一种二氧化碳诱导的纳米孔氢键有机框架材料及其制备方法与应用
1.本发明涉及一种二氧化碳诱导的纳米孔氢键有机框架材料及其制备方法与应用,属于氢键有机框架材料制备领域。
背景技术:
2.氢键有机框架(hydrogen-bonded organic framework,hof)作为一种由有机配体通过氢键相互作用自组装而成的新型微孔晶体材料,近年来受到了广泛的关注。hofs有很多固有的优点,如结晶度高、比表面积大、合成条件温和、毒性低、孔径可调、易重结晶再生等。由于这些独特的性质,hofs在气体储存和分离、手性分离、化学传感、质子传导、光致发光、药物输送、酶封装、催化等方面具有广泛的应用。尤其是hofs在催化方面的应用吸引了广泛关注。然而,由于其无金属的特性,单一hof的催化活性较低。因此,通常需要在hof中引入另一种催化活性相,如金属、半导体、氧化还原活性单元或金属配合物等,以提高其催化性能。但在hofs中引入第二相会导致催化剂合成和相分离的复杂。因此,开发具有高催化性能的单一hof催化剂具有重要的意义,但仍具有挑战性。
技术实现要素:
3.本发明的目的是提供一种co2诱导的纳米孔hof材料,所制备的纳米孔p-pfc-1材料能在温和条件下光催化胺氧化偶联为亚胺,具有优异的催化活性。
4.本发明所提供的纳米孔hof材料的制备方法,包括如下步骤:
5.s1、将有机配体溶于有机溶剂中,然后加入溶剂与水的混合液,得到反应体系;
6.s2、向所述反应体系中泵入co2,在搅拌条件下进行反应即得到所述纳米孔hof材料。
7.上述的制备方法中,步骤s1中,所述有机配体可为1,3,6,8-四(4-羧基苯)芘(h4tbapy)、2,4,6-三(4-羧基苯基)-1,3,5-三嗪、1,2,4,5-四(4-羧基苯基)苯或5,5
′‑
二(乙酰氨基)-二间苯甲酸;
8.当采用所述1,3,6,8-四(4-羧基苯)芘作为有机配体时,得到的纳米孔hof材料为p-pfc-1材料;
9.所述有机溶剂可为n,n-二甲基甲酰胺、四氢呋喃、二甲基亚砜或甲氧基苯;
10.所述溶剂可为乙醇、甲醇、异丙醇或正己烷,
11.所述混合液中,所述溶剂与所述水的体积比可为1:0.5~3,优选1:1;
12.所述混合液可使产物析出。
13.将所述反应体系转移至反应釜中再泵入所述co2。
14.上述的制备方法中,步骤s1中,所述有机溶剂的用量可为:10~120ml/1mol所述有机配体,优选:50~60ml/1mol所述有机配体;
15.所述混合液中所述溶剂的用量可为:10~120ml/1mol所述有机配体,优选:40~
50ml/1mol所述有机配体;
16.所述混合液中所述水的用量可为:10~120ml/1mol所述有机配体,优选:40~50ml/1mol所述有机配体。
17.上述的制备方法中,步骤s2中,泵入所述co2至体系内压力为0~10mpa,但不为零,优选:1~8.1mpa、1.53~8.02mpa、3.12~8.02mpa、4.50~8.02mpa、6.64~8.02mpa、1.53mpa、3.12mpa、4.50mpa、6.64mpa或8.02mpa;
18.所述反应的温度为0~50℃,时间为1~16h,优选在35℃下反应12h。
19.本发明方法制备得到的纳米孔hof材料能够作为光催化剂,在温和条件下光催化胺氧化偶联为亚胺化合物;
20.温和条件指的是:空气氛围、室温和无需额外氧化剂、光敏剂、牺牲剂和助催化剂的条件。
21.本发明还提供了一种由胺制备亚胺化合物的方法,包括如下步骤:
22.在所述纳米孔hof材料的催化下,在空气中胺经偶联反应即得所述亚胺化合物;
23.所述胺为苄胺或取代苄胺;
24.所述取代苄胺中的取代基可为c1~c5的烷基、c1~c5的烷氧基、c1~c5的氟代烷氧基、卤素或苄基;其中,烷基优选为c1~c4的烷基,烷氧基优选为c1~c3的烷氧基,氟代烷氧基优选为c1~c3的氟代烷氧基,卤素优选为氟或溴;
25.所述取代苄胺优选4-甲基苄胺、3-甲基苄胺、2-甲基苄胺、4-甲氧基苄胺、2-甲氧基苄胺、4-溴苄胺、4-氟苄胺、二苄胺、4-叔丁基苄胺和4-(三氟甲氧基)苄胺中任一种;
26.所述纳米孔hof材料与所述胺的配比为:1~5mg:0.80mmol;
27.所述偶联反应的温度为10~40℃,时间为0~2h,但不为零。
28.所述偶联反应采用的溶剂可为乙腈、n,n-二甲基甲酰胺、二甲基亚砜、正辛烷、二氯甲烷、四氢呋喃或水。
29.本发明提供的纳米孔hof材料(p-pfc-1材料),具有独特的周期性纳米孔结构(纳米孔宽度~13nm)、优秀的光吸收性能、有效的电子-空穴对分离效率和优异的光电性能,在温和条件下对光催化胺氧化偶联为亚胺具有优异的催化活性。
附图说明
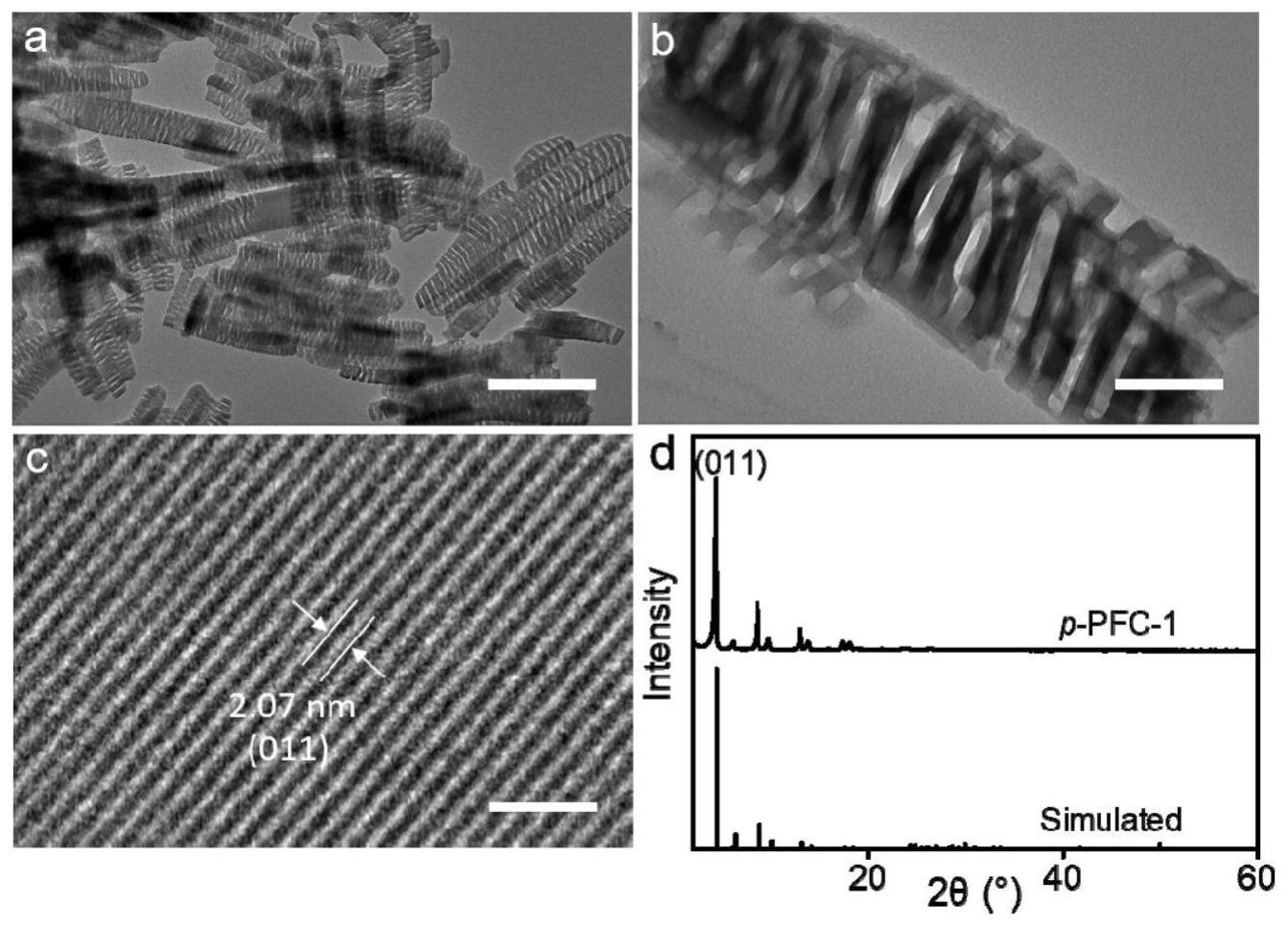
30.图1为本发明实施例1制备的p-pfc-1的透射电子显微镜图像(图1a和图1b,图1a的标尺为800nm,图1b的标尺为100nm)、冷冻高分辨透射电子显微镜图像(图1c,标尺为10nm)、x射线衍射图谱(图1d)。
31.图2为本发明对比例1制备的o-pfc-1的x射线衍射图谱(图2a)和透射电子显微镜图像(图2b的标尺为200nm)。
32.图3为本发明实施例1制备的p-pfc-1、对比例1制备的o-pfc-1和配体h4tbapy的傅里叶变换红外光谱图(图3a)、p-pfc-1和o-pfc-1的红外拟合曲线(图3b)、ck-边(图3c)和o k-边(图3d)的同步辐射软边x射线吸收近边结构光谱。
33.图4为本发明实施例1制备的p-pfc-1和对比例1制备的o-pfc-1光催化苄胺氧化偶联反应的转化率(线)和选择性(点)随时间变化的曲线(图4a)和不同时间所对应的n-苄烯丁胺生成速率(图4b)、p-pfc-1光催化氧化偶联不同苄胺衍生物的转化率和选择性(图4c)。
34.图5为本发明实施例3中反应时间固定为12h时、不同co2压力下得到样品的x射线衍射谱图。
35.图6为本发明实施例3中反应时间固定为12h时、不同co2压力下得到样品的透射电子显微镜图像(图6的标尺为100nm)。
36.图7为本实施例4中反应时间固定为12h时、不同co2压力下合成的pfc-1材料在温和条件下光催化苄胺氧化偶联为n-苄烯丁胺的转化率随co2压力变化的曲线。
具体实施方式
37.下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
38.下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
39.实施例1、纳米孔hof材料p-pfc-1的制备
40.将配体h4tbapy溶于n,n-二甲基甲酰胺中,接着加入乙醇和去离子水的混合溶液(v:v=1:1),在35℃下快速泵入co2到反应体系中,在搅拌条件下反应12h,即得。
41.具体操作步骤为:在超声条件下将h4tbapy(50mg,0.073mmol)溶于4ml n,n-二甲基甲酰胺中,得到均相溶液。在上述溶液中加入7ml乙醇和去离子水的混合溶液(v:v=1:1)。将上述混合物转移至15ml聚四氟乙烯内衬的不锈钢反应釜中,密封反应釜。快速将co2泵入系统,在35℃下边充气边搅拌,直至达到所需压力(8.02mpa)。在搅拌条件下反应12h后,泄压除去co2,离心收集黄色固体。用二氯甲烷和去离子水各洗涤3次,离心分离。最后在-70℃的真空冷冻干燥箱中冷冻干燥24h后即获得p-pfc-1。
42.将p-pfc-1材料分别进行透射电子显微镜和冷冻高分辨透射电子显微镜、x射线衍射分析,结果如图1所示。图1a和图1b所示的透射电子显微镜照片呈现出具有周期性纳米孔的纳米棒形貌,纳米棒的长度约为150~1500nm,宽度约为70~200nm,纳米孔的平均孔宽度~13nm。图1c的冷冻高分辨透射电子显微镜照片表明,晶格间距为2.07nm,对应于p-pfc-1的(011)晶面。图1d的x射线衍射图谱和衍射峰的相对强度与pfc-1模拟的衍射结果相吻合,说明成功合成了p-pfc-1。其在4.3
°
处有一个明显的主峰,对应p-pfc-1的(011)晶面,这与冷冻高分辨透射电子显微镜的结果一致。
43.对比例1、o-pfc-1材料的制备
44.将配体h4tbapy溶于n,n-二甲基甲酰胺中,接着加入乙醇和去离子水的混合溶液(v:v=1:1),在35℃,搅拌条件下反应12h,即得。
45.具体操作步骤为:在超声条件下将h4tbapy(50mg,0.073mmol)溶于4ml n,n-二甲基甲酰胺中,得到均相溶液。在上述溶液中加入7ml乙醇和去离子水的混合溶液(v:v=1:1)。将上述混合物转移至15ml聚四氟乙烯内衬的不锈钢反应釜中,密封反应釜。将反应釜置于35℃的水浴锅中搅拌反应12h,离心收集黄色固体。用二氯甲烷和去离子水各洗涤3次,离心分离。最后在-70℃的真空冷冻干燥箱中冷冻干燥24h后即获得o-pfc-1。
46.将o-pfc-1材料分别进行x射线衍射和透射电子显微镜表征,结果如图2所示。图2a的x射线衍射图谱和衍射峰的相对强度与pfc-1模拟的衍射图谱结果相吻合,说明成功合成了o-pfc-1。透射电子显微镜照片表明o-pfc-1呈现出纳米棒的形貌(图2b)。
47.将p-pfc-1和o-pfc-1材料分别进行傅里叶变换红外光谱、同步辐射软边x射线吸收近边结构光谱分析,结果如图3所示。
48.利用傅里叶变换红外光谱研究了p-pfc-1的化学和结构信息。以o-pfc-1和配体h4tbapy作为参考样品。p-pfc-1的傅里叶变换红外光谱分别在1381和1695cm-1
处分别表现出-oh(δ)的弯曲振动和羧基中c=o的伸缩振动特征,与h4tbapy(1383和1698cm-1
)的峰位置相比,这两个振动峰向低波数处移动(图3a),这归因于p-pfc-1的羧基中c=o和-oh之间形成了氢键,说明p-pfc-1的生成。p-pfc-1在1720cm-1
处的吸收峰对应于游离羧基中c=o的伸缩振动。通过对ir谱图进行拟合(图3b),可得p-pfc-1中缔合羧基与游离羧基的相对含量之比为2.0,远低于o-pfc-1(4.3)。结果表明,与o-pfc-1相比,p-pfc-1中羧基配体的缔合程度较低,自由羧基较多。
49.此外,采用同步辐射软边x射线吸收近边结构光谱(xanes)对材料的c k-边和o k-边进一步表征,结果分别如图3c和3d所示。同步辐射软k-边x射线吸收近边结构光谱(xanes)结果表明,p-pfc-1的c k-边xanes谱在284.9、288.3和291-293ev处表现出三个主要的特征峰(图3c)。284.9ev处的共振峰对应于芳香族碳的c1s
→
π*跃迁,291-293ev处的高能量特征峰来自于碳的1s
→
σ*跃迁。288.3ev处的共振峰对应于羧基碳(-cooh)的c1s
→
π*跃迁。p-pfc-1在288.3ev处的信号强度明显低于o-pfc-1,表明p-pfc-1的未占用态密度降低,电子密度增加。上述分析表明,p-pfc-1比o-pfc-1有更多的自由羧基。
50.对p-pfc-1的o k-边xanes谱(图3d),在534.5ev处的峰归因于羧基中1s
→
σ*(-oh)的跃迁,535ev以上的宽特征峰来自o1s
→
σ*(c=o)的跃迁。532.1ev处的特征峰来自与芳香环的边键合的羧基中o1s
→
π*(c=o)的跃迁。值得注意的是,与o-pfc-1相比,p-pfc-1在532.1ev处的峰强度显著降低。这表明p-pfc-1中羰基与芳香环的边键合能力减弱。
51.上述结果表明,p-pfc-1中的游离羧基含量高于o-pfc-1,这与ft-ir结果一致。
52.实施例2、pfc-1材料的光催化应用
53.实施例1制备的p-pfc-1材料和对比例1制备的的o-pfc-1材料光催化苄胺氧化偶联为n-苄烯丁胺的反应物随反应时间的转化率、选择性和产物n-苄烯丁胺的生成速率,实施例1的p-pfc-1材料光催化苄胺氧化偶联的底物拓展测试。
54.具体步骤为:
55.将实施例1制备的纳米孔p-pfc-1和对比例1制备的o-pfc-1材料用于温和条件下光催化苄胺氧化偶联为n-苄烯丁胺的反应。在100ml圆底烧瓶中,将2mg hof材料和0.80mmol苄胺分散在5ml有机溶剂乙腈中。混合物在空气中搅拌30min。用橡胶塞塞住圆底烧瓶,用注射器针头连接空气,用300w氙灯(380《λ《780nm)照射。然后通过循环水装置将反应系统温度保持在25℃。反应0~2h,产物用核磁共振氢谱检测。并在相同的实验条件下,与o-pfc-1的催化性能对比,反应结果如图4所示。
56.图4为p-pfc-1和o-pfc-1在光催化苄胺氧化偶联反应中不同时间所对应的转化率和选择性(图4a),产物n-苄烯丁胺的生成速率(图4b),实施例1的p-pfc-1材料光催化苄胺氧化偶联的底物拓展测试(图4c)。
57.图4a所示的o-pfc-1和p-pfc-1的光催化性能对比表明,两者都能在温和条件下催化苄胺的氧化偶联,对n-苄烯丁胺的选择性均》99%。很明显,p-pfc-1的催化性能显著优于o-pfc-1的催化性能。如p-pfc-1能够在1h内高效地将苄胺转化为产物n-苄烯丁胺,n-苄烯丁胺的生成速率为467.2mmol g-1
h-1
(图4b)。而此时o-pfc-1的转化率为71.7%,n-苄烯丁胺的生成速率为332.7mmol g-1
h-1
。同时,p-pfc-1可以在温和条件下催化各种苄胺衍生物
生成相应的亚胺,具有较高的转化率和选择性(图4c)。
58.实施例3、不同co2压力下制备p-pfc-1材料
59.本实施例的反应时间固定为12h时,不同co2压力下得到的各种pfc-1材料的合成方法如下:
60.在超声条件下将h4tbapy(50mg,0.073mmol)溶于4ml n,n-二甲基甲酰胺中,得到均相溶液。在上述溶液中加入7ml乙醇和去离子水的混合溶液(v:v=1:1)。将上述混合物转移至15ml聚四氟乙烯内衬的不锈钢反应釜中,密封反应釜。快速将co2泵入系统,在35℃下边充气边搅拌,直至达到所需压力(1.53、3.12、4.50、6.64mpa)。在搅拌条件下反应12h后,泄压除去co2,离心收集黄色固体。用二氯甲烷和去离子水各洗涤3次,离心分离。最后在真空冷冻干燥箱中冷冻干燥24h后即获得不同co2压力下制备的pfc-1材料。
61.将不同co2压力制得的pfc-1材料进行x射线衍射图谱表征,结果如图5所示。
62.图5为本实施例中不同co2压力下得到样品的x射线衍射图谱。
63.从图5可以看出,在1.53、3.12、4.50和6.64mpa压力下得到的产物x射线衍射图谱和衍射峰的相对强度与pfc-1模拟的衍射图谱结果一致,说明在不同co2压力下都能够形成pfc-1。
64.将不同co2压力下得到的pfc-1材料进行透射电子显微镜表征,结果如图6所示。
65.图6为本实施例中不同co2压力下得到样品的透射电子显微镜图像,从图6可以看出,随着co2压力的升高,hof纳米棒的多孔性增强。
66.实施例4、不同co2压力下制备的p-pfc-1材料的光催化性能
67.实施例3得到的不同co2压力下制备的pfc-1材料对光催化苄胺氧化偶联为n-苄烯丁胺反应转化率的影响。
68.具体步骤为:
69.将实施例3制备的不同co2压力下的pfc-1材料用于光催化苄胺氧化偶联为n-苄烯丁胺反应。在100ml圆底烧瓶中,将2mg不同co2压力下的各种pfc-1材料和0.80mmol苄胺分散在5ml有机溶剂乙腈中。混合物在空气中搅拌30min。用橡胶塞塞住圆底烧瓶,用注射器针头连接空气,用300w氙灯(380《λ《780nm)照射。然后通过循环水装置将反应系统温度保持在25℃。反应0~2h,产物用核磁共振氢谱检测。反应结果如图7所示。结果表明,随着co2压力的增大,其催化效率逐渐升高,在合成时间为12h时达到最大。
70.上述内容仅为对本发明所作的举例说明。本发明所述技术领域的技术人员可以对所描述的实施例进行修改、补充或替换,如果不偏离本发明的构思范围,均应视为属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1