,4-二芳基咪唑类化合物的方法,该方法需要钯催化(journal of heterocyclic chemistry,1994,31(6),1637-1639)。
技术实现要素:
5.针对现有技术存在的问题,本发明提供一种1-苄基-2,4-二芳基咪唑类化合物及其在药学上可接受的水合物或盐的1-苄基-2,4-二芳基咪唑类化合物的合成方法及其在抗肿瘤上的应用,1-苄基-2,4-二芳基咪唑类化合物可以用于治疗乳腺癌。
6.为了达到上述目的,本发明采用的技术方案为:
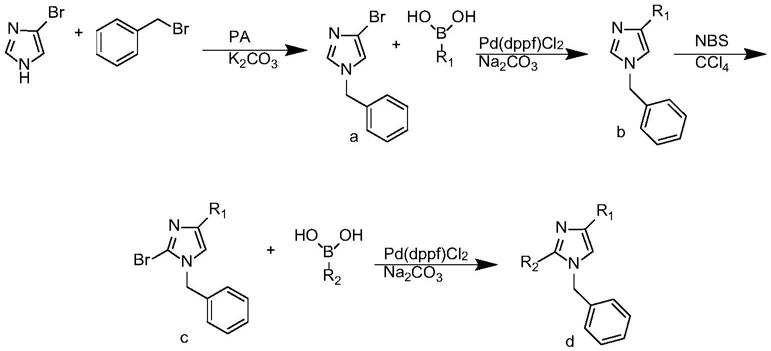
7.一种1-苄基-2,4-二芳基咪唑类化合物的合成方法,包括以下步骤:
8.所述化合物的合成路线如下:
[0009][0010]
所述化合物的制备方法如下:
[0011]
(1)制备上述化合物a
[0012]
将反应原料4-溴咪唑溶于丙酮中,再加入溴化苄和无水碳酸钾。室温反应条件下12-15h经萃取、过滤、重结晶得到a。所述的4-溴咪唑、溴化苄、无水碳酸钾的摩尔比为1:2~3:1~1.5;
[0013]
(2)制备上述化合物b
[0014]
将化合物a溶于溶剂1,4-二氧六环中,其中a的浓度为0.1-0.5mol/l,再加入催化剂pd(dppf)cl2和无水碳酸钠,于惰性气体下加热到100-110℃反应4~5h。反应结束后冷却至室温,加入水和乙酸乙酯萃取,取有机层浓缩柱层析(石油醚:乙酸乙酯的体积比=8~10:1)得到化合物b。所述化合物a、pd(dppf)cl2、无水碳酸钠的摩尔比为1:2~3:0.05~0.1:2~2.5;水与1,4-二氧六环的体积比为1:3~4。其中,r1选自苯基、4-氯苯基、4-甲基苯基、2-噻吩基、3-氯苯基或3-甲基苯基;
[0015]
(3)制备上述化合物c
[0016]
将化合物b溶于溶剂ccl4中,其中b的浓度为0.05~0.1mol/l,再加入n-溴代琥珀酰亚胺nbs于30~40℃下反应12~16h,反应结束后加入20ml水和30ml二氯甲烷,取有机层加入无水碳酸钠过滤,浓缩后柱层析(石油醚:乙酸乙酯的体积比=8~4:1)得到化合物c纯品。其中化合物b:nbs的摩尔比=1:1~1.5;
[0017]
(4)制备上述化合物d
[0018]
将化合物c溶于溶剂1,4-二氧六环中,其中c的浓度为0.1~0.5mol/l,再加入pd(dppf)cl2和无水碳酸钠,于惰性气体下加热到100~110℃反应4~5h,反应结束
后冷却至室温,加入水和乙酸乙酯萃取,取有机层浓缩柱层析(石油醚:乙酸乙酯=10~5:1)得到化合物d纯品。其中化合物c:pd(dppf)cl2:无水碳酸钠=1:2~4:0.05~0.1:2~2.5;水与1,4-二氧六环的体积比为1:3~4。
[0019]
采用上述方法制备得到一种1-苄基-2,4-二芳基咪唑类化合物,其在药学上可接受的水合物或盐,所述的1-苄基-2,4-二芳基咪唑类化合物的化学分子结构通式为:
[0020][0021]
其中,r1选自苯基、2-噻吩、3-氯苯基、4-氯苯基、3-甲基苯基、4-甲基苯基。r2选自苯基、3-甲基苯基、4-甲基苯基、4-氯苯基、3-氯苯基、4-乙酰基苯基。
[0022]
进一步的,所述的1-苄基-2,4-二芳基咪唑类化合物的结构式依次如下:
[0023][0024][0025]
其中,d1为1-苄基-2,4-二苯基-1h-咪唑;d2为1-苄基-4-苯基-2-(间甲基苯基)-1h-咪唑;d3为1-苄基-4-苯基-2-(对甲基苯基)-1h-咪唑;d4为1-苄基-2-(4-氯苯基)-4-苯基-1h-咪唑;d5为1-(4-(苄基-4-苯基-1h-咪唑-2-基)苯基)乙酰基;d6为1-苄基-4-(4-氯苯基)-2-苯基-1h-咪唑;d7为1-苄基-2,4-二(4-氯苯基)-1h-咪唑;d8为1-苄基-2-(4-氯苯基)-4-(4-甲基)-1h-咪唑;d9为1-苄基-2-苯基-4-(4-甲基)-1h-咪唑;d10为1-苄基-2-苯基-4-(噻吩-2-基)-1h-咪唑;d11为1-苄基-4-(3-氯苯基)-2-(4-氯苯基)-1h-咪唑;d12为1-苄基-2-(4-氯苯基)-4-(间甲基)-1h-咪唑。
[0026]
采用上述方法制备得到的一种1-苄基-2,4-二芳基咪唑类化合物及其在药学上可接受的水合物或盐在抗肿瘤上的应用,所述的1-苄基-2,4-二芳基咪唑类化合物及其在药学上可接受的水合物或盐具有治疗癌症的用途,所述的癌症包括乳腺癌。
[0027]
本发明的有益效果为:本发明公开了一系列结构新颖的小分子化合物,具有抗乳腺癌的作用,抗乳腺癌活性相当于或优于阳性对照17-aag。
附图说明
[0028]
图1为d1的氢谱;图2为d2的氢谱;图3为d3的氢谱;
[0029]
图4为d4的氢谱;图5为d5的氢谱;图6为d6的氢谱;
[0030]
图7为d7的氢谱;图8为d8的氢谱;图9为d9的氢谱;
[0031]
图10为d10的氢谱;图11为d11的氢谱;图12为d12的氢谱;
[0032]
图13为化合物d7对mcf-7细胞中hsp90客户蛋白及热休克蛋白hsp90和hsp70表达的影响。
[0033]
图14为mcf-7细胞给药(d1-d4)48h后对细胞的增殖抑制图,给药浓度0-100μm。
[0034]
图15为mcf-7细胞给药(d5-d8)48h后对细胞的增殖抑制图,给药浓度0-100μm。
[0035]
图16为mcf-7细胞给药(d9-d12)48h后对细胞的增殖抑制图,给药浓度0-100μm。
[0036]
图17为mda-mb-231细胞给药(d1-d4)48h后对细胞的增殖抑制图,给药浓度0-50μm。
[0037]
图18为mda-mb-231细胞给药(d5-d8)48h后对细胞的增殖抑制图,给药浓度0-50μm。
[0038]
图19为mda-mb-231细胞给药(d9-d12)48h后对细胞的增殖抑制图,给药浓度0-50μm。
具体实施方式
[0039]
下面结合实施例进一步说明本发明以及本发明所进行的方式。这些实施例仅是为了进一步阐述本发明而非本发明的保护仅限于此。
[0040]
现参考下列说明性实施例进一步描述本发明,其中除非另有说明:
[0041]
(1)以摄氏度给出温度,在室温或环境温度下进行操作。
[0042]
(2)有机溶液用无水硫酸钠干燥。
[0043]
(3)最终产物具有令人满意的质子核磁共振波谱和质谱。
[0044]
(4)所给出的收率仅用于说明,工艺开发可获得更高收率,如需更多,则重复制备。
[0045]
(5)柱色谱纯化应用自我填充的硅胶柱进行。
[0046]
(6)使用用四级杆轨道离子阱高分辨质谱仪(q exactive)核磁共振氢谱是用500mhz核磁共振仪(avance iii 500mhz)采集的,除特别标明外,均采用cdcl3或dmso-d6作溶剂,以tms为内标,耦合常数(j)的单位是赫兹(hertz)。
[0047]
在下列实施例中使用以下缩写:
[0048]
dmso为二甲基亚砜;二氯甲烷为dcm;etoac为乙酸乙酯;pe为石油醚;nbs为n-溴代琥珀酰亚胺;pd(dppf)cl2为[1,1'-双(二苯基膦基)二茂铁]二氯化钯;
[0049]
pa为丙酮。
[0050]
以下是一种1-苄基-2,4-二芳基咪唑类化合物结构式为d1~d12的制备方法:
[0051]
d1:1-苄基-2,4-二苯基-1h-咪唑
[0052]
中间体a的制备方法:1-苄基-4-溴咪唑
[0053]
将4-溴咪唑(8g,54.4mmol)溶于pa(70ml)的溶液加入到圆底烧瓶中,分别加入溴化苄(16.6ml,136mmol)和无水碳酸钾(7.52g,54.4mmol),在室温搅拌13h。反应结束后加入etoac(50ml),水(40ml)萃取。有机相用饱和食盐水(2*100ml)洗两次,然后干燥,过滤,减压旋转蒸发除去溶剂。粗产品用dcm(5ml)溶解,再缓慢加入适量石油醚剧烈摇晃至固体析出,真空泵抽滤得到中间体a(白色晶体,3.2g,35.26%)。
[0054]
中间体b1的制备方法:1-苄基-4苯基咪唑
[0055]
将中间体a(2.38g,10mmol)和1,4-二氧六环(18ml)溶液加入到圆底烧瓶中,分别加入苯硼酸(3.05g,25mmol)和pd(dppf)cl2(0.55g,0.75mmol),将无水碳酸钠(2.39g,22.5mmol)溶于水(6ml)中再加入到以上体系中,在110℃油浴中搅拌4h。反应结束后冷却至室温,加入etoac(50ml),水(40ml)萃取。有机相用饱和食盐水(2*50ml)洗两次,然后干燥,过滤,减压旋转蒸发除去溶剂。粗产品用硅胶柱层析(pe/etoac=8:1)提纯分离,得到中间体b1(白色固体,1.43g,60.85%)。
[0056]
中间体c1的制备方法:1-苄基-2-溴-4苯基咪唑
[0057]
将中间体b1(1.43g,6mmol)和ccl4(30ml)溶液加入到圆底烧瓶中,继续加入nbs(3.4g,1.2mmol)在35℃水浴中搅拌14h。反应结束后加入dcm(50ml),水(40ml)萃取,然后干燥,过滤,减压旋转蒸发除去溶剂。粗产品用硅胶柱层析(pe/etoac=5:1)提纯分离,得到中间体c1(白色固体,1.24g,65.84%)。
[0058]
将白色中间体c1(0.471g,1.5mmol)加入到1,4-二氧六环(10.5ml)溶液溶于圆底烧瓶中,分别加入苯硼酸(0.4575g,3.75mmol)和pd(dppf)cl2(0.11g,0.15mmol)。将无水碳酸钠(0.41g,3.8mmol)溶于水(3ml)中再加入到以上体系中,在110℃油浴中搅拌4h。反应结束后冷却至室温,加入etoac(30ml),水(20ml)萃取。有机相用饱和食盐水(2*50ml)洗两次,然后加入无水硫酸钠干燥,过滤,减压旋转蒸发除去溶剂。粗产品加入硅胶(约粗产物3倍量),然后加入etoac溶解,减压蒸发除去溶剂,将粗产品与硅胶的混合物再经过柱分离(pe/etoac=5:1)后,浓缩得到白色固体d1(142mg,29.13%)。核磁共振h谱如图1所示:1h nmr(500mhz,chloroform-d)δ7.65(d,j=2.0hz,1h),7.50(dd,j=7.2,1.8hz,2h),7.43
–
7.36(m,2h),7.30
–
7.25(m,4h),7.21(ddd,j=15.0,6.9,1.8hz,4h),7.14(dd,j=7.2,1.8hz,1h),6.97(dt,j=6.8,2.0hz,2h),4.97(d,j=2.0hz,2h).
13
c nmr(126mhz,chloroform-d)δ138.34,137.14,134.55,130.98,130.58,128.94,128.82(d,j=4.2hz),128.72,128.14,
127.92,126.94,126.54,126.35,48.78.hrms(esi,m/z)for c
22h18n2 calcd,311.1470[m+h]
+
;found,311.1560[m+h]
+
。
[0059]
d2:1-苄基-4-苯基-2-(间甲基苯基)-1h-咪唑
[0060]
将白色中间体c1(0.471g,1.5mmol)溶于1,4-二氧六环(9ml)溶液加入到圆底烧瓶中,分别加入3-甲基苯硼酸(0.41g,3mmol)和pd(dppf)cl2(0.05g,0.075mmol)。将无水碳酸钠(0.32g,3mmol)溶于水(3ml)中再加入到以上体系中,在100℃油浴中搅拌5h。反应结束后冷却至室温,加入etoac(30ml),水(20ml)萃取。有机相用饱和食盐水(2*50ml)洗两次,然后加入无水硫酸钠干燥,过滤,减压旋转蒸发除去溶剂。粗产品加入硅胶(约粗产物3倍量),然后加入etoac溶解,减压蒸发除去溶剂,将粗产品与硅胶的混合物再经过柱分离(pe/etoac=10:1)后,浓缩得到白色固体d2(172mg,35.28%)。核磁共振h谱如图2所示:1h nmr(500mhz,chloroform-d)δ7.64(d,j=2.9hz,1h),7.54
–
7.49(m,2h),7.28(dd,j=7.3,3.4hz,4h),7.23
–
7.17(m,3h),7.17
–
7.11(m,1h),7.03(d,j=7.7hz,1h),6.98(p,j=2.6hz,3h),4.94(d,j=2.9hz,2h),2.29(d,j=2.8hz,3h).
13
c nmr(126mhz,chloroform-d)δ138.57,137.97,136.95,136.62,134.48,131.58,130.35,129.51,129.03,128.76(d,j=3.3hz),128.12,127.90,127.05,126.45,126.31,48.85,21.33.hrms(esi,m/z)for c
23h20
n2calcd,325.1626[m+h]
+
;found,325.1715[m+h]
+
。
[0061]
d3:1-苄基-4-苯基-2-(对甲基苯基)-1h-咪唑
[0062]
将白色中间体c1(0.471g,1.5mmol)溶于1,4-二氧六环(12ml)溶液加入到圆底烧瓶中,分别加入4-甲基苯硼酸(0.61g,4.5mmol)和pd(dppf)cl2(0.09g,0.12mmol)。将无水碳酸钠(0.36g,3.4mmol)溶于水(3ml)中再加入到以上体系中,在105℃油浴中搅拌4.5h。反应结束后冷却至室温,加入etoac(30ml),水(20ml)萃取。有机相用饱和食盐水(2*50ml)洗两次,然后加入无水硫酸钠干燥,过滤,减压旋转蒸发除去溶剂。粗产品加入硅胶(约粗产物3倍量),然后加入etoac溶解,减压蒸发除去溶剂,将粗产品与硅胶的混合物再经过柱分离(pe/etoac=9:1)后,浓缩得到白色固体d3(356mg,48%)。核磁共振h谱如图3所示:1h nmr(500mhz,chloroform-d)δ7.62(s,1h),7.51(d,j=7.3hz,1h),7.27(s,1h),7.23
–
7.15(m,3h),7.11(t,j=7.9hz,2h),7.02
–
6.97(m,1h),4.95(s,1h),2.39(s,2h).
13
c nmr(126mhz,chloroform-d)δ138.62,138.12,136.99,136.76,134.69,130.81,129.68,128.92,128.79,128.11,127.88,127.44,126.93,126.48,126.26,48.64,21.40.hrms(esi,m/z)for c
23h20n2 calcd,325.1626[m+h]
+
;found,325.1714[m+h]
+
。
[0063]
d4:1-苄基-2-(4-氯苯基)-4-苯基-1h-咪唑
[0064]
将白色中间体c1(0.471g,1.5mmol)溶于1,4-二氧六环(9ml)溶液加入到圆底烧瓶中,分别加入4-氯苯硼酸(0.94g,6mmol)和pd(dppf)cl2(0.05g,0.075mmol)。将无水碳酸钠(0.41g,3.8mmol)溶于水(3ml)中再加入到以上体系中,在110℃油浴中搅拌4h。反应结束后冷却至室温,加入etoac(30ml),水(20ml)萃取。有机相用饱和食盐水(2*50ml)洗两次,然后加入无水硫酸钠干燥,过滤,减压旋转蒸发除去溶剂。粗产品加入硅胶(约粗产物3倍量),然后加入etoac溶解,减压蒸发除去溶剂,将粗产品与硅胶的混合物再经过柱分离(pe/etoac=8:1)后,浓缩得到白色固体d4(376mg,62.57%)。核磁共振h谱如图4所示:1h nmr(500mhz,chloroform-d)δ7.67(s,1h),7.50
–
7.45(m,2h),7.37
–
7.27(m,5h),7.25
–
7.11(m,5h),6.96(dd,j=7.4,2.2hz,2h),4.96(s,2h).
13
c nmr(126mhz,chloroform-d)δ138.88,
137.52,136.40,134.87,134.25,132.28,129.26,129.05,128.90,128.26,128.06,127.44,126.80,126.61,48.86.hrms(esi,m/z)for c
22h17
cln
2 calcd,345.1080[m+h]
+
;found,345.1171[m+h]
+
。
[0065]
d5:1-(4-(苄基-4-苯基-1h-咪唑-2-基)苯基)乙酰基
[0066]
将白色中间体c1(0.471g,1.5mmol)溶于1,4-二氧六环(12ml)溶液加入到圆底烧瓶中,分别加入3-氯苯硼酸(0.615g,3.75mmol)和pd(dppf)cl2(0.11g,0.15mmol)。将无水碳酸钠(0.41g,3.8mmol)溶于水(3ml)中再加入到以上体系中,在110℃油浴中搅拌4h。反应结束后冷却至室温,加入etoac(30ml),水(20ml)萃取。有机相用饱和食盐水(2*50ml)洗两次,然后加入无水硫酸钠干燥,过滤,减压旋转蒸发除去溶剂。粗产品加入硅胶(约粗产物3倍量),然后加入etoac溶解,减压蒸发除去溶剂,将粗产品与硅胶的混合物再经过柱分离(pe/etoac=5:1)后,浓缩得到白色固体d5(325mg,64.45%)。核磁共振h谱图如图5所示:1h nmr(500mhz,chloroform-d)δ7.97
–
7.91(m,2h),7.69(s,1h),7.48
–
7.43(m,2h),7.32(d,j=8.4hz,2h),7.27(d,j=2.0hz,3h),7.24
–
7.15(m,3h),7.00
–
6.93(m,2h),5.01(s,2h),2.62(s,3h).
13
c nmr(126mhz,chloroform-d)δ197.57,139.36,137.93,136.90,136.27,135.52,134.11,131.08,128.93,128.81,128.28,128.11,126.85,126.77,49.00,26.69.hrms(esi,m/z)for c
24h20
n2ocalcd,353.1576[m+h]
+
;found,353.1661[m+h]
+
。
[0067]
d6:1-苄基-4-(4-氯苯基)-2-苯基-1h-咪唑
[0068]
中间体a的制备方法:1-苄基-4-溴咪唑
[0069]
将4-溴咪唑(7.4g,50mmol)溶于pa(70ml)的溶液加入到圆底烧瓶中,分别加入溴化苄(10.2ml,100mmol)和无水碳酸钾(8.63g,62.5mmol),在室温搅拌12h。反应结束后加入etoac(50ml),水(40ml)萃取。有机相用饱和食盐水(2*100ml)洗两次,然后干燥,过滤,减压旋转蒸发除去溶剂。粗产品用dcm(5ml)溶解,再缓慢加入适量石油醚剧烈摇晃至固体析出,真空泵抽滤得到中间体a(白色晶体,2.9g,34.71%)。
[0070]
中间体b2的制备方法:1-苄基-4-(4-氯苯基)咪唑
[0071]
将中间体a(2.38g,10mmol)和1,4-二氧六环(24ml)溶液加入到圆底烧瓶中,分别加入4-氯苯硼酸(3.2g,20mmol)和pd(dppf)cl2(0.37g,0.5mmol),将无水碳酸钠(2.12g,20mmol)溶于水(6ml)中再加入到以上体系中,在100℃油浴中搅拌5h。反应结束后冷却至室温,加入etoac(50ml),水(40ml)萃取。有机相用饱和食盐水(2*50ml)洗两次,然后干燥,过滤,减压旋转蒸发除去溶剂。粗产品用硅胶柱层析(pe/etoac=9:1)提纯分离,得到中间体b2(白色固体,1.4g,51.85%)。
[0072]
中间体c2的制备方法:1-苄基-2-溴-4(4-氯苯基)咪唑
[0073]
将中间体b2(1.4g,5.5mmol)和ccl4(55ml)溶液加入到圆底烧瓶中,继续加入nbs(1.5g,8.3mmol)在30℃水浴中搅拌16h。反应结束后加入dcm(50ml),水(40ml)萃取,然后干燥,过滤,减压旋转蒸发除去溶剂。粗产品用硅胶柱层析(pe/etoac=4:1)提纯分离,得到中间体c2(白色固体,0.77g,40.21%)。
[0074]
将白色中间体c2(0.5g,1.5mmol)溶于1,4-二氧六环(9ml)溶液加入到圆底烧瓶中,分别加入苯硼酸(0.4575g,3.75mmol)和pd(dppf)cl2(0.11g,0.15mmol)。将无水碳酸钠(0.41g,3.8mmol)溶于水(3ml)中再加入到以上体系中,在110℃油浴中搅拌4h。反应结束后冷却至室温,加入etoac(30ml),水(20ml)萃取。有机相用饱和食盐水(2*50ml)洗两次,然后加入无水硫酸钠干燥,过滤,减压旋转蒸发除去溶剂。粗产品加入硅胶(约粗产物3倍量),然后加入etoac溶解,减压蒸发除去溶剂,将粗产品与硅胶的混合物再经过柱分离(pe/etoac=7:1)后,浓缩得到白色固体d6(144mg,27.83%)。核磁共振h谱如图6所示:1h nmr(500mhz,chloroform-d)δ7.67(s,1h),7.43
–
7.33(m,5h),7.27(dd,j=5.2,2.0hz,3h),7.22
–
7.12(m,4h),6.99
–
6.93(m,2h),4.96(s,2h).
13
c nmr(126mhz,chloroform-d)δ137.18,136.26,132.78,132.17,130.86,130.02,129.24,129.06,128.99,128.29,128.04(d,j=5.4hz),127.00,48.95.hrms(esi,m/z)for c
22h17
cln2calcd,345.1080[m+h]
+
;found,345.1172[m+h]
+
。
[0075]
d7:1-苄基-2,4-二(4-氯苯基)-1h-咪唑
[0076]
将白色中间体c2(0.5g,1.5mmol)溶于1,4-二氧六环(12ml)溶液加入到圆底烧瓶中,分别加入4-氯苯硼酸(0.7g,4.5mmol)和pd(dppf)cl2(0.05g,0.075mmol)。将无水碳酸钠(0.32g,3mmol)溶于水(3ml)中再加入到以上体系中,在100℃油浴中搅拌5h。反应结束后冷却至室温,加入etoac(30ml),水(20ml)萃取。有机相用饱和食盐水(2*50ml)洗两次,然后加入无水硫酸钠干燥,过滤,减压旋转蒸发除去溶剂。粗产品加入硅胶(约粗产物3倍量),然后加入etoac溶解,减压蒸发除去溶剂,将粗产品与硅胶的混合物再经过柱分离(pe/etoac=7:1)后,浓缩得到白色固体d7(76mg,13.37%)。核磁共振h谱如图7所示:1h nmr(500mhz,chloroform-d)δ7.65(s,1h),7.45
–
7.37(m,2h),7.36
–
7.31(m,2h),7.30
–
7.26(m,3h),7.21
–
7.14(m,2h),7.13
–
7.07(m,2h),6.99
–
6.91(m,2h),4.95(s,2h).
13
cnmr(126mhz,chloroform-d)δ137.86,137.62,135.12,132.81,132.30,129.40,128.93,128.70,128.43,128.13,127.83,127.69,126.82,48.92.hrms(esi,m/z)for c
22h16
cl2n2calcd,379.0691[m+h]
+
;found,379.0783[m+h]
+
。
[0077]
d8:1-苄基-2-(4-氯苯基)-4-(4-甲基)-1h-咪唑
[0078]
中间体a的制备方法:1-苄基-4-溴咪唑
[0079]
将4-溴咪唑(7.4g,50mmol)溶于pa(70ml)的溶液加入到圆底烧瓶中,分别加入溴化苄(18.3ml,150mmol)和无水碳酸钾(10.4g,75mmol),在室温搅拌15h。反应结束后加入etoac(50ml),水(40ml)萃取。有机相用饱和食盐水(2*100ml)洗两次,然后干燥,过滤,减压旋转蒸发除去溶剂。粗产品用dcm(5ml)溶解,再缓慢加入适量石油醚剧烈摇晃至固体析出,真空泵抽滤得到中间体a(白色晶体,4.1g,52.38%)。
[0080]
中间体b3的制备方法:1-苄基4-(4-甲基苯基)咪唑
[0081]
将中间体a(2.38g,10mmol)和1,4-二氧六环(21ml)溶液加入到圆底烧瓶中,分别加入4-甲基苯硼酸(3.4g,20mmol)和pd(dppf)cl2(0.73g,1mmol),将无水碳酸钠(2.65g,25mmol)溶于水(6ml)中再加入到以上体系中,在110℃油浴中搅拌4h。反应结束后冷却至室温,加入etoac(50ml),水(40ml)萃取。有机相用饱和食盐水(2*50ml)洗两次,然后干燥,过滤,减压旋转蒸发除去溶剂。粗产品用硅胶柱层析(pe/etoac=10:1)提纯分离,得到中间体b3(白色固体,1.4g,46.37%)。
[0082]
中间体c3的制备方法:1-苄基-2-溴-4(4甲基苯基)咪唑
[0083]
将中间体b3(1.4g,5.6mmol)和ccl4(30ml)溶液加入到圆底烧瓶中,继续加入nbs(1g,5.6mmol)在40℃水浴中搅拌12h。反应结束后加入dcm(50ml),水(40ml)萃取,然后干燥,过滤,减压旋转蒸发除去溶剂。粗产品用硅胶柱层析(pe/etoac=4:1)提纯分离,得到中间体c3(白色固体,0.86g,46.59%)。
[0084]
将白色中间体c3(0.494g,1.5mmol)溶于1,4-二氧六环(9ml)溶液加入到圆底烧瓶中,分别加入4-氯苯硼酸(0.7g,4.5mmol)和pd(dppf)cl2(0.05g,0.075mmol)。将无水碳酸钠(0.32g,3mmol)溶于水(3ml)中再加入到以上体系中,在110℃油浴中搅拌4h。反应结束后冷却至室温,加入etoac(30ml),水(20ml)萃取。有机相用饱和食盐水(2*50ml)洗两次,然后加入无水硫酸钠干燥,过滤,减压旋转蒸发除去溶剂。粗产品加入硅胶(约粗产物3倍量),然后加入etoac溶解,减压蒸发除去溶剂,将粗产品与硅胶的混合物再经过柱分离(pe/etoac=7:1)后,浓缩得到白色固体d8(256mg,47.54%)。核磁共振h谱如图8所示:1h nmr(500mhz,chloroform-d)δ7.67(s,1h),7.34(dd,j=12.7,8.4hz,4h),7.29(s,1h),7.26(s,2h),7.12(d,j=8.5hz,2h),7.03(d,j=8.0hz,2h),7.00
–
6.94(m,2h),4.96(s,2h),2.29(s,3h).
13
c nmr(126mhz,chloroform-d)δ138.77,137.33,134.81,132.28,131.15,129.22,129.00(d,j=5.3hz),128.89,128.05,127.00,126.82,126.55,48.90,21.14.hrms(esi,m/z)for c
23h19
cln2calcd,359.1237[m+h]
+
;found,359.1327[m+h]
+
。
[0085]
d9:1-苄基-2-苯基-4-(4-甲基)-1h-咪唑
[0086]
将白色中间体c3(0.494g,1.5mmol)溶于1,4-二氧六环(12ml)溶液加入到圆底烧瓶中,分别加入苯硼酸(0.73g,6mmol)和pd(dppf)cl2(0.05g,0.075mmol)。将无水碳酸钠(0.32g,3mmol)溶于水(3ml)中再加入到以上体系中,在110℃油浴中搅拌4h。反应结束后冷却至室温,加入etoac(30ml),水(20ml)萃取。有机相用饱和食盐水(2*50ml)洗两次,然后加入无水硫酸钠干燥,过滤,减压旋转蒸发除去溶剂。粗产品加入硅胶(约粗产物3倍量),然后加入etoac溶解,减压蒸发除去溶剂,将粗产品与硅胶的混合物再经过柱分离(pe/etoac=10:1)后,浓缩得到白色固体d9(196mg,43.1%)。核磁共振h谱如图9所示:1h nmr(500mhz,chloroform-d)δ7.64(s,1h),7.37(tq,j=8.9,2.5hz,5h),7.30
–
7.26(m,3h),7.25
–
7.18(m,2h),7.04
–
6.94(m,4h),4.96(s,2h),2.27(s,3h).
13
c nmr(126mhz,chloroform-d)δ
138.20,136.93,136.50,136.05,131.41,130.99,130.52,128.89(d,j=2.8hz),128.79,128.68,128.40,127.93,126.98,126.50,48.84,21.13.hrms(esi,m/z)for c
23h20
n2calcd,325.1626[m+h]
+
;found,325.1711[m+h]
+
。
[0087]
d10:1-苄基-2-苯基-4-(噻吩-2-基)-1h-咪唑
[0088]
中间体a的制备方法:1-苄基-4-溴咪唑
[0089]
将4-溴咪唑(7.4g,50mmol)溶于pa(70ml)的溶液加入到圆底烧瓶中,分别加入溴化苄(15.3ml,125mmol)和无水碳酸钾(5.3g,50mmol),在室温搅拌15h。反应结束后加入etoac(50ml),水(40ml)萃取。有机相用饱和食盐水(2*100ml)洗两次,然后干燥,过滤,减压旋转蒸发除去溶剂。粗产品用dcm(5ml)溶解,再缓慢加入适量石油醚剧烈摇晃至固体析出,真空泵抽滤得到中间体a(白色晶体,4.7g,55.46%)。
[0090]
中间体b4的制备方法:1-苄基-4-噻吩咪唑
[0091]
将中间体a(0.8g,3.36mmol)和1,4-二氧六环(16ml)溶液加入到圆底烧瓶中,分别加入2-噻吩硼酸(1.3g,10mmol)和pd(dppf)cl2(0.123g,0.168mmol),将无水碳酸钠(0.712g,6.72mmol)溶于水(4ml)中再加入到以上体系中,在110℃油浴中搅拌4h。反应结束后冷却至室温,加入etoac(30ml),水(20ml)萃取。有机相用饱和食盐水(2*30ml)洗两次,然后干燥,过滤,减压旋转蒸发除去溶剂。粗产品用硅胶柱层析(pe/etoac=8:1)提纯分离,得到中间体b4(白色固体,0.4614g,57%)。
[0092]
中间体c4的制备方法:1,苄基-2-溴-4(2-噻吩)咪唑
[0093]
将中间体b4(0.46g,2mmol)和ccl4(30ml)溶液加入到圆底烧瓶中,继续加入nbs(0.43g,2.5mmol)在30℃水浴中搅拌16h。反应结束后加入dcm(30ml),水(20ml)萃取,然后干燥,过滤,减压旋转蒸发除去溶剂。粗产品用硅胶柱层析(pe/etoac=8:1)提纯分离,得到中间体c4(白色固体,0.3919g,61.04%)。
[0094]
将白色中间体c4(0.46g,1.5mmol)溶于1,4-二氧六环(9ml)溶液加入到圆底烧瓶中,分别加入苯硼酸(0.4575g,3.75mmol)和pd(dppf)cl2(0.05g,0.075mmol)。将无水碳酸钠(0.32g,3mmol)溶于水(3ml)中再加入到以上体系中,在105℃油浴中搅拌4.5h。反应结束后冷却至室温,加入etoac(30ml),水(20ml)萃取。有机相用饱和食盐水(2*50ml)洗两次,然后加入无水硫酸钠干燥,过滤,减压旋转蒸发除去溶剂。粗产品加入硅胶(约粗产物3倍量),然后加入etoac溶解,减压蒸发除去溶剂,将粗产品与硅胶的混合物再经过柱分离(pe/etoac=7:1)后,浓缩得到白色固体d10(168mg,35.22%)。核磁共振h谱如图10所示:1h nmr(500mhz,chloroform-d)δ7.60(s,1h),7.47
–
7.37(m,3h),7.29
–
7.26(m,5h),7.08(d,j=4.8hz,1h),6.96(dd,j=6.8,2.8hz,2h),6.84(d,j=5.4hz,2h),4.94(s,2h).
13
c nmr
(126mhz,chloroform-d)δ138.19,136.99,136.35,134.14,131.13,129.63,129.15,128.95,128.82,128.01,127.77,127.17,126.95,123.32,122.25,48.91.hrms(esi,m/z)for c
20h16
n2scalcd,317.1034[m+h]
+
;found,317.1125[m+h]
+
。
[0095]
d11:1-苄基-4-(3-氯苯基)-2-(4-氯苯基)-1h-咪唑
[0096]
中间体a的制备方法:1-苄基-4-溴咪唑
[0097]
将4-溴咪唑(7.4g,50mmol)溶于pa(70ml)的溶液加入到圆底烧瓶中,分别加入溴化苄(15.3ml,125mmol)和无水碳酸钾(5.3g,50mmol),在室温搅拌13h。反应结束后加入etoac(50ml),水(40ml)萃取。有机相用饱和食盐水(2*100ml)洗两次,然后干燥,过滤,减压旋转蒸发除去溶剂。粗产品用dcm(5ml)溶解,再缓慢加入适量石油醚剧烈摇晃至固体析出,真空泵抽滤得到中间体a(白色晶体,4.2g,51.25%)。
[0098]
中间体b5的制备方法:1-苄基-4-(3-氯苯基)咪唑
[0099]
将中间体a(2.38g,10mmol)和1,4-二氧六环(18ml)溶液加入到圆底烧瓶中,分别加入3-氯苯硼酸(3.9g,25mmol)和pd(dppf)cl2(0.37g,0.5mmol),将无水碳酸钠(2.12g,20mmol)溶于水(6ml)中再加入到以上体系中,在110℃油浴中搅拌4h。反应结束后冷却至室温,加入etoac(50ml),水(40ml)萃取。有机相用饱和食盐水(2*50ml)洗两次,然后干燥,过滤,减压旋转蒸发除去溶剂。粗产品用硅胶柱层析(pe/etoac=8:1)提纯分离,得到中间体b5(白色固体,1.1g,40.9%)。
[0100]
中间体c5的制备方法:1-苄基-2-溴-4(3-氯苯基)咪唑
[0101]
将中间体b5(1.1g,4.35mmol)和ccl4(30ml)溶液加入到圆底烧瓶中,继续加入nbs(0.98g,5.5mmol)在30℃水浴中搅拌4h。反应结束后加入dcm(50ml),水(40ml)萃取,然后干燥,过滤,减压旋转蒸发除去溶剂。粗产品用硅胶柱层析(pe/etoac=8:1)提纯分离,得到中间体c5(白色固体,0.693g,42.16%)。
[0102]
将白色中间体c5(0.46g,1.5mmol)溶于1,4-二氧六环(9ml)溶液加入到圆底烧瓶中,分别加入4-氯苯硼酸(0.585g,3.75mmol)和pd(dppf)cl2(0.05g,0.075mmol)。将无水碳酸钠(0.32g,3mmol)溶于水(6ml)中再加入到以上体系中,在110℃油浴中搅拌4h。反应结束后冷却至室温,加入etoac(30ml),水(20ml)萃取。有机相用饱和食盐水(2*50ml)洗两次,然后加入无水硫酸钠干燥,过滤,减压旋转蒸发除去溶剂。粗产品加入硅胶(约粗产物3倍量),然后加入etoac溶解,减压蒸发除去溶剂,将粗产品与硅胶的混合物再经过柱分离(pe/etoac=5:1)后,浓缩得到白色固体d11(268mg,56.18%)。核磁共振h谱如图11所示:1h nmr(500mhz,chloroform-d)δ7.67(s,1h),7.58(t,j=1.9hz,1h),7.39
–
7.33(m,2h),7.29(dd,j=5.1,2.0hz,3h),7.22(dt,j=7.0,1.7hz,1h),7.15
–
7.08(m,4h),6.98
–
6.92(m,2h),
4.96(s,2h).
13
c nmr(126mhz,chloroform-d)δ137.64,137.52,136.11,136.01,135.20(d,j=5.5hz),134.29,132.15,129.40(d,j=3.2hz),128.94,128.45,128.13(d,j=6.9hz),126.81,126.71,126.62,124.51,48.95.hrms(esi,m/z)for c
22h16
cl2n2calcd,379.0691[m+h]
+
;found,379.0786[m+h]
+
。
[0103]
d12:1-苄基-2-(4-氯苯基)-4-(间甲基)-1h-咪唑
[0104]
中间体a的制备方法:1-苄基-4-溴咪唑
[0105]
将4-溴咪唑(7.4g,50mmol)溶于pa(70ml)的溶液加入到圆底烧瓶中,分别加入溴化苄(15.3ml,125mmol)和无水碳酸钾(5.3g,50mmol),在室温搅拌13h。反应结束后加入etoac(50ml),水(40ml)萃取。有机相用饱和食盐水(2*100ml)洗两次,然后干燥,过滤,减压旋转蒸发除去溶剂。粗产品用dcm(5ml)溶解,再缓慢加入适量石油醚剧烈摇晃至固体析出,真空泵抽滤得到中间体a(白色晶体,4.2g,51.25%)。
[0106]
中间体b6的制备方法:1-苄基-4-(3-甲基苯基)咪唑
[0107]
将中间体a(2.38g,10mmol)和1,4-二氧六环(18ml)溶液加入到圆底烧瓶中,分别加入3-甲基苯硼酸(3.4g,25mmol)和pd(dppf)cl2(0.367g,0.5mmol),将无水碳酸钠(2.12g,20mmol)溶于水(6ml)中再加入到以上体系中,在110℃油浴中搅拌4h。反应结束后冷却至室温,加入etoac(50ml),水(40ml)萃取。有机相用饱和食盐水(2*50ml)洗两次,然后干燥,过滤,减压旋转蒸发除去溶剂。粗产品用硅胶柱层析(pe/etoac=10:1)提纯分离,得到中间体b6(白色固体,1.15g,46.37%)。
[0108]
中间体c6的制备方法:1-苄基-2-溴-4(3-甲基苯基)咪唑
[0109]
将中间体b6(1.15g,4.9mmol)和ccl4(30ml)溶液加入到圆底烧瓶中,继续加入nbs(1.3g,7.35mmol)在35℃水浴中搅拌4.5h。反应结束后加入dcm(50ml),水(40ml)萃取,然后干燥,过滤,减压旋转蒸发除去溶剂。粗产品用硅胶柱层析(pe/etoac=6:1)提纯分离,得到中间体c6(白色固体,0.72g,43.2%)。
[0110]
将白色中间体c6(0.5g,1.5mmol)溶于1,4-二氧六环(9ml)溶液加入到圆底烧瓶中,分别加入4-氯苯硼酸(0.585g,3.75mmol)和pd(dppf)cl2(0.05g,0.075mmol)。将无水碳酸钠(0.32g,3mmol)溶于水(3ml)中再加入到以上体系中,在100℃油浴中搅拌5h。反应结束后冷却至室温,加入etoac(30ml),水(20ml)萃取。有机相用饱和食盐水(2*50ml)洗两次,然后加入无水硫酸钠干燥,过滤,减压旋转蒸发除去溶剂。粗产品加入硅胶(约粗产物3倍量),然后加入etoac溶解,减压蒸发除去溶剂,将粗产品与硅胶的混合物再经过柱分离(pe/etoac=6:1)后,浓缩得到白色固体d12(112mg,23%)。核磁共振h谱如图12所示:1h nmr(500mhz,chloroform-d)δ7.66(s,1h),7.48(d,j=1.8hz,1h),7.36
–
7.31(m,2h),7.30
–
7.27(m,2h),7.15
–
7.03(m,5h),7.00
–
6.93(m,3h),4.96(s,2h),2.28(s,3h).
13
cnmr
(126mhz,chloroform-d)δ138.94,137.92,137.44,136.41,134.81,134.07,132.28,129.19,129.08,128.89,128.03(d,j=2.7hz),127.47,127.40,126.78,123.62,48.86,21.46.hrms(esi,m/z)for c
23h19
cln2calcd,359.1237[m+h]
+
;found,359.1324[m+h]
+
。
[0111]
图13为化合物d7对mcf-7细胞中hsp90客户蛋白及热休克蛋白hsp90和hsp70表达的影响。上述1-苄基-2,4-二芳基咪唑类化合物能够抑制人乳腺癌mcf-7细胞中hsp90客户蛋白akt的表达,并且会引起hsp70和hsp90表达水平的上调。这和经典hsp90抑制剂17-aag的特征相符,是hsp90 n-末端抑制剂的显著特征。蛋白免疫印迹实验方法如下:
[0112]
细胞总蛋白的提取:以每皿106个细胞的密度接种mcf-7细胞,在co2恒温培养箱中孵育过夜,吸弃培养基,将待测化合物配成0μm、8μm、24μm三个浓度的溶液,将17-aag配成10μm的溶液,一次加入孔板中,置于二氧化碳培养箱继续培养24h。用pbs洗3次随后加入150μl的ripa裂解液(含有1%pmsf),在冰上裂解15min后将细胞和裂解液全部收集并转移到1.5ml离心管中,12000g、4℃离心10min。吸取上清转移到新离心管中,用bca试剂盒进行蛋白定量。
[0113]
1-苄基-2,4-二芳基咪唑类化合物对相关蛋白表达量的影响实验:设定总蛋白的质量为20μg。以浓度为10%的分离胶和5%的浓缩胶进行实验。将待测蛋白用移液枪转移至上样孔道中,右边的孔道加入蛋白marker,接通电源设置电压150v,电泳1h。将提前活化好的pvdf膜和切好的滤纸转移到转膜缓冲液中。取出胶,用切胶刀根据marker指示的位置找到目标蛋白并切出需要的胶块,依次按照棉花-三层滤纸-胶-膜的顺序组装,按正负极接通电泳,设置电流为250ma。转膜完毕后取出pvdf膜,在tbst中洗膜2次,每次5min。用5%的脱脂牛奶封闭1h。一抗按照抗体说明书中建议的稀释比例稀释,pvdf膜放置孵育盒中置于4℃冰箱孵育过夜。将pvdf膜转移至tbst中洗3次,每次10min。二抗用1:4000的比例用tbst进行稀释,pvdf膜放至孵育盒中常温条件孵育1h,将pvdf膜转移至tbst中洗3次,每次10min。凝胶成像系统显现出相应的蛋白条带。
[0114]
上述1-苄基-2,4-二芳基咪唑类化合物在抗肿瘤上的应用,1-苄基-2,4-二芳基咪唑类化合物具有治疗乳腺癌的用途。图14-图19为mcf-7细胞给药(d1-d4)48h,细胞的增殖抑制情况示意图。1-苄基-2,4-二芳基咪唑类化合物的体外抗肿瘤活性如表1所示。
[0115]
表1.对乳腺癌细胞的体外抗肿瘤活性
[0116]
[0117][0118]
由表1可知,1-苄基-2,4-二芳基咪唑类化合物具有抗肿瘤的用途。阳性对照17-aag在乳腺癌细胞mcf-7和乳腺癌细胞mda-mb-231中的ic
50
值分别为10.57μm和41.6μm。1-苄基-2,4-二芳基咪唑类化合物d7、d8、d11在mcf-7乳腺癌细胞中的抗肿瘤增殖抑制活性均优于17-aag,1-苄基-2,4-二芳基咪唑类化合物d1-d12在mda-mb-231乳腺癌细胞中的抗肿瘤抑制活性均优于17-aag。
[0119]
化合物对肿瘤细胞的生长抑制:
[0120]
mcf-7和mda-mb-231细胞系的培养基为89%dmem,10%fetal bovine serum(fbs)以及1%antibiotics(gibco life technologies)放在37℃in5%co2/95%air的培养箱中。当细胞占满培养瓶80-90%时,即可用于实验。以17-aag为阳性对照进行实验。
[0121]
对mcf-7细胞生长抑制测试方法:
[0122]
将mcf-7细胞以5000cell/孔的细胞浓进行铺到96孔板上,每孔200μl细胞悬液,铺完后培养12h。吸弃培养基,将化合物配成12.5μm、50μm、75μm、和100μm,4个浓度梯度的溶液,每个样品做5个复孔,一次加入孔板中,置于二氧化碳培养箱继续培养48h后,吸弃废液每孔加100μl无血清培养基和10μl cck8继续培养1h后,用多功能酶标仪在450nm处测定各孔的od值并计算抑制率:
[0123]
成活率=(给药组od值-空白孔od值)/(阴性对照组od值-空孔od值)
×
100%。并利用相关软件计算样品的ic
50
值,如图13-15为d1-d12对mcf-7乳腺癌细胞作用的细胞增长抑制图。
[0124]
对mda-mb-231细胞生长抑制测试方法:
[0125]
将mda-mb-231细胞以6000cell/孔的细胞浓进行铺到96孔板上,每孔200μl细胞悬液,铺完后培养15h。吸弃培养基,将化合物配成10μm、20μm、30μm、40μm、和50μm,5个浓度梯度的溶液,每个样品做5个复孔,一次加入孔板中,置于二氧化碳培养箱继续培养48h后,吸弃废液每孔加100μl无血清培养基和10μl cck8继续培养1h后,用多功能酶标仪在450nm处测定各孔的od值并计算抑制率:
[0126]
成活率=(给药组od值-空白孔od值)/(阴性对照组od值-空孔od值)
×
100%.。并利用相关软件计算样品的ic
50
值,如图16-18为d1-d12对mda-mb-231乳腺癌细胞作用的细胞增长抑制图。
[0127]
以上所述实施例仅表达了本发明的实施方式,但并不能因此而理解为对本发明专利的范围的限制,应当指出,对于本领域的技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些均属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1