一种采用少量简并引物全面扩增humanTCRβ链文库的制备方法与流程
一种采用少量简并引物全面扩增humantcr
β
链文库的制备方法
技术领域
1.本发明涉及免疫组库测序文库的扩增制备技术领域,具体为一种采用少量简并引物全面扩增humantcrβ链文库的制备方法。
背景技术:
2.免疫组库(immune repertoire,ir)是指在任何指定时间,个体的循环系统内所有功能多样性b细胞和t细胞的总和,免疫系统的多样性对机体健康至关重要,其多样性越丰富,机体的免疫系统就越健康,识别并清除抗原的能力就越强,机体的获得性免疫能识别无数种抗原分子,并随之做出反应,其分子基础为t细胞表面受体和b细胞表面受体(tcr和bcr)特异性识别抗原区段,激活免疫应答,免疫系统中t细胞库和b细胞库分别包含了所有特异性不同的t细胞克隆和b细胞克,这种识别受体的多样性在基因重排过程中产生,多样性体现为受体基因v(d)j片段重组,tcrβ基因群可变区包括v β、dβ、jβ三类基因片段,tcrα基因群包括vα、jα两类基因片段,不同基因片段及不同链的组合可产生1016种可能性,因此分析多样性主要是分析 v(d)j片段的序列特征。
3.目前关于免疫组库ngs制备技术包括多重pcr与5’race(rapidamplification of cdna ends),其中race法在逆转录、tdt加尾、pcr扩增这三个连接的酶促反应过程中,任何一步的失败都会导致前功尽弃,即便是上述反应平稳顺利,但结果也通常会出现一些非特异性产物或非全长的产物,且成本较高,相比之下,多重pcr建库简单易行、成本低等优点便体现出来,成为免疫组库建库的优选方法,但是多重pcr存在一个弊端就是不同引物之间会形成引物二聚体,且引物对数越多,二聚体现象越严重,这就大大降低了引物的扩增效率。而且,引物对数越多,实验成本就越高,难度也越大,目前国内外关于免疫组库的制备无非两种,一种是全引物扩增,而tcr 的可变区v区基因有152个,采用传统的多重pcr扩增技术路线,需要上百对引物,这无疑为免疫组库的扩增制备增添了很多困难和成本,后一种就是局部扩增,采用一部分v区引物扩增,利用局部检测整体,而这种方法很容易对结果造成偏差,且随机性太强,可重复性差,实验结果不稳定,因此本发明利用一种独特的计算机算法针对152个v基因设计出43对简并引物,通过一系列的实验条件优化,实现了利用少量引物全覆盖扩增humantcrβ链测序文库。
技术实现要素:
4.本发明提供的发明目的在于提供一种采用少量简并引物全面扩增 humantcrβ链文库的制备方法。
5.通过独特的计算机算法针对152个v基因设计出43对简并引物,并对实验条件优化,实现了利用少量引物全覆盖扩增humantcrβ链测序文库。
6.为了实现低成本、全面扩增humantcrβ链cdr3文库。
7.本发明提供如下技术方案:一种采用少量简并引物全面扩增humantcrβ链文库的
制备方法,包括以下步骤:
8.s1、淋巴细胞裂解和总rna提取:将样本预处理后,进行rna提取;
9.s2、逆转录:对获得的淋巴细胞的总rna进行逆转录,获得cdna;
10.s3、多重pcr:通过对cdna进行第一轮pcr和第二轮pcr,对cdna的tcrβ链基因进行特异性扩增;
11.s4、添加测序接头序列及index;
12.s5、测序结果分析:高通量测序完成后,使用fastq文件进行数据分析,采用以mixcr为核心的独特的分析工具将测序结果比对到t细胞受体的v、d、j、c基因参考序列上,利用前一步骤获得的比对结果拼接clonotypes,输出比对结果,最后生成免疫组库多样性图谱。
13.进一步的,根据s1中的操作步骤,包括以下步骤:
14.s101、取250μl血液转移至1.5ml的rnase-free离心管中,加入750μl裂解液,反复吹打,并剧烈振荡混匀后,室温静置5min;
15.s102、加入200μl氯仿,剧烈振荡15sec混匀后,制得样品,室温静置2min;
16.s103、将样品置于离心机中,使混合溶液分层,取上层水相转移至1.5ml的rnase-free离心管中,加入0.5倍体积无水乙醇,颠倒混匀,获得预处理混合液;
17.s104、将rna吸附柱套入2ml收集管中,将预处理混合液加入到rna吸附柱中,离心,弃掉废液;
18.s105、加入500μl去蛋白液,离心,弃废液;
19.s106、将rna吸附柱放回收集管,加入500μl漂洗液,室温离心30sec,弃废液,重复循环一次;
20.s107、将rna吸附柱放回收集管,空柱室温离心2min,除去残留的漂洗液;
21.s108、将rna吸附柱放入新的1.5mlrnase-free离心管中,rnase-freeh2o洗脱,室温放置2min,然后通过离心1min,收集滤液,即为rna溶液,然后通过离心1min,收集滤液,即为rna溶液。
22.进一步的,所述离心机设置温度为4℃,转速为12000rpm,离心10min。
23.进一步的,根据s2中的操作步骤,包括以下步骤:
24.s201、在rnase-free离心管中配置混合液,将配置的混合液,在温度为65℃的温度下,加热5min,迅速置于冰上骤冷;
25.s202、配制第一链cdna合成反应液,用移液器轻轻吹打混匀后,进行第一链cdna合成反应;
26.进一步的,所述混合液为rnase-freeddh2o、gene-specific-primers和total-rna混合制得混合液。
27.进一步的,所述第一链cdna合成反应液为混合液、2
×
rtmix和enzymemix混合制得。
28.进一步的,根据s3中的操作步骤,包括以下步骤:
29.s301、第一轮pcr将25.25μlmastermix、5μlv-primermix(seqidno:1-seqidno:43)、5μlc区引物(seqidno:44)、cdna模板和ddh2o,混合后,配置成多重pcr反应液;
30.s302、将配置成的多重pcr反应液经过电泳检测和磁珠纯化,将pcr产物使用dna clean beads磁珠进行纯化;
31.s303、进行第二轮pcr,将25μl polymerase mix、1μ lv-primeradmix(seqidno:47-seqidno:90)、1μlc-primerad、第一轮pcr纯化产物和50μlddh2o混合,制得pcr反应液配制;
32.s304、然后将pcr反应液配制进行胶回收纯化,通过电泳条带验证。
33.进一步的,根据s302中的操作步骤,包括以下步骤:
34.s3021、将磁珠由冰箱中取出,室温平衡至少30min,配制80%乙醇,通过涡旋振荡或充分颠倒磁珠;
35.s3022、吸取1.0
×
的dna clean beads至dna溶液中,室温孵育5min;
36.(≥20μl),涡旋振荡或使用移液器轻轻吹打至充分混匀,室温静置5min。
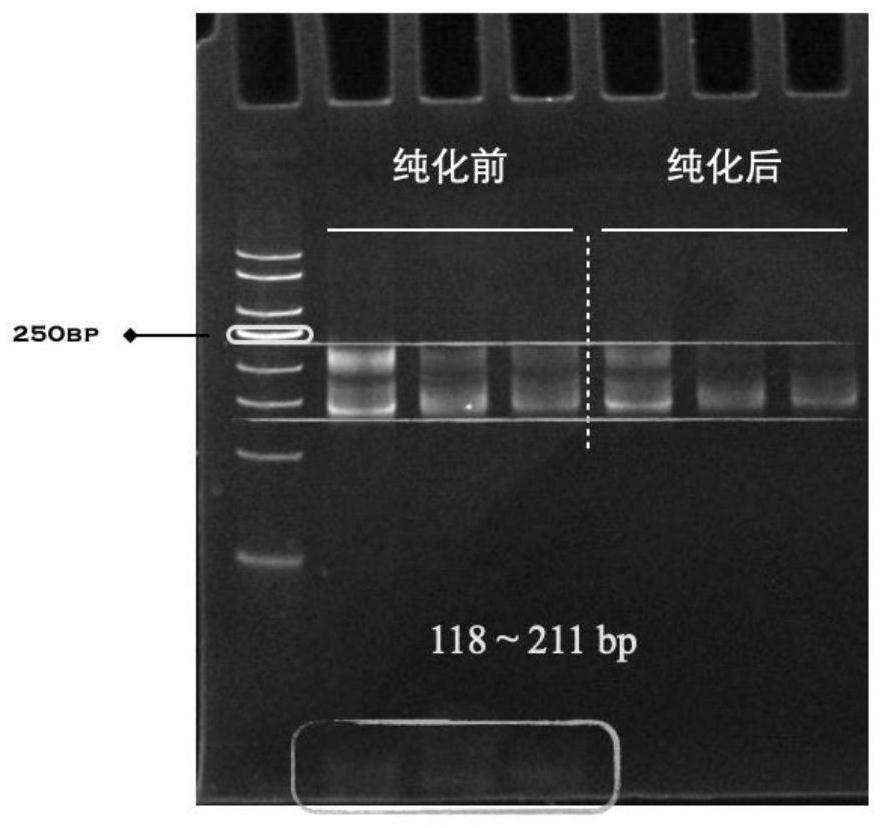
37.进一步的,根据s304中的操作步骤,包括以下步骤:
38.s3041、将带有目的片段的凝胶块转移至1.5ml离心管中,称重得出凝胶块的重量,近似地确定其体积,加入等体积的binding buffer,在50-60℃水浴中温浴至凝胶完全融化,每2-3min振荡或涡旋混合物;
39.s3042、取一个dna结合柱装在2ml收集管内,将获得的dna熔胶液全部转移至dna结合柱中,室温下10000xg离心1min,弃收集管中的滤液,将柱子套回2ml收集管内收集管中;
40.s3043、收集管中的滤液,将dna结合柱套回2ml收集管内收集管,转移300μl binding buffer至柱子中,室温下,最大速度(≥13000)离心1min,弃滤液;
41.s3044、将dna结合柱套回2ml收集管内收集管,转移700μl洗脱buffer 至dna结合柱中,室温下10000xg离心1min,弃滤液,后重复一次;
42.s3045、将dna结合柱套回2ml收集管内收集管,室温下≥13000xg离心 2min以甩干dna结合柱基质残余的液体;
43.s3046、将dna结合柱装在一个干净的1.5ml离心管上,加入15~30μl 的elution buffer到dna结合柱的基质上,室温放置1min,13000xg离心1min 以洗脱dna。
44.进一步的,根据s4中的操作步骤,包括以下步骤:
45.s401、25μl polymerase、1μl primer—p5、1μl primer—p7、第二轮pcr纯化产物cdna、ddh2o混合制得pcr反应液配;
46.s402、胶回收纯化后,建库终产物电泳条带验证。
47.本发明提供了一种采用少量简并引物全面扩增humantcrβ链文库的制备方法,具备以下有益效果:构建文库较短,但包含完整的cdr3区段,可以获得更高的通量,更低的价格,每条特异性引物的添加量与扩增体系都是经过大量实验优化后确定的,采用本发明的方法可以极大降低引物二聚体的数量,获得高质量文库。
附图说明
48.图1为本发明一种采用少量简并引物全面扩增humantcrβ链文库的第一轮pcr电泳条带;
49.图2为本发明一种采用少量简并引物全面扩增humantcrβ链文库的第二轮pcr产物;
50.图3为本发明一种采用少量简并引物全面扩增humantcrβ链文库的建库终产物电泳条带;
51.图4为本发明一种采用少量简并引物全面扩增humantcrβ链文库的免疫多样性图谱;
52.图5为本发明一种采用少量简并引物全面扩增humantcrβ链文库的克隆型频谱对比。
具体实施方式
53.请参阅图1-5,本发明提供一种技术方案:一种采用少量简并引物全面扩增humantcrβ链文库的制备方法,包括以下步骤:
54.s1、淋巴细胞裂解和总rna提取:将样本预处理后,进行rna提取;
55.s2、逆转录:对获得的淋巴细胞的总rna进行逆转录,获得cdna;
56.s3、多重pcr:通过对cdna进行第一轮pcr和第二轮pcr,对cdna的tcrβ链基因进行特异性扩增;
57.s4、添加测序接头序列及index;
58.s5、测序结果分析:高通量测序完成后,使用fastq文件进行数据分析,采用以mixcr为核心的独特的分析工具将测序结果比对到t细胞受体的v、d、 j、c基因参考序列上,利用前一步骤获得的比对结果拼接clonotypes,输出比对结果,最后生成免疫组库多样性图谱。
59.具体的,根据s1中的操作步骤,包括以下步骤:
60.s101、取250μl血液转移至1.5ml的rnase-free离心管中,加入750 μl裂解液,反复吹打,并剧烈振荡混匀后,室温静置5min;
61.s102、加入200μl氯仿,剧烈振荡15sec混匀后,制得样品,室温静置 2min;
62.s103、将样品置于离心机中,使混合溶液分层,取上层水相转移至1.5ml的rnase-free离心管中,加入0.5倍体积无水乙醇,颠倒混匀,获得预处理混合液;
63.s104、将rna吸附柱套入2ml收集管中,将预处理混合液加入到rna吸附柱中,离心,弃掉废液;
64.s105、加入500μl去蛋白液,离心,弃废液;
65.s106、将rna吸附柱放回收集管,加入500μl漂洗液,室温离心30sec,弃废液,重复循环一次;
66.s107、将rna吸附柱放回收集管,空柱室温离心2min,除去残留的漂洗液;
67.s108、将rna吸附柱放入新的1.5ml rnase-free离心管中,rnase-freeh2o洗脱,室温放置2min,然后通过离心1min,收集滤液,即为rna溶液,然后通过离心1min,收集滤液,即为rna溶液。
68.具体的,离心机设置温度为4℃,转速为12000rpm,离心10min。
69.具体的,根据s2中的操作步骤,包括以下步骤:
70.s201、在rnase-free离心管中配置混合液,将配置的混合液,在温度为 65℃的温度下,加热5min,迅速置于冰上骤冷;
71.s202、配制第一链cdna合成反应液,用移液器轻轻吹打混匀后,进行第一链cdna合
成反应;
72.具体的,混合液为rnase-freeddh2o、gene-specific-primers和totalrna混合制得混合液。
73.具体的,第一链cdna合成反应液为混合液、2
×
rt mix和enzyme mix混合制得。
74.具体的,根据s3中的操作步骤,包括以下步骤:
75.s301、第一轮pcr将25.25μl mastermix、5μl v-primer mix(seqidno:1-seqidno:43)、5μl c区引物(seqidno:44)、cdna模板和ddh2o,混合后,配置成多重pcr反应液;
76.s302、将配置成的多重pcr反应液经过电泳检测和磁珠纯化,将pcr产物使用dna clean beads磁珠进行纯化;
77.s303、进行第二轮pcr,将25μl polymerase mix、1μl v-primeradmix(seqidno:47-seqidno:90)、1μl c-primerad、第一轮pcr纯化产物和50 μlddh2o混合,制得pcr反应液配制;
78.s304、然后将pcr反应液配制进行胶回收纯化,通过电泳条带验证。
79.具体的,根据s302中的操作步骤,包括以下步骤:
80.s3021、将磁珠由冰箱中取出,室温平衡至少30min,配制80%乙醇,通过涡旋振荡或充分颠倒磁珠;
81.s3022、吸取1.0
×
的dna clean beads至dna溶液中,室温孵育5min;
82.(≥20μl),涡旋振荡或使用移液器轻轻吹打至充分混匀,室温静置5min。
83.具体的,根据s304中的操作步骤,包括以下步骤:
84.s3041、将带有目的片段的凝胶块转移至1.5ml离心管中,称重得出凝胶块的重量,近似地确定其体积,加入等体积的binding buffer,在50-60℃水浴中温浴至凝胶完全融化,每2-3min振荡或涡旋混合物;
85.s3042、取一个dna结合柱装在2ml收集管内,将获得的dna熔胶液全部转移至dna结合柱中,室温下10000xg离心1min,弃收集管中的滤液,将柱子套回2ml收集管内收集管中;
86.s3043、收集管中的滤液,将dna结合柱套回2ml收集管内收集管,转移 300μl binding buffer至柱子中,室温下,最大速度(≥13000)离心1min,弃滤液;
87.s3044、将dna结合柱套回2ml收集管内收集管,转移700μl洗脱buffer至dna结合柱中,室温下10000xg离心1min,弃滤液,后重复一次;
88.s3045、将dna结合柱套回2ml收集管内收集管,室温下≥13000xg离心 2min以甩干dna结合柱基质残余的液体;
89.s3046、将dna结合柱装在一个干净的1.5ml离心管上,加入15~30μl 的elution buffer到dna结合柱的基质上,室温放置1min,13000xg离心1min以洗脱dna。
90.具体的,根据s4中的操作步骤,包括以下步骤:
91.s401、25μl polymerase、1μl primer—p5、1μl primer—p7、第二轮pcr纯化产物cdna、ddh2o混合制得pcr反应液配;
92.s402、胶回收纯化后,建库终产物电泳条带验证。
93.实施例的方法进行检测分析,并与现有技术进行对照,得出如下数据:
[0094] 文库质量价格实施例较高较低
现有技术较低较高
[0095]
根据上述表格数据可以得出,当使用实施例时,构建文库较短,但包含完整的cdr3区段,可以获得更高的通量,更低的价格,本发明提供的pcr扩增体系可以很大程度消减引物二聚体的产生,获得高质量文库。
[0096]
本发明提供了一种采用少量简并引物全面扩增humantcrβ链文库的制备方法,包括以下步骤:
[0097]
s1、淋巴细胞裂解和总rna提取:将样本预处理后,进行rna提取;
[0098]
s2、逆转录:对获得的淋巴细胞的总rna进行逆转录,获得cdna;
[0099]
s3、多重pcr:通过对cdna进行第一轮pcr和第二轮pcr,对cdna的tcrβ链基因进行特异性扩增;
[0100]
s4、添加测序接头序列及index;
[0101]
s5、测序结果分析:高通量测序完成后,使用fastq文件进行数据分析,采用以mixcr为核心的独特的分析工具将测序结果比对到t细胞受体的v、d、 j、c基因参考序列上,利用前一步骤获得的比对结果拼接clonotypes,输出比对结果,最后生成免疫组库多样性图谱,见图4,图中每一个色块代表一种clonotype,色块大小代表每一种clonotype的数量,色块的种类越多,大小越接近,则反映出受试者的免疫组库多样性就越丰富,其免疫力就越健康,为了验证方法的稳定可靠性,把同一个供试者的血样分成5份,每份血样各做一次平行实验,将得到的5份文库分别添加不同的index一同上机测序,对测序结果进行分析,发现5份血样的克隆型频谱高度一致,clonotypes的种类及数量都十分接近,表明使用该方法进行扩增humantcrβ链测序文库是具有高度稳定可靠性的,克隆型频谱对比见图5。
[0102]
根据s1中的操作步骤,包括以下步骤:
[0103]
s101、取250μl血液转移至1.5ml的rnase-free离心管中,加入750 μl裂解液,反复吹打,并剧烈振荡混匀后,室温静置5min;
[0104]
s102、加入200μl氯仿,剧烈振荡15sec混匀后,制得样品,室温静置 2min;
[0105]
s103、将样品置于离心机中,使混合溶液分层,取上层水相转移至1.5ml的rnase-free离心管中,加入0.5倍体积无水乙醇,颠倒混匀,获得预处理混合液;
[0106]
s104、将rna吸附柱套入2ml收集管中,将预处理混合液加入到rna吸附柱中,离心,弃掉废液;
[0107]
s105、加入500μl去蛋白液,离心,弃废液;
[0108]
s106、将rna吸附柱放回收集管,加入500μl漂洗液,室温离心30sec,弃废液,重复循环一次;
[0109]
s107、将rna吸附柱放回收集管,空柱室温离心2min,除去残留的漂洗液;
[0110]
s108、将rna吸附柱放入新的1.5ml rnase-free离心管中,rnase-freeh2o洗脱,室温放置2min,然后通过离心1min,收集滤液,即为rna溶液,然后通过离心1min,收集滤液,即为rna溶液,rna溶液可置于-80℃长期保存。
[0111]
离心机设置温度为4℃,转速为12000rpm,离心10min。
[0112]
根据s2中的操作步骤,包括以下步骤:
[0113]
s201、在rnase-free离心管中配置混合液,将配置的混合液,在温度为 65℃的温度下,加热5min,迅速置于冰上骤冷;
[0114][0115]
s202、配制第一链cdna合成反应液,将8μl混合液、10μlrtmix和2μlenzyme mix混合,用移液器轻轻吹打混匀后,在50℃条件下反应45min后,在85℃的条件下反应2min,进行第一链cdna合成反应,产物可立即用于pcr反应,或在-20℃保存,并在半年内使用,长期存放建议分装后在-70℃保存,cdna应避免反复冻融;
[0116]
混合液为rnase-freeddh2o、gene-specific-primers和totalrna混合制得混合液。
[0117]
第一链cdna合成反应液为混合液、rtmix和enzyme mix混合制得。
[0118]
根据s3中的操作步骤,包括以下步骤:
[0119]
s301、第一轮pcr将25μl polymerase mix、5μl v-primer mix(seqidno:1-seqidno:43)、5μl c区引物(seqidno:44)、cdna模板和ddh2o,混合后,配置成多重pcr反应液,在(1)、温度94℃下反应1min,(2)、温度94℃下反应30s,(3)、温度60℃下反应1min,(4)、温度72℃下反应 10min,重复(2)和(3)30次,其中v-primer mix为5’端多重pcr引物,各引物名称及序列见下表所列;
[0120]
[0121][0122]
s302、将配置成的多重pcr反应液经过电泳检测和磁珠纯化,将pcr产物使用dna clean beads磁珠进行纯化,电泳检测扩增结果,条带在100~200 bp之间;
[0123]
s303、进行第二轮pcr,将25μl polymerase mix、1μl v-primeradmix(seqidno:47-seqidno:90)、1μl c-primerad、第一轮pcr纯化产物和50 μl ddh2o混合,制得pcr反应液配制,pcr反应液配制在(5)、温度94℃下反应1min,(6)、温度94℃下反应30s,(7)、温度60℃下反应1min, (8)、温度72℃下反应10min,重复(6)和(7)10次,其中v-primeradmix为5’端多重pcr建库引物,各引物按照1:1的体积比混合,终浓度为10 μm,各引物名称及序列见下表所列;
[0124]
[0125]
[0126]
[0127][0128]
s304、然后将pcr反应液配制进行胶回收纯化,通过电泳条带验证。
[0129]
根据s302中的操作步骤,包括以下步骤:
[0130]
s3021、将磁珠由冰箱中取出,室温平衡至少30min,配制80%乙醇,通过涡旋振荡或充分颠倒磁珠;
[0131]
s3022、吸取1.0
×
的dna clean beads至dna溶液中,室温孵育5min;
[0132]
(≥20μl),涡旋振荡或使用移液器轻轻吹打至充分混匀,室温静置5min。
[0133]
根据s304中的操作步骤,包括以下步骤:
[0134]
s3041、将带有目的片段的凝胶块转移至1.5ml离心管中,称重得出凝胶块的重量,
近似地确定其体积,加入等体积的binding buffer,在50-60℃水浴中温浴至凝胶完全融化,每2-3min振荡或涡旋混合物;
[0135]
s3042、取一个dna结合柱装在2ml收集管内,将获得的dna熔胶液全部转移至dna结合柱中,室温下10000xg离心1min,弃收集管中的滤液,将柱子套回2ml收集管内收集管中;
[0136]
s3043、收集管中的滤液,将dna结合柱套回2ml收集管内收集管,转移 300μl binding buffer至柱子中,室温下,最大速度(≥13000)离心1min,弃滤液;
[0137]
s3044、将dna结合柱套回2ml收集管内收集管,转移700μl洗脱buffer至dna结合柱中,室温下10000xg离心1min,弃滤液,后重复一次;
[0138]
s3045、将dna结合柱套回2ml收集管内收集管,室温下≥13000xg离心 2min以甩干dna结合柱基质残余的液体;
[0139]
s3046、将dna结合柱装在一个干净的1.5ml离心管上,加入15~30μl 的elution buffer到dna结合柱的基质上,室温放置1min,13000xg离心1min以洗脱dna。
[0140]
根据s4中的操作步骤,包括以下步骤:
[0141]
s401、25μl polymerase、1μl primer—p5、1μl primer—p7、第二轮pcr纯化产物cdna、ddh2o混合制得pcr反应液配,pcr反应液配在(9)、温度98℃下反应1min,(10)、温度98℃下反应10s,(11)、温度60℃下反应5s,(12)、温度72℃下反应5s,(13)、温度72℃下反应3min,重复(10)、(11)和(12)15次,引物序列中,i5序列与p5序列嵌合一起, i7序列与p7序列嵌合一起,具体序列如下;
[0142][0143][0144]
s402、胶回收纯化后,建库终产物电泳条带验证。
[0145]
尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1