甲苯磺酸艾多沙班的中间体及其制备方法与流程
1.本发明涉及有机化学合成领域,尤其涉及甲苯磺酸艾多沙班的中间体及其制备方法。
背景技术:
2.甲苯磺酸艾多沙班是一种直接抗凝血xa因子抑制剂,用于治疗全膝关节置换术、全髋关节置换术或髋关节骨折手术后患者的静脉血栓栓塞症;其药理上可接受盐或者它们的水合物的制备方法。
3.甲苯磺酸艾多沙班由第一三共(daiichi sankyo)株式会社研发,于2011年4月22日获日本医药品医疗器械综合机构(pmda)批准上市,之后于2015年1月8日获美国食品药品管理局(fda)批准上市,于2015年6月19日获欧洲药物管理局(ema)批准上市,于2018年12月25日获得国家药品监督管理局(nmpa)批准上市。由第一三共株式会社在日本上市销售,商品名为为口服片剂。
4.目前市场中,对于甲苯磺酸艾多沙班的合成工艺不少,但是绝大部分工艺路线都是通过其关键中间体4,5,6,7-四氢-5-甲基-噻唑并[5,4-c]吡啶-2-羧酸进行合成的,因此,对于该甲苯磺酸艾多沙班中间体的合成研究至关重要,该关键中间体结构式如下所示:
[0005]
目前文献报道4,5,6,7-四氢-5-甲基-噻唑并[5,4-c]吡啶-2-羧酸的制备方法主要路线如下:1)109c6x合成路线一(us2005119486a1)
[0006]
该路线以4-氨基吡啶为起始原料,与boc-酸酐反应得到109h1-00,经正丁基锂与s单质作用下得到化合物109h2-00,与甲酸进行关环反应得到化合物109h3-00,再与碘甲烷反应得到相应吡啶季铵盐类化合物109h4-00,硼氢化合物进行还原反应得到化合物109c4-00,用正丁基锂与二氧化碳气体制备得到相应的羧酸锂盐化合物109c6-10,最后经盐酸中和并成盐得到化合物109c6-20。该路线两次用到昂贵且容易着火的正丁基锂,还有易燃易爆的s单质,期间还用了沸点低、昂贵且毒性大的碘甲烷试剂,从生产安全的角度讲存在的隐患较多,且不利于控制成本,不适合工业化生产。
[0007]
2)109c6x合成路线(us9233980b2)
[0008]
该路线以n-甲基-4-哌啶酮为起始原料与单氰胺水溶液和s单质环合得到相应的噻唑胺化合物109c2-00,用亚硝酸钠与氢溴酸通过重氮化反应得到溴代物109c3-00,低温条件下与正丁基氯、二氧化碳气体反应得到羧酸锂盐,最后经盐酸中和并成盐得到化合物109c6-20。该路线用到易燃易爆的s单质,还有价格昂贵且容易着火的正丁基锂,存在较多的生产安全隐患,且不利于控制成本,不适合于工业化规模的生产。
[0009]
有鉴于此,本发明的目的在于提供一种制备工艺简便、反应条件温和、成本低、原料易得的4,5,6,7-四氢-5-甲基-噻唑并[5,4-c]吡啶-2-羧酸的制备方法,以克服现有技术中存在的上述缺陷。
技术实现要素:
[0010]
本发明提供了一种用于制备甲苯磺酸艾多沙班的中间体及其制备方法,本方法具有工艺简便、反应条件温和、成本低、原料易得的优点。
[0011]
为实现上述发明目的,本发明通过以下技术方案实现:第一方面,本发明提供了一种制备甲苯磺酸艾多沙班式为(ii)的中间体,其结构式如下:
[0012]
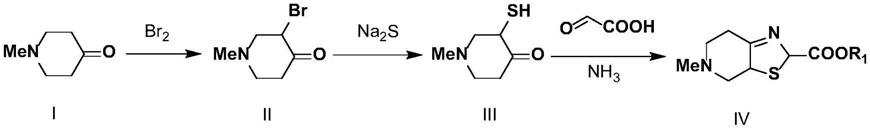
第二方面,本发明提供了一种制备甲苯磺酸艾多沙班式为(ii)的中间体的制备方法,所述式(ii)化合物的合成步骤包括:以化合物n-甲基-4-哌啶酮为原料,与溴素发生取代反应,生成式(ii)所示化合物3-溴-n-甲基-4-哌啶酮。
[0013]
作为优选,在与溴素反生取代反应时的反应温度为20~25℃,在此温度下能够有效防止副反应的发生,使得最终式(ii)所示化合物的收率保持在较高值。
[0014]
第三方面,本发明提供了一种制备甲苯磺酸艾多沙班式为(iii)的中间体,其结构式如下:
[0015]
第四方面,本发明提供了一种制备甲苯磺酸艾多沙班式为(iii)的中间体的制备
方法,所述式(iii)化合物的合成步骤包括:式(ii)所示化合物与硫化钠进行硫代反应,得到式(iii)所示化合物,反应式如下:
[0016]
作为优选,硫代反应过程中保持反应温度为-5~0℃,反应时间20~60min。
[0017]
第五方面,本发明提供了一种制备甲苯磺酸艾多沙班式为(iv)的中间体,其结构式如下:其中,r1=nh4或h。
[0018]
第六方面,本发明提供了一种制备甲苯磺酸艾多沙班式为(iv)的中间体的制备方法,以化合物n-甲基-4-哌啶酮为原料,与溴素发生取代反应,生成式(ii)所示化合物;式(ii)所示化合物与硫化钠进行硫代反应,得到式(iii)所示化合物;式(iii)所示化合物与乙醛酸进行环合反应,得到式(iv)所示化合物,反应式如下:下:其中,r1=nh4或h。
[0019]
第七方面,本发明提供了一种制备甲苯磺酸艾多沙班式为(iv-1)的中间体,其结构式如下:
[0020]
第八方面,本发明提供了一种制备甲苯磺酸艾多沙班式为(iv-1)的中间体的制备方法,所述(iv-1)所示化合物的合成步骤包括:式(iii)所示化合物与乙醛酸以及氨进行环合反应,得到式(iv-1)所示化合物n-甲基-4-哌啶并二氢噻唑甲酸铵,反应式如下:
[0021]
第九方面,一种制备甲苯磺酸艾多沙班式为(iv-2)的中间体,其结构式如下:
[0022]
第十方面,一种制备甲苯磺酸艾多沙班式为(iv-2)的中间体的制备方法,所述式(iv-2)所示化合物的合成步骤包括:式(iv-1)所示化合物酸化后,得到式(iv-2)所示化合物。
[0023]
作为优选,环合反应过程中,保持反应温度为20~25℃,搅拌反应18~36h。
[0024]
第十一方面,本发明提供了一种制备甲苯磺酸艾多沙班式为(v)的中间体,其特征在于,其结构式如下:其中,r2选自c1-c6的脂肪烃、苯基或取代苯基。
[0025]
第十二方面,本发明提供了一种制备甲苯磺酸艾多沙班式为(v)的中间体的制备方法,所述式(v)所示化合物的合成步骤包括:式(iv)所示化合物与醇在酸催化下发生酯化反应,得到式(v)所示化合物,其反应式如下:其中,r1选自nh4或h;r2选自c1-c6的脂肪烃、苯基或取代苯基。
[0026]
作为优选,所述醇为甲醇、乙醇、丙醇、异丙醇、苯酚等中的任意一种。
[0027]
作为优选,所述酸为浓硫酸。
[0028]
作为优选,酯化反应过程中,保持反应温度为20~25℃,搅拌反应8~12h,反应结束后使用碳酸氢钠中和至ph为7-8。
[0029]
第十三方面,本发明提供了一种制备甲苯磺酸艾多沙班式为(v-1)所述的中间体,其结构式如下:
[0030]
第十四方面,本发明提供了一种制备甲苯磺酸艾多沙班式为(v-1)的中间体的制备方法,所述(v-1)所示化合物的合成步骤包括:式(iv-1)所示化合物与乙醇在酸催化下发
生酯化反应,得到式(v-1)化合物,其反应式如下:
[0031]
第十五方面,本发明提供了一种制备甲苯磺酸艾多沙班式为(vi)的中间体,其结构式如下:其中,r2选自c1-c6的脂肪烃、苯基或取代苯基。
[0032]
第十六方面,本发明提供了一种制备甲苯磺酸艾多沙班式为(vi)的中间体的制备方法,所述式(vi)所示化合物的合成步骤包括:由式(iv)所示化合物与醇发生酯化反应得到式(v)所示化合物,再反应得到式(vi)所示化合物;或者直接由式(iv)所示化合物在醇与酸的作用下反应得到式(vi)所示化合物,其反应式如下:或者,其中,r1选自nh4或h;r2选自c1-c6的脂肪烃、苯基或取代苯基。
[0033]
第十七方面,本发明提供了一种制备甲苯磺酸艾多沙班式为(vi-1)的中间体,其结构式如下:
[0034]
第十八方面,本发明提供了一种制备甲苯磺酸艾多沙班式为(vi-1)的中间体的制备方法,所述式(vi-1)所示化合物的合成步骤包括:由式(iv-1)所示化合物与乙醇酯化得到式(v-1)所示化合物,再反应得到式(vi-1)所示化合物;或者直接由式(iv-1)所示化合物在乙醇与酸的作用下反应得到式(vi-1)所示化合物,其反应式如下:
或者,
[0035]
第十九方面,本发明提供了一种制备甲苯磺酸艾多沙班式为(vii)的中间体的制备方法,所述式(vii)所示化合物的合成步骤包括:以化合物n-甲基-4-哌啶酮为原料,与溴素发生取代反应,生成式(ii)所示化合物;式(ii)所示化合物与硫化钠进行硫代反应,得到式(iii)所示化合物;式(iii)所示化合物与乙醛酸进行环合反应,得到式(iv)所示化合物;式(iv)所示化合物与醇发生酯化反应得到式(v)所示化合物,再反应得到式(vi)所示化合物;或者直接由式(iv)所示化合物在醇与酸的作用下反应得到式(vi)所示化合物;式(vi)所示化合物经碱水解、酸中和后得到式(vii)所示化合物;其反应式如下:其中,r1选自nh4或h;r2选自c1-c6的脂肪烃、苯基或取代苯基。
[0036]
第二十方面,本发明提供了一种制备甲苯磺酸艾多沙班式为(vii)的中间体的制备方法,所述式(vii)所示化合物的合成步骤包括:以化合物n-甲基-4-哌啶酮为原料,与溴素发生取代反应,生成式(ii)所示化合物;式(ii)所示化合物与硫化钠进行硫代反应,得到式(iii)所示化合物;式(iii)所示化合物与乙醛酸进行环合反应,得到式(iv-1)所示化合物;式(iv)所示化合物与醇发生酯化反应得到式(v-1)所示化合物,再反应得到式
(vi-1)所示化合物;或者直接由式(iv-1)所示化合物在醇与酸的作用下反应得到式(vi-1)所示化合物;式(vi-1)所示化合物经碱水解、酸中和后得到式(vii)所示化合物;其反应式如下:
[0037]
因此,本发明具有以下有益效果:(1)简单易行:本发明所采用合成路线的各个原料易制备,且各步产物的稳定性大大增加,在工厂生产时的运输稳定性、储存稳定性、使用稳定性都得到大幅增加,这使得反应中各个中间体的质量控制更加方便且降低了对设备的要求,加上在合成路线中的无复杂反应,这得整个合成路线在兼具简单的同时还具有易操作的优点;(2)成本低:本发明合成路线的各个原料及反应中都没有使用贵重的催化剂或辅助试剂,保证了各步工艺产生的三废污染少且易于处理,能使得反应在兼具绿色环保的同时也大大降低了成本;(3)收率高:采用本发明甲苯磺酸艾多沙班的新的中间体及其制备方法中,每步反应的收率都较高,后处理简单方便,更利于大批量生产时的调控。
附图说明
[0038]
图1为化合物(ii)的h-nmr谱图。
[0039]
图2为化合物(iii)的质谱谱图。
[0040]
图3为化合物(iv-1)的h-nmr谱图。
[0041]
图4为化合物(v-1)的h-nmr谱图。
[0042]
图5为化合物(vi-1)的h-nmr谱图。
[0043]
图6为化合物(vi-1)的质谱谱图。
[0044]
图7为化合物(vii)的h-nmr谱图。
具体实施方式
[0045]
下面结合具体实施例对本发明做进一步描述。本领域普通技术人员在基于这些说明的情况下将能够实现本发明。此外,下述说明中涉及到的本发明的实施例通常仅是本发明一部分的实施例,而不是全部的实施例。因此,基于本发明中的实施例,本领域普通技术
人员在没有做出创造性劳动的前提下所获得的所有其他实施例,都应当属于本发明保护的范围。
[0046]
实施例1本甲苯磺酸艾多沙班的中间体4,5,6,7-四氢-5-甲基-噻唑并[5,4-c]吡啶-2-羧酸采用以下工艺步骤得到。
[0047]
(1)化合物(ii)的合成反应瓶中加入40g冰醋酸,温度25℃以下,滴加n-甲基-4-哌啶酮22.6g(199.7mmol),滴加48%氢溴酸水溶液34g(201.7mmol)与冰醋酸40g配制成的溶液;温度20℃以下,滴加溴素32g(200.2mmol)与冰醋酸30g配制成的溶液,滴完20-25℃保温搅拌过夜,过滤、滤饼用乙酸乙酯淋洗,收集固体,烘干,得到52g化合物ii,收率95.4%。
[0048]
(2)化合物(iii)的合成反应瓶中加入10g水和3.7g(22.0mmol)的na2s.5h2o,搅拌溶解,温度降至-5-0℃,分批加入3-溴-n-甲基-4-哌啶酮5g(18.3mmol),滴完-5-0℃保温搅拌反应0.5小时,过滤、滤饼用水淋洗,收集固体,烘干,得到2.4g化合物iii,收率90%。
[0049]
(3)化合物(iv-1)的合成反应瓶中加入甲醇45g,乙醛酸一水合物2.5g(27mmol)和4g(27mmol)的化合物iii,搅拌溶解,温度控制30℃以下,滴加7m的氨/甲醇溶液16ml(108mmol),滴完20-25℃保温搅拌反应24小时,反应液于40℃减压浓缩,得到6.4g固体粗品化合物iv-1,收率50%。
[0050]
(4)化合物(v-1)的合成反应瓶中加入乙醇10ml,化合物iv粗品0.6g,搅拌溶解,温度控制0-5℃,滴加浓硫酸0.5ml(5mmol),滴完缓慢升到20-25℃保温搅拌反应10小时,用饱和碳酸氢钠溶液中和ph=7-8,反应于50℃减压浓缩,浓缩物加入甲醇10ml搅拌、过滤、滤液于40℃减压浓缩,得到
化合物v-1粗品0.6g。
[0051]
(5)化合物(vi-1)的合成反应瓶中加入甲醇20ml,化合物iv-1粗品0.6g,fe2(so4)3固体0.6g,搅拌溶解,加热升温回流反应10小时,过滤、滤液于50℃减压浓缩,浓缩物装上硅胶柱,溶剂甲醇/二氯甲烷=1:20比例洗脱纯化,得到0.2g化合物vi-1。
[0052]
(6)化合物(vii)的合成反应瓶中加入1.3g(5.8mmol)化合物vi-1,甲醇10ml搅拌溶解,温度降至0-5℃,滴加1m的氢氧化钠溶液6ml(6mmol),滴完0-5℃保温搅拌1.5小时,反应液用0.5m的稀盐酸调ph=3-4,反应液于40-45℃减压浓缩,蒸出甲醇,浓缩物过滤、滤饼用乙醇3ml水6ml的混合溶液淋洗,过滤、滤饼烘干,得到化合物vii。
[0053]
实施例2本甲苯磺酸艾多沙班的中间体4,5,6,7-四氢-5-甲基-噻唑并[5,4-c]吡啶-2-羧酸采用以下工艺步骤得到。
[0054]
(1)化合物(ii)的合成反应瓶中加入30g冰醋酸,温度25℃以下,滴加n-甲基-4-哌啶酮17.0g(149.8mmol),滴加48%氢溴酸水溶液25.5g(151.3mmol)与冰醋酸30g配制成的溶液;温度20℃以下,滴加溴素24g(150.2mmol)与冰醋酸22.5g配制成的溶液,滴完20-25℃保温搅拌过夜,过滤、滤饼用乙酸乙酯淋洗,收集固体,烘干,得到40g化合物ii,收率97.9%。
[0055]
(2)化合物(iii)的合成反应瓶中加入10g水和3.36g(20.0mmol)的na2s.5h2o,搅拌溶解,温度降至-5-0℃,分批加入3-溴-n-甲基-4-哌啶酮4.55g(16.7mmol),滴完-5-0℃保温搅拌反应1小时,过滤、滤饼用水淋洗,收集固体,烘干,得到2.21g化合物iii,收率91.2%。
[0056]
(3)化合物(iv-1)的合成反应瓶中加入甲醇45g,乙醛酸一水合物2.5g(27mmol)和4g(27mmol)的化合物iii,搅拌溶解,温度控制30℃以下,滴加7m的氨/甲醇溶液16ml(108mmol),滴完20-25℃保温搅拌反应36小时,反应液于40℃减压浓缩,得到6.4g固体粗品化合物iv-1,收率50%。
[0057]
(4)化合物(v-2)的合成
反应瓶中加入甲醇10ml,化合物iv粗品0.5g,搅拌溶解,温度控制0-5℃,滴加浓硫酸0.5ml(5mmol),滴完缓慢升到20-25℃保温搅拌反应8小时,用饱和碳酸氢钠溶液中和ph=7-8,反应于50℃减压浓缩,浓缩物加入甲醇10ml搅拌、过滤、滤液于40℃减压浓缩,得到化合物v-2粗品0.5g。
[0058]
(5)化合物(vi-2)的合成反应瓶中加入甲醇20ml,化合物iv-2粗品0.6g,fe2(so4)3固体0.6g,搅拌溶解,加热升温回流反应8小时,过滤、滤液于50℃减压浓缩,浓缩物装上硅胶柱,溶剂甲醇/二氯甲烷=1:20比例洗脱纯化,得到0.2g化合物vi-2。
[0059]
(6)化合物(vii)的合成反应瓶中加入1.17g(5.5mmol)化合物vi-2,甲醇10ml搅拌溶解,温度降至0-5℃,滴加1m的氢氧化钠溶液6ml(6mmol),滴完0-5℃保温搅拌1.5小时,反应液用0.5m的稀盐酸调ph=3-4,反应液于40-45℃减压浓缩,蒸出甲醇,浓缩物过滤、滤饼用乙醇3ml水6ml的混合溶液淋洗,过滤、滤饼烘干,得到化合物vii。
[0060]
实施例3本甲苯磺酸艾多沙班的中间体4,5,6,7-四氢-5-甲基-噻唑并[5,4-c]吡啶-2-羧酸采用以下工艺步骤得到。
[0061]
化合物(ii)、化合物(iii)以及化合物(iv-1)如实施例1所示。
[0062]
(4)化合物(v-3)的合成反应瓶中加入异丙醇10ml,化合物iv-1粗品0.6g,搅拌溶解,温度控制0-5℃,滴加浓硫酸0.5ml(5mmol),滴完缓慢升到20-25℃保温搅拌反应12小时,用饱和碳酸氢钠溶液中和ph=7-8,反应于50℃减压浓缩,浓缩物加入甲醇10ml搅拌、过滤、滤液于40℃减压浓缩,得到化合物v-3粗品0.55g。
[0063]
(5)化合物(vi-3)的合成
反应瓶中加入甲醇20ml,化合物iv-3粗品0.6g,fe2(so4)3固体0.6g,搅拌溶解,加热升温回流反应12小时,过滤、滤液于50℃减压浓缩,浓缩物装上硅胶柱,溶剂甲醇/二氯甲烷=1:20比例洗脱纯化,得到0.22g化合物vi-3。
[0064]
(6)化合物(vii)的合成反应瓶中加入1.32g(5.5mmol)化合物vi-3,甲醇10ml搅拌溶解,温度降至0-5℃,滴加1m的氢氧化钠溶液6ml(6mmol),滴完0-5℃保温搅拌1.5小时,反应液用0.5m的稀盐酸调ph=3-4,反应液于40-45℃减压浓缩,蒸出甲醇,浓缩物过滤、滤饼用乙醇3ml水6ml的混合溶液淋洗,过滤、滤饼烘干,得到化合物vii。
[0065]
实施例4本甲苯磺酸艾多沙班的中间体4,5,6,7-四氢-5-甲基-噻唑并[5,4-c]吡啶-2-羧酸采用以下工艺步骤得到。
[0066]
化合物(ii)、化合物(iii)以及化合物(iv-1)如实施例1所示。
[0067]
(1)化合物(vi-1)的合成反应瓶中加入乙醇20ml,含量47%的化合物iv-1粗品1.7g(4mmol),搅拌溶解,温度控制20-25℃,滴加浓硫酸1.1g(11mmol),滴完20-25℃保温搅拌反应24小时,用饱和碳酸氢钠溶液中和ph=7-8,过滤、滤液于50℃减压浓缩,浓缩物加入乙酸乙酯和水,搅拌分层,有机相于40℃减压浓缩,得到0.7g化合物vi-1,收率77.8%。
[0068]
(2)化合物(vii)的合成反应瓶中加入1.3g(5.8mmol)化合物vi-1,甲醇10ml搅拌溶解,温度降至0-5℃,滴加1m的氢氧化钠溶液6ml(6mmol),滴完0-5℃保温搅拌1.5小时,反应液用0.5m的稀盐酸调ph=3-4,反应液于40-45℃减压浓缩,蒸出甲醇,浓缩物过滤、滤饼用乙醇3ml水6ml的混合溶液淋洗,过滤、滤饼烘干,得到化合物vii。
[0069]
实施例5本甲苯磺酸艾多沙班的中间体4,5,6,7-四氢-5-甲基-噻唑并[5,4-c]吡啶-2-羧酸采用以下工艺步骤得到。
[0070]
化合物(ii)、化合物(iii)的合成如实施例1所示。
[0071]
(1)化合物(iv-2)的合成
反应瓶中加入甲醇45g,乙醛酸一水合物2.5g(27mmol)和4g(27mmol)的化合物iii,搅拌溶解,温度控制30℃以下,滴加7m的氨/甲醇溶液16ml(108mmol),滴完20-25℃保温搅拌反应24小时,反应结束后滴加盐酸调节ph值至6.5,反应液于40℃减压浓缩,得到6.2g固体粗品化合物iv-2,收率48.5%。
[0072]
(2)化合物(v-1)的合成反应瓶中加入乙醇10ml,化合物iv-2粗品0.6g,搅拌溶解,温度控制0-5℃,滴加浓硫酸1ml(10mmol),滴完缓慢升到20-25℃保温搅拌反应12小时,用饱和碳酸氢钠溶液中和ph=7-8,反应于50℃减压浓缩,浓缩物加入甲醇10ml搅拌、过滤、滤液于40℃减压浓缩,得到化合物v-1粗品0.6g。
[0073]
(3)化合物(vi-1)以及化合物(vii)的合成同实施例1所示。
[0074]
实施例6本甲苯磺酸艾多沙班的中间体4,5,6,7-四氢-5-甲基-噻唑并[5,4-c]吡啶-2-羧酸采用以下工艺步骤得到。
[0075]
化合物(iv-2)的合成如实施例5所示。
[0076]
(2)化合物(vi-1)的合成反应瓶中加入乙醇20ml,含量50%的化合物iv-2粗品1.6g(4mmol),搅拌溶解,温度控制20-25℃,滴加浓硫酸1.5g(15mmol),滴完20-25℃保温搅拌反应24小时,用饱和碳酸氢钠溶液中和ph=7-8,过滤、滤液于50℃减压浓缩,浓缩物加入乙酸乙酯和水,搅拌分层,有机相于40℃减压浓缩,得到0.73g化合物vi-1,收率79.9%。
[0077]
化合物(vii)的合成同实施例1所示。
[0078]
上述对本技术中涉及的发明的一般性描述和对其具体实施例的描述不应理解为是对该发明技术方案构成的限制。本领域所属技术人员根据本技术的公开,可以在不违背所涉及的发明构成要素的前提下,对上述一般性描述或/和实施例中的公开技术特征进行增加、减少或组合,形成属于本技术保护范围之内的其它的技术方案。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1