一种环β-1,2-葡聚糖与姜黄素包合物及其制备方法
一种环
β-1,2-葡聚糖与姜黄素包合物及其制备方法
技术领域
1.本发明涉及一种环β-1,2-葡聚糖与姜黄素包合物及其制备方法,属于生物技术领域。
背景技术:
2.环β-1,2-葡聚糖(cyclicβ-1,2-glucans)是指由葡萄糖单体组成,通过β-1,2糖苷键连接而成的一类环状多糖,聚合度分布为17~25,在某些菌株中聚合度可达到40。环β-1,2-葡聚糖分为无支链结构和有支链结构,有支链结构的取代基包括磷酸甘油酯基,甲基丙二酰基以及琥珀酰基。并且聚合度和取代基在不同的细菌种类中有很大差异。因为环β-1,2-葡聚糖与环糊精的结构极其相似,均是由葡萄糖单体组成的环状结构。
3.姜黄素(curcumin),其结构式如下:是从姜科姜黄属的一些植物的根茎中提取的一种天然的多酚类化合物,为橙黄色结晶粉末,难溶于水,易溶于甲醇、乙醇、丙酮等有机溶剂。具有抗氧化作用、抗炎作用、抗肿瘤作用等多种生理生化活性,日益受到国内外学者的关注。
4.姜黄素具有抗氧化、抗肿瘤、抗肿瘤和降血脂等作用,但姜黄素溶解性差、口服生物利用度低等缺点,导致它无论在医药保健品领域或者化妆品行业的应用受到了限制。目前,市场可以买到的姜黄根粉和含有姜黄素的保健产品多是进口产品。产品种类丰富,有姜黄素胶囊、片剂、茶和功能饮料,主要功效为抗氧化及保肝等作用。但由于姜黄根粉和姜黄素的溶水性较差,并容易分解等特点,大大阻碍了姜黄素在功能性食品以及保健品中的应用。同时中国已经进入老龄化社会,养生保健,提高生命质量越来越成为人们对保健品和功能性食品的要求。姜黄素是目前公认的对预防和治疗老年痴呆有效的天然化合物。因此,该类保健品和功能性食品的推出一定会受到市场的欢迎。
5.但姜黄素的水溶性低、生物利用度低、易降解、稳定性差等特性,限制其在临床上的广泛应用。因此,增加姜黄素的水溶性从而提高其生物利用度显得尤为重要。
技术实现要素:
6.为了得到一种增加姜黄素的水溶性的方法,本发明采用环β-1,2-葡聚糖与姜黄素进行包合,由于环β-1,2-葡聚糖具有较强水溶性(环β-1,2-葡聚糖溶解度为250g/l;环糊精为18g/l),且聚合度较环糊精聚合度大(环β-1,2-葡聚糖聚合物为17-25,最大者为40;环糊精的聚合度为6-12),环状空腔直径更大(环β-1,2-葡聚糖空腔直径为0.88~1.30nm;环糊精为0.85nm)。单位体积的环β-1,2-葡聚糖可以包合更多的物质、携带更大的分子。
7.本发明首先提供了一种环β-1,2-葡聚糖的制备方法,所述方法包括以下步骤:
8.(1)以放射型根瘤菌(rhizobium radiobacter)atcc 1333为发酵菌株,将其种子液接种至发酵培养基中,发酵制备得到发酵上清液;
9.(2)对发酵上清液进行纯化,制备得到环β-1,2-葡聚糖。
10.在本发明的一种实施方式中,步骤(1)为:将种子液以体积比10%~15%的接种量接种于发酵培养基中,转化温度为30~33℃。
11.在本发明的一种实施方式中,步骤(1)为,将放射型根瘤菌(rhizobium radiobacter)atcc1333在种子培养基中活化,获得种子液;将种子液以体积比为10%~15%的接种量接种于7-l发酵罐中,发酵获得发酵液;将发酵液离心并纯化获得环β-1,2-葡聚糖。
12.在本发明的一种实施方式中,步骤(1)为:将放射型根瘤菌(rhizobium radiobacter)atcc1333在种子培养基中活化,获得种子液;将种子液以体积比10%~15%的接种量接种于7-l发酵罐中,转化温度30℃,两阶段控制ph,设置初始通气比1vvm,初始转速400r/min为100%do值,发酵获得发酵液;将发酵液离心获得发酵上清液。
13.在本发明的一种实施方式中,所述发酵的反应条件为:30~33℃,200rpm的条件下培养24h。
14.在本发明的一种实施方式中,所述两阶段控制ph为,发酵前期菌种生长期(发酵0~24h),控制ph为7.0,发酵24h后,为产糖期,控制ph为7.0。
15.在本发明的一种实施方式中,所述种子培养基包括(g/l):蛋白胨10,酵母膏2,mgso4·
7h2o 1,调ph 7.0。
16.在本发明的一种实施方式中,所述发酵培养基包括:甘露醇10~30g/l,谷氨酸1.0~5.0g/l,nacl 0.2~0.6g/l,k2hpo
4 1.0~3.0g/l,mgso4·
7h2o 0.2~0.6g/l,cacl2·
2h2o 0.02~0.06g/l,微量元素0.1l/l;
17.微量元素母液:fecl3·
6h2o 2.5g/l,mncl2·
4h2o 1g/l,na2moo4·
2h2o 0.01g/l,znso4·
7h2o 0.01g/l,cuso4·
5h2o 0.01g/l,h3bo
3 0.01g/l,cocl2·
6h2o 0.01g/l,生物素0.01g/l,硫胺素0.01g/l。
18.在本发明的一种实施方式中,所述发酵培养基为:甘露醇20g/l,谷氨酸1.0g/l;nacl 0.2g/l,k2hpo
4 1.0g/l,mgso4·
7h2o 0.2g/l,cacl2·
2h2o 0.04g/l,微量元素0.1l/l;
19.微量元素母液:fecl3·
6h2o 2.5g/l,mncl2·
4h2o 1g/l,na2moo4·
2h2o 0.01g/l,znso4·
7h2o 0.01g/l,cuso4·
5h2o 0.01g/l,h3bo
3 0.01g/l,cocl2·
6h2o 0.01g/l,生物素0.01g/l,硫胺素0.01g/l。
20.在本发明的一种实施方式中,所述步骤(2)的纯化方法具体为:
21.步骤1:将步骤(1)制备得到的发酵上清液旋转蒸发浓缩,取上清液,并加入无水乙醇进行孵育后,取上清液,浓缩后,得到浓缩溶液;将无水乙醇添加到浓缩溶液中,进行醇沉后,取上清液,并浓缩至原体积的1/5~1/10,得到浓缩液;
22.步骤2:向步骤1得到的浓缩液中加入十倍体积无水乙醇,得到混合物,并将混合物进行醇沉后,取沉淀,去除有机试剂后,冻干得到粗寡糖粉末;
23.步骤3:将步骤2中的粗寡糖粉末加水配制成溶液,除蛋白后,使用石墨化碳固相萃取小柱将石墨化碳固相萃取小柱采用乙腈活化,并用水淋洗后上样,采用不同浓度的乙腈对样品进行洗脱,取10-20%的乙腈洗脱液作为固相萃取纯化的样品;
24.步骤4:收集步骤3经过固相萃取纯化的样品,旋蒸浓缩去除乙腈,冷冻干燥浓缩,得到预处理好的样品;
25.步骤5:将步骤4得到的样品溶于水中,制备得到寡糖超纯水溶液,将该溶液注入deae-sepharose ff离子交换柱层析柱进行纯化,得到中性寡糖;
26.步骤6:将步骤5中得到中性寡糖通过2000d透析袋透析去除盐离子后,真空冷冻干燥机中冻干得到中性寡糖纯品。
27.在本发明的一种实施方式中,所述步骤(2)的纯化方法具体为:
28.步骤1:将实施例1获得的发酵上清液旋转蒸发浓缩后,在10000rpm条件下离心10min,取上清液,向上清液中加入3倍体积的无水乙醇,并在4℃下孵育12h后,在10000rpm离心10min,取上清液;
29.将上清液浓缩至原体积的1/10,得到浓缩溶液;将3倍体积的无水乙醇添加到浓缩溶液中,以使乙醇的终浓度达到75%(v/v)后,在4℃条件下放置12h后,10000rpm离心10min,取上清液,将上清液浓缩至原体积的1/5,得到浓缩液。
30.步骤2:向步骤1中的浓缩液中加入十倍体积无水乙醇,以使乙醇的最终浓度达到91%,得到混合物,并将混合物在4℃乙醇醇沉12h后,10000rpm离心10min,取沉淀,氮吹去除有机试剂,冻干得到粗寡糖粉末。
31.步骤3:将步骤2中的粗寡糖粉末配制溶液,经sevag试剂除蛋白后,使用石墨化碳固相萃取小柱进行纯化,将石墨化碳固相萃取小柱采用乙腈活化,并用水淋洗后上样;采用不同浓度的乙腈(5%、10%、15%、20%、25%、30%)依次对样品进行洗脱,收集不同浓度的乙腈对样品进行洗脱后的洗脱液;
32.分别将上述洗脱液采用tlc薄层色谱进行目标糖的分离检测;tlc薄层色谱的检测方法为:
33.样品(2μl)采用硅胶-60板(20cm
×
10cm)作为固定阶段,以优化后的正丁醇-乙醇-水(5:5:9v/v/v)作为流动相进行分离,显色剂(3,5二羟基甲苯900mg+无水乙醇375ml+蒸馏水25ml+浓硫酸50ml)浸润薄层板后,立即至于105℃干燥箱,显色5min,出现暗红色斑点。
34.tlc薄层色谱结果显示:目标糖在15%和20%乙腈浓度下洗脱下来。
35.收集合并含有目标糖的洗脱液即:将15%和20%乙腈浓度洗脱液进行合并后得到纯化样品。
36.步骤4:将步骤3得到的纯化样品旋蒸浓缩去除乙腈,在-40℃预冷冻好进行冷冻干燥浓缩,然后将其过0.22μm滤膜得到预处理好的样品。
37.步骤5:将步骤4得到的样品溶于超纯水中,制备得到寡糖超纯水溶液(终浓度为20g/l),将该溶液注入deae-sepharose ff离子交换柱层析柱(1.9cm
×
20cm)。采用超纯水和0.1mol/l、0.2mol/l、0.3mol/l梯度kcl水溶液进行洗脱,流速为1.0ml/min,每5min收集一次洗脱液,将收集到的洗脱液,采用蒽酮硫酸法及tlc薄层色谱法检测收集得到的洗脱液中是否含有目标产物,将含有目标产物的洗脱液进行合并后,浓缩得到目标中性寡糖。
38.结果显示:目标中性寡糖的糖含量占比为92.9%。
39.采用考马斯亮蓝检测目标中性糖的蛋白浓度0g/l。
40.步骤6:将步骤5中得到中性寡糖通过2000d透析袋透析去除盐离子,最后在真空冷冻干燥机中冻干得到环β-1,2-葡聚糖纯品。
41.本发明还提供了一种利用上述方法制备得到环β-1,2-葡聚糖,所述环β-1,2-葡聚糖的结构式如下所示(如图7所示):
[0042][0043]
本发明还提供了上述环β-1,2-葡聚糖在制备姜黄素促溶剂中的应用。
[0044]
本发明还提供了一种姜黄素促溶剂,所述促溶剂中含有上述环β-1,2-葡聚糖。
[0045]
本发明还提供了一种包合物,所述包合物中含有上述环β-1,2-葡聚糖与姜黄素。
[0046]
在本发明的一种实施方式中,所述包含物是按照下述方法制备得到的:
[0047]
将姜黄素溶液和环β-1,2-葡聚糖溶液按照摩尔比为(10:1)~(5:2)的比例进行混合后搅拌,时间为48~72h,得到混合物,将混合物过滤后冷冻干燥,得到包合物。
[0048]
在本发明的一种实施方式中,将姜黄素溶液和环β-1,2-葡聚糖溶液的按照摩尔比为1:1的比例进行混合。
[0049]
本发明还提供了上述包合物在医药保健品、食品饮料中制备含有姜黄素的产品中的应用。
[0050]
本发明还提供了上述环β-1,2-葡聚糖的结构分析。
[0051]
在本发明的一种实施方式中,通过基质辅助激光解吸电离飞行时间质谱(maldi-tof-ms)分析环β-1,2-葡聚糖的分子量及其聚合度。
[0052]
在本发明的一种实施方式中,通过离子色谱分析环β-1,2-葡聚糖的单糖组成。
[0053]
在本发明的一种实施方式中,通过傅立叶红外光谱分析环β-1,2-葡聚糖的官能团组成。
[0054]
本发明还提供了环β-1,2-葡聚糖与姜黄素包埋体系的建立及结构表征。
[0055]
在本发明的一种实施方式中,是环β-1,2-葡聚糖与姜黄素包埋体系的建立及结构表征。
[0056]
在本发明的一种实施方式中,通过dsc分析环β-1,2-葡聚糖与姜黄素包合物。
[0057]
在本发明的一种实施方式中,通过fi-tr分析环β-1,2-葡聚糖与姜黄素包合物。
[0058]
在本发明的一种实施方式中,通过1h nmr分析环β-1,2-葡聚糖与姜黄素包合物。
[0059]
在本发明的一种实施方式中,通过sem分析环β-1,2-葡聚糖与姜黄素包合物。
[0060]
在本发明的一种实施方式中,聚合度范围为17-23,以19聚合度为主,仅由葡萄糖单体组成的葡聚糖;具有典型的多糖的吸收峰,为吡喃糖构型;在891.20cm-1
处的吸收峰是β-糖苷键的特征吸收峰。
[0061]
本发明还提供了所述环β-1,2-葡聚糖与姜黄素在制备生物活性产品方面的应用。
[0062]
有益效果
[0063]
(1)本发明通过放射性根瘤菌的7-l生物反应器上罐发酵,使环β-1,2-葡聚糖的产量由1.9g/l提高至2.79g/l,产量提高了46.8%。
[0064]
(2)本发明提供的环β-1,2-葡聚糖的水溶性好(250g/l),而且可以包裹更多的姜
黄素,是一种良好的包埋材料。本发明制备得到的姜黄素/环β-1,2-葡聚糖包合物经dsc、ft-ir、1hnmr和sem鉴定证明已形成,相比传统的姜黄素/环糊精包合物其稳定性更好。
[0065]
(3)本发明将姜黄素药物分子制成环β-1,2-葡聚糖的包合物后,可以为姜黄素提供一种新的包合材料。不仅阻止姜黄素的有效成分的氧化分解,使姜黄素原有的功效得到保证,而且药物的理化性质将得到显著改善,解决了生物利用度低、制剂困难等一些问题。该包合物具有制备过程简单,无毒副作用,食用安全,可长期服用及保健功效显著等特点。
附图说明
[0066]
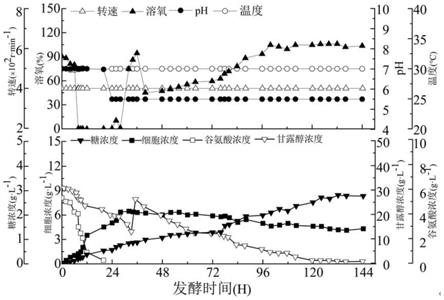
图1:ph控制(生长期控制7.0,产糖期控制5.5)条件下环β-1,2-葡聚糖发酵过程曲线。
[0067]
图2:姜黄素与环β-1,2-葡聚糖、环糊精及其类似物的相溶解度曲线。
[0068]
图3:姜黄素、环β-1,2-葡聚糖、两者的对照混合物和包合物的差示扫描量热图;a:姜黄素;b:环β-1,2-葡聚糖;c:环β-1,2-葡聚糖与姜黄素包埋物;d:环β-1,2-葡聚糖与姜黄素对照混合物。
[0069]
图4:姜黄素、环β-1,2-葡聚糖、两者的对照混合物和包合物的红外光谱图;a:姜黄素;b:环β-1,2-葡聚糖;c:环β-1,2-葡聚糖与姜黄素对照混合物;d:环β-1,2-葡聚糖与姜黄素包埋物。
[0070]
图5:姜黄素、环β-1,2-葡聚糖和包合物的氢谱图;a:姜黄素;b:环β-1,2-葡聚糖;c:环β-1,2-葡聚糖与姜黄素包埋物。
[0071]
图6:姜黄素、环β-1,2-葡聚糖、两者的对照混合物和包合物的扫描电子显微镜谱图;a:姜黄素;b:环β-1,2-葡聚糖;c:环β-1,2-葡聚糖与姜黄素对照混合物;d:环β-1,2-葡聚糖与姜黄素包埋物。
[0072]
图7:环β-1,2-葡聚糖结构式。
[0073]
图8:环β-1,2-葡聚糖核磁共振图谱(a:1h;b:13c)。
具体实施方式
[0074]
下面结合具体的实例和附图对本发明做进一步的阐述,但具体实例并不对本发明做任何的限定。以下实例中所述培养基、试剂等均为本领域的普通技术人员通过购买可以得到。
[0075]
下述实施例中涉及的rhizobium radiobacter atcc 1333来源于江南大学的糖生物制造与生物反应器研究室。下述实施例中所涉及的姜黄素、α-环糊精、β-环糊精、2羟丙基-β-环糊精购自国药集团。
[0076]
下述实施例中所涉及的检测方法如下:
[0077]
胞外中性寡糖产量测定:
[0078]
胞外多糖含量=总糖含量—酸性糖含量。
[0079]
总糖含量测定采用蒽酮-硫酸比色法测定;
[0080]
酸性糖含量采用间苯二酚法测定,剩下的糖含量约为胞外中性寡糖糖的含量。
[0081]
tlc薄层色谱法鉴定环β-1,2-葡聚糖
[0082]
环β-1,2-葡聚糖可以在展开剂(正丁醇:乙醇:水=5:5:9)的作用下达到分离,糖
反应显色剂(3,5二羟基甲苯900mg+无水乙醇375ml+蒸馏水25ml+浓硫酸50ml),立即至于105℃干燥箱,显色3-5min,直至出现暗红色斑点。
[0083]
溶解度检测方法:采用高效液相色谱法进行检测包合物中姜黄素的含量,色谱柱为zorbax sb-c18(4.6
×
150mm,agilent)。流动相:0.1%磷酸水溶液(a)-乙腈(b),梯度洗脱(0~10min,30%~60%b;10min~12min,60%~65%b;12~15min,65%~70%b;15~20min,70%~100%b)。检测波长:280nm。进样量:15μl,流速:1ml/min,检测温度:30℃。得到姜黄素的回归方程为:y=18229x-2.8;相关系数0.9994;保留时间:11.215min;范围(mm/l):0.1~5。以包合物的主体浓度对包合物中的姜黄素浓度作图,即得到相溶解度图
[0084]
稳定常数的计算:
[0085][0086]
其中s0为不含宿主分子的姜黄素浓度,约为1.65
×
10-2
mm/l。式中的斜率为相溶解度图的斜率。
[0087]
下述实施例中所涉及的培养基如下:
[0088]
斜面培养基(g/l):蛋白胨10,酵母膏2,mgso4·
7h2o 1,琼脂粉15,调ph 7.0。
[0089]
种子培养基(g/l):蛋白胨10,酵母膏2,mgso4·
7h2o 1,调ph 7.0。
[0090]
发酵培养基(g/l):甘露醇20;谷氨酸1.0;nacl 0.2;k2hpo
4 1.0;mgso4·
7h2o 0.2;cacl2·
2h2o 0.04;微量元素0.1l/l;
[0091]
微量元素母液(g/l):fecl3·
6h2o 2.5;mncl2·
4h2o 1;na2moo4·
2h2o 0.01;znso4·
7h2o0.01;cuso4·
5h2o 0.01;h3bo
3 0.01;cocl2·
6h2o 0.01;生物素0.01;硫胺素0.01。
[0092]
实施例1:环β-1,2-葡聚糖的制备
[0093]
具体步骤如下:
[0094]
1、rhizobium radiobacter atcc 1333的摇瓶发酵制备胞外中性寡糖
[0095]
(1)将rhizobium radiobacter atcc 1333接种于斜面培养基,在30℃,倒置培养,培养48-72h后,在超净工作台,挑取2~3环单菌落接种至含有50ml的种子培养基中,30℃,200rpm的条件培养24h,即得到活化后的种子液;
[0096]
(2)将步骤(1)中获得的种子液,以10%(v/v)的接种量接种于发酵培养基中,30℃,200rpm,发酵培养144h,制备得到发酵液,检测发酵液中的胞外中性寡糖(发酵液中的总糖含量减去酸性糖含量,粗估算为中性糖含量),其含量为1.9g/l。
[0097]
2、发酵罐制备胞外中性寡糖
[0098]
(1)将rhizobium radiobacter atcc 1333接种于斜面培养基,在30℃,倒置培养培养48~72h后,在超净工作台,挑取2~3环单菌落接种至含有50ml的种子培养基中,30℃,200rpm的条件培养24h,即得到活化后的种子液。
[0099]
(2)7-l发酵罐水平体系:
[0100]
发酵罐总装液量为3.5l的发酵培养基,将步骤(1)中获得的种子液以10%(v/v)接种量接种于发酵培养基,细胞生长期间,每隔4h,外源补充谷氨酸,至谷氨酸加入总浓度为5g/l;
[0101]
碳源补料采用分批补料,当发酵时间为36h,补入甘露醇至终浓度为20g/l。
[0102]
培养温度为30℃,初始ph为7.0。通过补加2mol/l的盐酸以及20%nh3·
2h2o水溶液控制ph在0~24h在7.0,后期(24h后)ph控制为5.5(如图1所示),至144h发酵结束获得发酵液,将发酵液离心获得发酵上清液;设置通气比1vvm,转速400r/min为100%do值。
[0103]
检测发酵液中的胞外中性寡糖,结果显示菌株产胞外中性寡糖含量达到最大为2.79g/l。而摇瓶发酵得到的发酵液中胞外中性寡糖含量为1.9g/l,较上罐水平发酵低了48%。
[0104]
实施例2:环β-1,2-葡聚糖的纯化
[0105]
具体步骤如下:
[0106]
步骤1:将实施例1获得的发酵上清液旋转蒸发浓缩后,在10000rpm条件下离心10min,取上清液,向上清液中加入3倍体积的无水乙醇,并在4℃下孵育12h后,在10000rpm离心10min,取上清液;
[0107]
将上清液浓缩至原体积的1/10,得到浓缩溶液;将3倍体积的无水乙醇添加到浓缩溶液中,以使乙醇的终浓度达到75%(v/v),得到混合物,将混合物在4℃条件下放置12h后,10000rpm离心10min,取上清液,将上清液浓缩至原体积的1/5,得到浓缩液。
[0108]
步骤2:继续向步骤1中的浓缩液中加入十倍体积无水乙醇,以使最终浓度达到91%的乙醇,并将混合物在4℃乙醇醇沉12h后,10000rpm离心10min,取沉淀,氮吹去除有机试剂,冻干得到粗寡糖粉末。
[0109]
步骤3:将步骤2中的粗寡糖粉末加水配制成溶液,经sevag试剂除蛋白后,使用石墨化碳固相萃取小柱进行纯化,将石墨化碳固相萃取小柱采用乙腈活化,并用水淋洗后上样;采用不同体积分数的乙腈(5%、10%、15%、20%、25%、30%)依次对样品进行洗脱,收集不同体积分数的乙腈对样品进行洗脱后的洗脱液;
[0110]
采用tlc薄层色谱进行分别检测上述洗脱液中是否含有目标产物;tlc薄层色谱的检测方法为:样品(2μl)采用硅胶-60板(20cm
×
10cm)作为固定阶段,以优化后的正丁醇-乙醇-水(5:5:9v/v/v)作为流动相进行分离,显色剂(3,5二羟基甲苯900mg+无水乙醇375ml+蒸馏水25ml+浓硫酸50ml)浸润薄层板后立即至于105℃干燥箱,显色5min,出现暗红色斑点。
[0111]
tlc薄层色谱结果显示:目标糖在15%和20%乙腈浓度下洗脱下来。收集合并含有目标糖的洗脱液(15%和20%乙腈浓度下洗脱下来的洗脱液)。
[0112]
步骤4:将步骤3得到的洗脱液旋蒸浓缩去除乙腈,在-40℃预冷冻好进行冷冻干燥浓缩,然后将其过0.22μm滤膜得到预处理好的样品。
[0113]
步骤5:将步骤4得到的样品溶于超纯水中,制备得到寡糖超纯水溶液(终浓度为20g/l),将该溶液注入deae-sepharose ff离子交换柱层析柱(1.9cm
×
20cm)。采用超纯水和0.1mol/l、0.2mol/l、0.3mol/l梯度kcl水溶液进行洗脱,流速为1.0ml/min,每5min收集一次洗脱液,将收集到的洗脱液,采用蒽酮硫酸法及tlc薄层色谱法检测收集得到的洗脱液中是否含有目标产物,将含有目标产物的洗脱液进行合并后,浓缩得到目标中性寡糖。
[0114]
其中:蒽酮硫酸法:
[0115]
蒽酮试剂:精确称取0.1g蒽酮溶解于100ml稀硫酸中(稀硫酸溶液是由76ml的浓硫酸(1.84)稀释至100ml),蒽酮试剂需要现配现用。
[0116]
葡萄糖标准溶液:称取烘干至恒重的葡萄糖100mg,加水溶解,定容至500ml,浓度为0.2mg
·
ml-1
。
[0117]
参照表1的顺序加入各溶液,立即振荡混匀,在沸水浴中煮沸10min,取出室温下冷却10min,振荡后于620nm处测定吸光度值。得到标准曲线为y=7.2858x-0.0004,其中r2=0.9994。
[0118]
表1葡萄糖标准浓度曲线的测定
[0119][0120]
tlc薄层色谱法:采用硅胶-60板(20cm
×
10cm;)作为固定阶段,以优化后的正丁醇-乙醇-水(5:5:9v/v/v)作为流动相进行分离,显色剂(3,5二羟基甲苯900mg+无水乙醇375ml+蒸馏水25ml+浓硫酸50ml)浸润薄层板后立即至于105℃干燥箱,显色5min,出现暗红色斑点。
[0121]
结果显示:目标中性寡糖的糖含量占比为92.9%。
[0122]
采用考马斯亮蓝检测目标中性糖的蛋白浓度0g/l。
[0123]
步骤6:将步骤5中得到中性寡糖通过2000d透析袋透析去除盐离子,最后在真空冷冻干燥机中冻干得到纯的目标中性寡糖,经检测,纯度为:92.9%。
[0124]
对比例1:
[0125]
具体实施步骤同实施例2,区别在于,调整步骤1为:
[0126]
将实施例1获得的发酵上清液旋转蒸发浓缩后,在10000rpm条件下离心10min,取上清液,分别向上清液中加入3倍、5倍、7倍体积的无水乙醇,并在4℃下孵育12h后,在10000rpm离心10min,分别取上清液1~3;
[0127]
分别将上清液1~3浓缩至原体积的1/10,得到浓缩溶液1~3;将3倍体积的无水乙醇分别添加到浓缩溶液1~3中,以使最终浓度分别达到75%的乙醇,在4℃条件下放置12h后,10000rpm离心10min,取上清液,将上清液浓缩至原体积的1/5,分别得到浓缩液1~3;
[0128]
结果显示:分别加入3倍、5倍、7倍体积的乙醇得到的目标中性寡糖纯度分别为:2.5%、31.3%、65.4%。
[0129]
对比例2:
[0130]
具体实施例同实施例2,区别在于,调整步骤2的石墨化碳固相萃取小柱为离子交换柱,结果显示,采用离子交换柱得到的目标中性寡糖纯度为:73.8%。
[0131]
对比例3:
[0132]
具体实施例同实施例2,区别在于,调整步骤6的2000d透析袋为:500d、1000d、3500d,结果显示,分别采用500d、1000d、3500d透析袋得到的目标中性寡糖纯度分别为:81.5%、86.4%、12.3%。
[0133]
对比例4:
[0134]
具体实施方式同实施例2,区别在于,调整步骤3中的tlc薄层色谱的检测方法为:采用硅胶-60板(20cm
×
10cm)作为固定阶段,以正丁醇-乙酸-水(2:1:1v/v/v)作为流动相进行分离,显色剂(3,5二羟基甲苯900mg+无水乙醇375ml+蒸馏水25ml+浓硫酸50ml)。
[0135]
结果显示:目标糖与其他成分不能达到有效的分离,无法检测到目标糖的存在。
[0136]
实施例3:环β-1,2-葡聚糖的结构分析
[0137]
将实施例2中得到的中性寡糖进行结构分析。
[0138]
(1)maldi-tof-ms
[0139]
测试条件:通过基质辅助激光解吸电离飞行时间质谱(maldi-tof-ms)分析纯化的中性寡糖的分子量。具体方法为:将1μl的寡糖样品(1g/l)点在靶上,在干燥器中负压干燥。在相同的位置点1μl的dhb基质溶液,在干燥器中负压干燥。样品与基质混合结晶后进行测量。以反射器模式在m/z 2000-4500的质量范围内进行分析。结果显示中性寡糖的聚合度为17~23。
[0140]
(2)单糖组成
[0141]
测试条件:将得到的中性寡糖在100℃,10h下完全酸水解后经过离子色谱检测单糖组成。具体方法为:精确称取样品5mg,加入300μl浓度为2mol/l的三氟乙酸,100℃恒温酸解12h,结束后氮吹仪吹干,并加入甲醇操作三次用于除去三氟乙酸,然后将水解产物用超纯水溶解后,吸出,定容5ml,通过美国戴安公司的ics-5000离子色谱仪(脉冲安培检测器)进行色谱分析。采用carbopac pa20色谱柱,结果显示,实施例2中得到的中性寡糖为仅由葡萄糖单体组成的葡聚糖。
[0142]
(3)fi-tr
[0143]
测试条件:傅立叶红外光谱(fi-tr)是测定物质的官能团结构的质谱手段。取4-5mg样品,与kbr晶体适量混合,研磨充分后,制片,经傅立叶红外光谱仪对测定样品进行红外波长(400~4000cm-1
)扫描。结果显示其具体典型的多糖的吸收峰,为吡喃糖构型。在891.20cm-1
处的吸收峰是β-糖苷键的特征吸收峰。
[0144]
(4)核磁共振鉴定
[0145]
测试条件:样品的1d nmr(1h和
13
c nmr)光谱在25℃下用avance iii 400mhz光谱仪(bruker,瑞士)测定。大约50mg样品溶解在d2o(99.9%)中。结果显示(如图8所示):异头质子h-1化学位移小于5.0ppm,说明该纯化物中只含有β-型吡喃糖。目标葡聚糖中104.30ppm对应的是非还原性c-1末端,化学位移出现在83ppm表示β-1,2-糖苷键链接多糖的c-2的吸收峰,并且表明该样品不含有非还原性的残基,呈现环状结构。另外,c-1,c-2,c-3,c-4的有多个化学位移数值,表明样品是不同聚合度的聚合物。
[0146]
实施例4:环β-1,2-葡聚糖与姜黄素包埋体系的建立
[0147]
具体步骤如下:
[0148]
步骤1:配制5mm的姜黄素溶液:将购买得到的姜黄素溶液配置终浓度为5mm、体积为2ml(含有200μl的无水乙醇)的姜黄素溶液超纯水中;
[0149]
同时,采用实施例2制备得到的环β-1,2-葡聚糖纯品溶于水后,配制体积为2ml不同浓度的环β-1,2-葡聚糖溶液(0.0mm、0.5mm、1.0mm、1.5mm、2.0mm)。
[0150]
将制备得到的姜黄素溶液分别与浓度为0.0mm、0.5mm、1.0mm、1.5mm、2.0mm的环β-1,2-葡聚糖溶液混合,分别制备得到混合物。
[0151]
步骤2:分别将步骤1制备得到的混合物置于黑色离心管中,在30
±
2℃磁力搅拌机上搅拌48h后,反应达到平衡,分别将混合物在10000g离心20min后,采用0.45μm滤膜过滤去除未包埋的姜黄素,冷冻冻干,即为包合物。
[0152]
即:分别采用浓度为0.0mm、0.5mm、1.0mm、1.5mm、2.0mm的环β-1,2-葡聚糖与姜黄素包合之后,分别制备得到包合物1~5。
[0153]
对比例5:
[0154]
具体方法同步骤1~2,区别在于,将环β-1,2-葡聚糖更换为α-环糊精、β-环糊精、2羟丙基-β-环糊精与姜黄素进行混合,分别制备得到包合物;具体为:
[0155]
采用不同浓度(0.0mm、0.5mm、1.0mm、1.5mm、2.0mm)的α-环糊精、β-环糊精、2羟丙基-β-环糊精与步骤1的姜黄素溶液进行混合,分别制备得到α-环糊精与姜黄素的包合物(包合物6~10)、β-环糊精与姜黄素的包合物(包合物11~15)、2羟丙基-β-环糊精与姜黄素的包合物(包合物16~20)。
[0156]
分别对包合物1~20进行相溶解度检测,包合物1~20的溶解度结果如表2及图2所示。
[0157]
表2包合物的溶解度结果
[0158][0159]
结合higuchi和connors理论,以客体分子对主体分子的浓度作图,可得到一条曲线,即相溶解度图(图2)。若相溶解度曲线有良好的线性关系,则可由此直线的斜率求出包合物的稳定常数。环β-1,2-葡聚糖和环糊精及其衍生物的相溶解度曲线,属于a
l
型,即体系中形成了1:1包合物。同时,可根据图2计算得到稳定常数。稳定常数越大,包合物越稳定。
[0160]
经计算,环β-1,2-葡聚糖,α-环糊精、β-环糊精、2羟丙基-β-环糊精的稳定常数分别是930、121、444、358m-1
,即环葡聚糖是α-环糊精的7.68倍,β-环糊精的2.09倍。
[0161]
结果显示,环β-1,2-葡聚糖的包埋效果显著高于环糊精及其衍生物,原因是环β-1,2-葡聚糖的空腔尺寸以及溶解度大于环糊精及其衍生物,因此,接下来选择环β-1,2-葡聚糖与姜黄素进行包合。
[0162]
对照混合物:
[0163]
对照混合物制备:
[0164]
将姜黄素粉末与环β-1,2-葡聚糖粉末(实施例2制备得到的环β-1,2-葡聚糖溶液冻干之后得到的)按照摩尔比为1:1的比例混合,形成了姜黄素与环β-1,2-葡聚糖的物理混合物,将物理混合物放置在陶瓷坩埚中,研磨15min,以获得均匀的共混物,即制备得到对照混合物。
[0165]
实施例5:包合物的结构鉴定
[0166]
将实施例4获得的包合物(姜黄素-环β-1,2-葡聚糖)及相应的对照品(姜黄素、环β-1,2-葡聚糖以及姜黄素与环β-1,2-葡聚糖的对照混合物)进行结构表征。
[0167]
详细方案如下:
[0168]
(1)dsc
[0169]
测试条件:样品(4-5mg)在50℃至250℃,铝质坩埚,以10℃/min的升温速率在流动
的n2气体中以50ml/min的流速进行测量,采用dsc q200(ta instruments,usa)进行。将适量的样品(姜黄素、环β-1,2-葡聚糖、两者的对照混合物(摩尔比为1:1)以及包合物)放在一个铝锅中按上述条件在同步热分析仪下进行测试,一个空白的平底锅被用作参考。
[0170]
实验结果如图3所示。
[0171]
由图3可知,姜黄素的熔点在171℃处存在一个尖锐的相变吸热峰,为姜黄素的熔点;环β-1,2-葡聚糖在130~150℃之间有一个宽广的吸热峰;对照混合物的dsc曲线出现了一个尖峰;而在包合物中,有一个浅而宽的吸热峰,同时姜黄素的相变吸热峰消失,表明姜黄素和环β-1,2-葡聚糖的包合后,形成了新的物相。
[0172]
(2)fi-tr
[0173]
测试条件:取样品(姜黄素、环β-1,2-葡聚糖、两者的对照混合物(摩尔比为1:1)以及包合物)些许,与kbr晶体适量混合,研磨充分后,制片,经傅立叶红外光谱仪对测定样品进行红外波长(400~4000cm-1
)扫描。
[0174]
实验结果如图4所示。
[0175]
图4中的a和b分别是姜黄素和环β-1,2-葡聚糖的红外色谱图,具有其典型的特征吸收峰;对照混合物中,可以明显看出其是姜黄素和环β-1,2-葡聚糖红外光谱的简单叠加,表明物理混合过程中存在微弱或不存在相互作用;包合物的显示,与环β-1,2-葡聚糖类似,几乎完全覆盖了姜黄素800cm-1
到1600cm-1
范围内的典型特征峰,表明包合物的形成。
[0176]
(3)1h nmr
[0177]
测试条件:样品的1h nmr光谱在25℃下用avance iii 400mhz光谱仪测定。将环β-1,2-葡聚糖溶于d2o中;姜黄素在dmso-d6中溶解,包合物在80%d2o中溶解。
[0178]
实验结果如图5a~图5c所示。
[0179]
由图5可知,经环β-1,2-葡聚糖络合后,包合物的1h nmr谱显示,其姜黄素的h-1、h-2、h-3和h-6的化学位移(δδ)分别发生了显著变化,分别为0.033、0.041、0.018和0.073ppm。同时,检测到环β-1,2-葡聚糖的h-1(0.1172ppm)和h-2(0.1254ppm)的向下化学位移,这直接决定了包合物的形成。
[0180]
(4)sem
[0181]
测试条件:采用扫描电子显微镜(sem)研究了姜黄素、环β-1,2-葡聚糖以及包合物的表面形貌。将样品粉末用双面胶碳带固定,然后喷涂金,使其在低真空条件下导电,获得sem照片。
[0182]
实验结果如图6a~图6d所示。
[0183]
由图6a可知,姜黄素以重复棒状晶体的形式出现;环β-1,2-葡聚糖(图6b)由相当无定形的平面薄片组成;对照混合物(图6c)呈混合形状,姜黄素的杆状晶体明显吸附在环β-1,2-葡聚糖表面;包合后,包合物(图6d)呈现非晶态体结构,没有姜黄素和环β-1,2-葡聚糖的原始形态。
[0184]
虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权利要求书所界定的为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1