一种维格列汀关键中间体的连续制备方法与流程
1.本发明涉及一种维格列汀关键中间体(s)-1-(2-氯乙酰基)吡咯烷-2-甲腈的合成方法,属于有机合成技术领域。
背景技术:
2.维格列汀(vildagliptin),化学式为(2s)-1-{2-[((3-羟基-1-金刚烷基)氨基]乙酰基}-吡咯烷-2甲腈,临床药物是口服给药的二胎基肽酶
‑ⅳ
(dpp
‑ⅳ
),原研为瑞士诺华制药有限公司,于2008年获准在欧盟上市,用于治疗2-型糖尿病。(s)-1-(2-氯乙酰基)吡咯烷-2-甲腈(1)是合成维格列汀的关键中间体,其结构式如下:
[0003][0004]
该中间体目前的合成路线主要分为两类:
[0005]
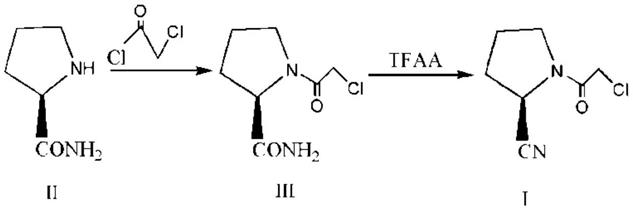
第一类以原研专利wo2000034241、wo2010022690为代表的的工艺路线,以l-脯氨酰胺(化合物ⅱ)为起始原料,直接进行氯乙酰化和腈化脱水,三步反应制得关键中间体ⅰ,工艺合成路线如下:
[0006][0007]
该工艺的起始原料l-脯氨酰胺通常由l-脯氨酸(化合物ⅳ)进行制得,由于l-脯氨酸吡咯环上的氨基的活泼性较高,导致酰胺化过程收率低,也因此导致l-脯氨酰胺的价格昂贵,而且腈化过程使用的脱水剂三氟乙酸酐(tfaa)也较为昂贵,使整体工艺成本较高。专利wo2006100181、wo20080167479等报道了直接以l-脯氨酸为起始原料,先以氯乙酰氯与氨基反应后,再进行酰胺化和腈化反应制得关键中间体ⅰ,工艺合成路线如下:
[0008][0009]
该路线步骤简单,避免了因氨基的活泼性导致的杂质问题,但是氯乙酰基在酰胺化过程中容易部分氨解,且仍然使用了三氟乙酸酐(tfaa)作为脱水剂,工艺收率仅有40%左右,产品杂质也较多,且最终产品的产率和纯度随生产规模的扩大会受到更大影响。
[0010]
第二种方法是专利wo20111101861、wo2014020462a1等报道的先保护l-脯氨酰胺的亚氨基后,再进行脱水及脱保护,最后氯乙酰化制备中间体ⅰ,路线工艺合成路线如下:
[0011][0012]
该路线通过氨基的保护,有效的减少了工艺杂质,使用了价格便宜的氰脲酰氯作为脱水剂,便于工业化生产。但该路线仍使用了昂贵的l-脯氨酰胺作为起始原料,而且后续脱保护过程使用了甲基磺酸,而磺酸类属于基因毒性杂质,会导致最终产品存在潜在的基因毒性残留的风险,不利于药品的安全性。若以l-脯氨酸计算,则工艺过程更加复杂,多次的产品结晶纯化造成了大量的收率损失,不具备工业化条件。
[0013]
目前一种高收率、高质量、成本较低且适合于工业化的(s)-1-(2-氯乙酰基)吡咯烷-2-甲腈的合成工艺亟待开发。
技术实现要素:
[0014]
针对现有技术中存在的技术问题,本发明要解决的技术问题在于提供维格列汀中间体(s)-1-(2-氯乙酰基)吡咯烷-2-甲腈的合成方法,通过实验发现l-脯氨酸中的吡咯环的氨基被保护后,各步反应均有较高的转化率,且各步的副产物均能通过萃取和蒸馏的方法除去,开发一种以l-脯氨酸为起始原料,直接一锅法合成boc-l-脯氨酰胺(化合物ⅶ)的方法,极大简化了boc-l-脯氨酰胺(化合物ⅶ)制备过程,无需中间结晶纯化过程,可以直接合成至中间体ⅰ;还开发了在二氯甲烷体系中高效脱除boc保护基的方法,进一步简化了工艺,而且避免了磺酸类杂质引入。本发明方法所得产品杂质少,纯度高达99.7%,收率可达60%以上(以l-脯氨酸计),适合工业化生产。
[0015]
本发明采取的技术方案如下:一种维格列汀关键中间体的连续制备方法,包括以下步骤:
[0016]
1)制备boc-l-脯氨酰胺:在氮气保护下,向反应容器中l-脯氨酸和二氯甲烷,至l-脯氨酸全部溶解,再加入碱,搅拌控温至0-20℃,滴加二碳酸二叔丁酯至体系溶清,继续控温至0-20℃并滴加氯甲酸乙酯,快速搅拌下,滴加氨水至体系溶清后继续反应3-5h,加入浓
盐酸调节体系至中性,分出有机相用无水硫酸钠干燥,蒸干溶剂,得到boc-l-脯氨酰胺;
[0017]
2)制备(s)-1-(2-氯乙酰基)吡咯烷-2-甲腈:向步骤(1)中所得boc-l-脯氨酰胺中加入二甲基甲酰胺,至boc-l-脯氨酰胺全部溶解后,加入氰脲酰氯,体系大量析出后,继续反应1-3h,降温至0-20℃,加入碳酸氢钠调节体系至中性,滤除固体,真空蒸除溶剂,加入二氯甲烷至全部溶解,加入脱保护溶剂,控温0-20℃滴入氯化亚砜,反应0.5-2h,加入纯化水至体系溶清后,加入碳酸钠调节体系至中性,分离得到有机相和水相,并反萃水相,有机相中加入三乙胺,控温0-10℃滴加氯乙酰氯,搅拌至反应完全,依次使用3-8%碳酸氢钠水溶液和纯化水洗涤有机相,蒸除溶剂,加入乙酸乙酯和环己烷重结晶,得到(s)-1-(2-氯乙酰基)吡咯烷-2-甲腈。
[0018]
工艺路线如下:
[0019][0020]
所述步骤(1)中的碱为氢氧化钠或三乙胺,其用量为l-脯氨酸摩尔量的1-2倍,优选地,温控为10-15℃。
[0021]
所述步骤(1)中氯甲酸乙酯用量为l-脯氨酸摩尔量的1-2倍,优选地,温控为10-15℃,反应时间4h。
[0022]
所述步骤(2)中氰脲酰氯与l-脯氨酸的质量比为0.4-0.8:1,优选地,温控为10-15℃,反应时间1h。
[0023]
所述步骤(2)中二氯甲烷的体积与l-脯氨酸的质量比为7-10:1,氯化亚砜的用量为l-脯氨酸摩尔量的0.9-1.5倍。
[0024]
所述脱保护溶剂为甲醇、乙醇和/或丁醇。
[0025]
所述脱保护溶剂用量为氯化亚砜摩尔量的1-3倍,加入氯化亚砜前温度优选0-10℃;加入氯化亚砜后反应时间优为1h。
[0026]
所述步骤(2)中的三乙胺用量为l-脯氨酸摩尔量的1-2倍,氯乙酰氯l-脯氨酸摩尔量0.8-1.2倍。
[0027]
所述步骤(2)中每100ml乙酸乙酯和环己烷的混合溶液中加入15-30gl-脯氨酸,其中乙酸乙酯:环己烷的体积比为1:4。
[0028]
本发明的创新点在于:
[0029]
1、使用一锅法,以l-脯氨酸为起始原料,合成boc-l-脯氨酰胺(化合物ⅶ)的方法,极大简化了boc-l-脯氨酰胺(化合物ⅶ)制备过程,无需中间结晶纯化过程,可以直接合成至中间体ⅰ。
[0030]
2、开发了在二氯甲烷体系中高效脱除boc保护基的方法,进一步简化了工艺,而且避免了磺酸类杂质引入。
[0031]
本发明的有益效果在于:
[0032]
1、以l-脯氨酸连续制备中间体ⅰ,工艺杂质明显减少,纯度达99.5%以上,总收率达到60%以上,收率及质量优势明显。
[0033]
2、工艺过程原料价格低廉,且工艺操作过程简单,条件温和,适于工业放大。
[0034]
3、脱保护过程采用廉价的氯化亚砜体系,无基因毒杂质引入,规避了下游药品安全风险。
附图说明
[0035]
图1为实施例1的(s)-1-(2-氯乙酰基)吡咯烷-2-甲腈(化合物ⅰ)的hplc图。
具体实施方式
[0036]
以下通过具体实施例进一步说明本发明,但本领域技术人员应当知晓本发明的具体实施例并不以任何方式限制本发明,且在本发明基础上所作出的任何等同替换均落入本发明的保护范围。
[0037]
实施例1:
[0038]
氮气保护下,圆底烧瓶中依次加入100ml二氯甲烷,23g l-脯氨酸,8.8g氢氧化钠,搅拌控温至10-15℃,滴加48g二碳酸二叔丁酯至体系溶清,继续控温10~15℃滴加26g氯甲酸乙酯,快速搅拌下滴加42g 25%氨水,体系溶清后继续反应4小时,加入浓盐酸调节体系至中性,分出有机相用无水硫酸钠干燥,蒸干溶剂,得37.7g无色油状物boc-l-脯氨酰胺。
[0039]
向所得boc-l-脯氨酰胺中加入100ml二甲基甲酰胺,室温加入14.8g氰脲酰氯,体系大量析出后,继续反应2h,降温至0-10℃加入碳酸氢钠中和体系后,滤除固体,真空蒸除溶剂,加入200ml二氯甲烷溶解,加入13g甲醇,控温0~10℃滴入24g氯化亚砜,反应1h,加入100ml纯化水至体系溶清后,低温加入碳酸钠调节体系ph至7~7.5,分离并反萃水相,二氯相中加入24g三乙胺,控温5-10℃滴加14g氯乙酰氯,控温搅拌2h至反应完全,依次使用5%碳酸氢钠水溶液和纯化水洗涤有机相,蒸除溶剂,加入20ml乙酸乙酯和环80ml己烷重结晶,得白色固体(s)-1-(2-氯乙酰基)吡咯烷-2-甲腈21g,纯度99.7%(见图1),收率61%(以l-脯氨酸计)。
[0040]
实施例2:
[0041]
氮气保护下,圆底烧瓶中依次加入100ml二氯甲烷,23g l-脯氨酸,22.3g三乙胺,搅拌控温至5-10℃,滴加48g二碳酸二叔丁酯至体系溶清,继续控温10~15℃滴加26g氯甲酸乙酯,快速搅拌下滴加42g 25%氨水,体系溶清后继续反应4小时,加入浓盐酸调节体系至中性,分出有机相用无水硫酸钠干燥,蒸干溶剂,得37.7g无色油状物boc-l-脯氨酰胺。
[0042]
向所得boc-l-脯氨酰胺中加入100ml二甲基甲酰胺,室温加入14.8g氰脲酰氯,体系大量析出后,继续反应2h,降温至0-10℃加入碳酸氢钠中和体系后,滤除固体,真空蒸除
溶剂,加入200ml二氯甲烷溶解,加入13g甲醇,控温0~10℃滴入24g氯化亚砜,反应1h,加入100ml纯化水至体系溶清后,低温加入碳酸钠调节体系ph至7~7.5,分离并反萃水相,二氯相中加入24g三乙胺,控温5-10℃滴加14g氯乙酰氯,控温搅拌2h至反应完全,依次使用5%碳酸氢钠水溶液和纯化水洗涤有机相,蒸除溶剂,加入20ml乙酸乙酯和环80ml己烷重结晶,得白色固体(s)-1-(2-氯乙酰基)吡咯烷-2-甲腈20.7g,纯度99.5%,收率60%(以l-脯氨酸计)。
[0043]
实施例3:
[0044]
氮气保护下,圆底烧瓶中依次加入100ml二氯甲烷,23g l-脯氨酸,8.8g氢氧化钠,搅拌控温至10-15℃,滴加48g二碳酸二叔丁酯至体系溶清,继续控温10~15℃滴加22.3g氯甲酸乙酯,快速搅拌下滴加42g 25%氨水,体系溶清后继续反应3小时,加入浓盐酸调节体系至中性,分出有机相用无水硫酸钠干燥,蒸干溶剂,得37.7g无色油状物boc-l-脯氨酰胺。
[0045]
向所得boc-l-脯氨酰胺中加入100ml二甲基甲酰胺,室温加入14.8g氰脲酰氯,体系大量析出后,继续反应2h,降温至0-10℃加入碳酸氢钠中和体系后,滤除固体,真空蒸除溶剂,加入200ml二氯甲烷溶解,加入13g甲醇,控温0~10℃滴入24g氯化亚砜,反应1h,加入100ml纯化水至体系溶清后,低温加入碳酸钠调节体系ph至7~7.5,分离并反萃水相,二氯相中加入24g三乙胺,控温5-10℃滴加14g氯乙酰氯,控温搅拌2h至反应完全,依次使用5%碳酸氢钠水溶液和纯化水洗涤有机相,蒸除溶剂,加入20ml乙酸乙酯和环80ml己烷重结晶,得白色固体(s)-1-(2-氯乙酰基)吡咯烷-2-甲腈19.3g,纯度99.7%,收率56%(以l-脯氨酸计)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1