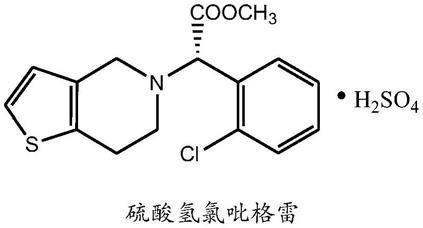
一种硫酸氢氯吡格雷球形晶型I的制备方法与流程
一种硫酸氢氯吡格雷球形晶型i的制备方法
一、技术领域
1.本发明属于药物合成领域,具有涉及一种硫酸氢氯吡格雷晶型i球形颗粒的制备方法。
二、
背景技术:
2.硫酸氢氯吡格雷,是氯吡格雷的硫酸氢盐,英文名clopidogrel hydrogen sulfate,化学名为:(+)-(s)-α-(2-氯苯基)-6,7-二氢噻吩并[3,2-c]吡啶-5(4h)-乙酸甲酯硫酸氢盐。硫酸氢氯吡格雷是一种血小板抑制剂,主要用于预防动脉粥样硬化血栓形成,进而防止缺血性中风和心脏病发作。该产品由法国制药企业赛诺菲-安万特公司研制,于1998年首次在美国和英国上市,并于2001年进入中国市场。硫酸氢氯吡格雷作为临床口服抗凝血一线用药,毒副作用少,其市场占有率远远高于其他药物。
[0003][0004]
硫酸氢氯吡格雷的药用晶型有i晶型和ii晶型两种,其中i晶型为热力学亚稳定晶型,ii晶型为热力学稳定晶型。ⅰ晶型的稳定性低于ii晶型,但是i晶型的溶解度和生物利用度优于ii晶型。目前市售硫酸氢氯吡格雷制剂产品大多为i晶型,而i晶型粉末静电效应强,易吸潮,在制剂粉末直压过程中易发生粘冲,因此可将其制备成流动性好的球形晶体来解决此类问题。
[0005]
专利cn201180072203.6公布了一种硫酸氢氯吡格雷i晶型球颗粒的制备方法,其采用2-丁醇、环己烷的混合溶剂体系制备ⅰ晶型球颗粒。该技术将硫酸的环己烷溶液在冷却条件下缓慢滴加到氯吡格雷游离碱的醇溶液中,由于硫酸需加入到环己烷中,有可能使环己烷变色,从而影响最终产品的颜色。
[0006]
专利wo2011083955公布了一种硫酸氢氯吡格雷i晶型球颗粒的制备方法,其使用2-丁醇、水的混合溶剂体系制备ⅰ晶型球颗粒。实验证明溶剂中含水极易导致硫酸氢氯吡格雷i晶型出胶,不利于i晶型的合成且收率会较低。另外,水的沸点较高,后续干燥需要较长时间和较高的温度进行干燥。
[0007]
专利cn102432625a公布了一种硫酸氢氯吡格雷i晶型球颗粒的制备方法,其采用乙酸乙酯作为溶剂来制备ⅰ晶型球颗粒,结晶时间均较长。采用乙酸乙酯作为结晶溶剂时,溶剂量大,结晶时间较长,生产成本高;而且容易在生产中团聚形成有粘性的黄色油状物,工艺不稳定,难以重复。
[0008]
因此,寻找一种简单高效、可控、晶型稳定、质量优的硫酸氢氯吡格雷晶型i球颗粒仍然是现有技术未解决的技术问题。
三、
技术实现要素:
[0009]
本发明的目的是提供一种成本低、产品质量优的硫酸氢氯吡格雷i晶型球颗粒的制备方法。
[0010]
一种硫酸氢氯吡格雷球形晶型i的制备方法,所述方法包含如下步骤:
[0011]
(1)将氯吡格雷游离碱溶解在溶剂a中,再加入溶剂b,搅拌并控制溶液温度在5~15℃,在0.5~1h内滴加浓硫酸;滴加完毕,控制溶液温度在在10~20℃,在1.5~2.5h内缓慢滴加混合溶剂d;
[0012]
所述溶剂a选自1-丁醇、2-丁醇、叔丁醇中的一种,溶剂a与氯吡格雷游离碱的质量比为7.7~8.3倍;溶剂b选自甲酸、乙酸中的一种,溶剂b与氯吡格雷游离碱的质量比为0.9~1.3倍;混合溶剂d为溶剂b和溶剂c的混合溶液,所述溶剂c选自正己烷、环己烷中的一种,其中混合溶剂d中含有的溶剂b的质量为氯吡格雷游离碱质量的0.05~0.2倍,混合溶剂d中含有的溶剂c的质量为氯吡格雷游离碱质量的0.85~1.2倍;
[0013]
(2)混合溶剂d滴毕后加入晶种,在10~20℃保温结晶6~8h,保温结束后缓慢降温至2℃以下(优选-5~2℃),总降温时间控制了24~36h;
[0014]
(3)在2℃以下(优选-5~2℃)进行过滤,滤饼用溶剂c洗涤,干燥,得到硫酸氢氯吡格雷i晶型球形晶体。
[0015]
所述步骤(1)中,所述浓硫酸的质量为氯吡格雷游离碱质量的0.28-0.31倍。
[0016]
所述步骤(1)中,所述的溶剂a优选为2-丁醇。
[0017]
所述步骤(1)中,所述的溶剂b优选为乙酸。
[0018]
所述步骤(1)中,所述的溶剂c优选为环己烷。
[0019]
所述步骤(1)中,所述混合溶剂d优选为环己烷和乙酸。
[0020]
所述步骤(1)中,特别优选:所述的溶剂a为2-丁醇,所述的溶剂b为乙酸,所述的溶剂c为环己烷,所述混合溶剂d为环己烷和乙酸。
[0021]
所述步骤(2)中,晶种选自硫酸氢氯吡格雷i型晶种,晶型的质量用量为氯吡格雷游离碱质量的0.01~0.05倍;所述的缓慢降温为梯度降温,所述梯度降温是为每2~3h温度下降1~3℃直至温度在2℃以下。
[0022]
所述步骤(3)中,所述溶剂c的质量优选为氯吡格雷游离碱质量的0.8~1.6倍。
[0023]
所述步骤(3)中,干燥温度在55~75℃,真空度在-0.07~-0.10mpa,时间在6~10h。
[0024]
在工业化生产过程中,前述技术方案中的氯吡格雷游离碱可由对应的氯吡格雷盐制备得到,其步骤如下:
[0025]
①
将氯吡格雷盐(优选其hplc纯度在99%以上)加入到有机溶剂中,加入5%~15%的无机碱水溶液;
[0026]
②
分相后,有机相用水洗涤,所得有机相加入脱色剂和干燥剂进行脱水除水;
[0027]
③
过滤、减压浓缩得到氯吡格雷游离碱;
[0028]
所述步骤
①
中,氯吡格雷盐为硫酸盐、盐酸盐、樟脑磺酸盐、苯磺酸盐中的至少一种。所述有机溶剂为二氯甲烷、三氯甲烷、环己烷、甲苯、乙酸乙酯等与水不互溶的有机溶剂中的至少一种。所述无机碱为碳酸钠、碳酸氢钠、碳酸钾、碳酸氢钾、氢氧化钠、氢氧化钾、磷酸氢二钾、磷酸氢二钠中的至少一种;所述氯吡格雷盐与无机碱的质量比为1:0.15~1:
0.45。所述脱色剂为活性炭、吸附树脂中的至少一种,脱色剂质量用量优选为氯吡格雷盐质量的0.01~0.04倍。所述干燥剂为硫酸镁、硫酸钠、硫酸钙、氯化钙中的至少一种,干燥剂质量用量优选为氯吡格雷盐质量的0.1~0.4倍。
[0029]
本发明涉及的溶剂可使用工业级以上的溶剂。
[0030]
与现有技术相比,本发明的有益效果在于:
[0031]
(1)本发明由于加入了对氯吡格雷游离碱溶解度较好的溶剂b与溶剂a制备成混合溶剂,加大了对氯吡格雷游离碱的溶解度,从而
①
大大减少了仲丁醇及总体溶剂的用量,降低了溶剂使用成本;同时在相同设备条件下时,本工艺的投料量更大,产能更高;
②
使氧化杂质等杂质的生成量减少,能得到质量更优的产品。
[0032]
(2)现有技术中通常先将浓硫酸用环己烷等有机溶剂稀释再将其加入到氯吡格雷游离碱溶液中,以减少杂质、加快反应速度以及使产品颜色更好,但在本发明中,不需要先将浓硫酸和有机溶剂混合,而是直接滴加完浓硫酸,再缓慢滴加溶解度不佳的混合溶剂d,使反应液逐渐缓慢趋向于过饱和,然后加晶种缓慢析料,使析晶过程缓慢进行,这保证了得到的晶型产品是球形颗粒且具有良好的颗粒度和质量。混合溶剂d中溶剂b的存在,避免了单独滴加不良溶剂c时,出现滴下去的液滴与反应液液面接触的那点快速析料、从而影响晶型产品的颗粒度和质量的问题。
四、附图说明
[0033]
图1是实施例3制备的硫酸氢氯吡格雷i晶型球颗粒的xrd图谱;
[0034]
图2是实施例3制备的硫酸氢氯吡格雷i晶型球颗粒的电镜照片;
[0035]
图3是实施例4制备的硫酸氢氯吡格雷i晶型球颗粒的显微镜照片(放大100倍)。
五、具体实施方式
[0036]
为使本技术实施例的目的、技术方案和优点更加清楚,下面将对本技术实施例中的技术方案进行清楚、完整地描述,但本发明的保护范围不限于此。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
[0037]
本发明所述的氯吡格雷盐由邻氯苯甘氨酸为原料进行制备,具体制备步骤如下:
[0038]
①
将邻氯苯甘氨酸、甲醇、浓硫酸混合后升温至回流,反应至少3h后,减压蒸馏甲醇至反应液浓稠,再加入水以及二氯甲烷或甲苯,搅拌溶清后分层,有机层浓缩得p1油。
[0039]
②
将酒石酸、甲醇、丙酮、p1油在30~50℃混合溶清后,降温,缓慢析料至5℃以下,离心烘干得p2。p2可进一步加甲醇精制后得到高质量的p2。
[0040]
③
将2-噻吩乙醇和对甲苯磺酰氯、苄基三乙基氯化铵或四丁基溴化胺溶解在二氯甲烷或甲苯中,在0~20℃滴加20%~50%浓度的氢氧化钠溶液。滴毕保温反应5h以上,再加入盐酸或升温至30℃以上终止反应。分层,有机溶剂浓缩后得p3油。p3油可进一步加甲醇精制后得到高质量的p3。
[0041]
④
取上述p2,溶解在水和二氯甲烷或水和甲苯中,再缓慢加入等当量的5%~15%浓度的碳酸钠或碳酸氢钠溶液。分层,有机层浓缩得p2油。
[0042]
将上述p3、p2油、苄基三乙基氯化铵或四丁基溴化胺、碳酸钠或磷酸氢二钾加入到
乙腈或甲苯中,在80℃~120℃反应20h以上,蒸除有机溶剂,再加入水和二氯甲烷或水和甲苯搅拌溶清,萃取分层得有机层。再加入适量乙酸乙酯、甲醇,在40℃以下滴加盐酸,降温至10℃以下,过滤烘干得p4。
[0043]
⑤
将p4加入到甲醛水溶液中,在20℃~30℃反应25~35h,反应再加入环己烷或甲苯,加入碳酸钠溶液,萃取分层,有机溶剂浓缩至干得油。然后加入丙酮,在10℃以下加入硫酸或樟脑磺酸或苯磺酸或盐酸成盐,搅拌析料5h以上,过滤得氯吡格雷盐。
[0044]
实施例1:
[0045]
①
将200g邻氯苯甘氨酸、400g甲醇、180g浓硫酸混合后升温至回流,反应4h后,减压蒸馏甲醇至反应液浓稠,再加入200g水、200g二氯甲烷,搅拌溶清后分层,有机层浓缩得200g p1油。
[0046]
②
将160g酒石酸、1000g甲醇、300g丙酮、200gp1油在40℃混合溶清后,降温,缓慢析料至5℃以下,过滤烘干得300g p2。300g p2中加800g甲醇,保温30℃精制再降温至5℃以下得260g p2。
[0047]
③
将100g 2-噻吩乙醇和200g对甲苯磺酰氯、15g苄基三乙基氯化铵溶解在700g二氯甲烷中,在10℃滴加30%浓度的氢氧化钠溶液300g。滴毕保温反应6h,再加入5kg盐酸终止反应。分层,有机溶剂浓缩后得250g p3油。p3油加250g甲醇,降温至5℃以下析料,过滤得220g p3。
[0048]
④
取上述合成的p2 200g,溶解在500g水和600g二氯甲烷中,再缓慢加入10%浓度的碳酸钠溶液800g。分层,有机层浓缩得p2油,再加入200g p3、10g苄基三乙基氯化铵、120g碳酸钠、120g乙腈,在90℃反应30h,蒸除有机溶剂,再加入500g水和500g二氯甲烷搅拌溶清,萃取分层得有机层,浓缩至干。再加入450g乙酸乙酯、100g甲醇,在40℃滴加70g盐酸,滴毕降温至10℃,过滤烘干得150g p4。
[0049]
⑤
将150g p4加入到900g 37%浓度的甲醛水溶液中,在25℃反应33h,再加入500g环己烷,加入250g10%的碳酸钠溶液,萃取分层,有机溶剂浓缩至干得油。然后加入500g丙酮,在5℃加入39g浓硫酸成盐,搅拌析料6h,过滤得135g氯吡格雷盐,hplc纯度99.0%以上。
[0050]
实施例2:
[0051]
①
将200g邻氯苯甘氨酸、400g甲醇、180g浓硫酸混合后升温至回流,反应4h后,减压蒸馏甲醇至反应液浓稠,再加入200g水、200g甲苯,搅拌溶清后分层,有机层浓缩得200g p1油。
[0052]
②
将160g酒石酸、1000g甲醇、300g丙酮、200gp1油在35℃混合溶清后,降温,缓慢析料至5℃以下,过滤烘干得300g p2。300g p2中加800g甲醇,保温30℃精制再降温至5℃以下得260g p2。
[0053]
③
将100g 2-噻吩乙醇和200g对甲苯磺酰氯、15g四丁基溴化胺溶解在700g二甲苯中,在10℃滴加30%浓度的氢氧化钠溶液300g。滴毕保温反应6h,再加入升温至35℃终止反应。分层,有机溶剂浓缩后得250g p3油。p3油加250g甲醇,降温至5℃以下析料,过滤得220g p3。
[0054]
④
取上述合成的p2 200g,溶解在500g水和600g甲苯中,再缓慢加入10%浓度的碳酸氢钠溶液800g。分层,有机层浓缩得p2油,再加入200g p3、10g四丁基溴化胺、120g磷酸氢二钾、120g甲苯,在110℃反应35h,反应结束再加入100g甲醇,在40℃滴加70g盐酸,滴毕降
温至10℃,过滤烘干得150g p4。
[0055]
⑤
将150g p4加入到900g37%浓度的甲醛水溶液中,在30℃反应27h,反应再加入500g甲苯,加入250g10%的碳酸钠溶液,萃取分层,有机溶剂浓缩至干得油。然后加入500g丙酮,在3℃以下加入90g樟脑磺酸成盐,搅拌析料6.5h,过滤得175g氯吡格雷盐,hplc纯度99.0%以上。
[0056]
实施例3:
[0057]
1、取碳酸钠55g,加入1045g水,搅拌,配制成5%的碳酸钠溶液。
[0058]
2、取根据实施例1步骤制备的硫酸氢氯吡格雷210g,加入900g二氯甲烷,加入上述5%浓度的碳酸钠溶液,直至水层ph在7~9。静置分层,有机层水洗2次,再加入6g活性炭,50g硫酸镁,搅拌后过滤,真空浓缩至氯吡格雷游离碱质量不再变化为止,此时氯吡格雷游离碱质量为160g。
[0059]
3、在上述的氯吡格雷游离碱中加入2-丁醇1300g,搅拌溶清,再加入乙酸180g,搅拌降温至15℃,再滴加48.5g浓硫酸,50min滴毕。滴毕控温在20℃,再滴加混合溶剂220g(其中乙酸30g,环己烷190g),2h滴毕。
[0060]
4、滴加完毕,加入8g硫酸氢氯吡格雷ⅰ晶型晶种,在20℃保温6h后,每隔3h降温2℃,当温度为0℃时,过滤,滤饼用200g环己烷淋洗,在75℃,真空度-0.08mpa下真空干燥6h,得159g硫酸氢氯吡格雷i晶型球颗粒。实施例3制备的硫酸氢氯吡格雷i晶型球颗粒的xrd图谱、电镜照片分别如图1、图2所示。
[0061]
实施例4:
[0062]
1、取碳酸氢钠25g,加入375g水,搅拌,配制成约6%的碳酸氢钠溶液。
[0063]
2、取实施例2制备的樟脑磺酸氯吡格雷150g,加入600g二氯甲烷,加入上述6%浓度的碳酸氢钠溶液,直至水层ph在7~9。静置分层,有机层水洗2次,再加入3g活性炭,25g硫酸镁,搅拌后过滤,真空浓缩至氯吡格雷游离碱质量不再变化为止,此时氯吡格雷游离碱质量为85g。
[0064]
3、在上述的氯吡格雷游离碱中加入2-丁醇700g,搅拌溶清,再加入乙酸90g,搅拌降温至15℃,再滴加25g浓硫酸,32min滴毕。滴毕控温在20℃,再滴加混合溶剂100g(其中乙酸7g,环己烷93g),1.5h滴毕。
[0065]
4、滴加完毕,加入3g硫酸氢氯吡格雷ⅰ晶型晶种,在20℃保温6h后,每隔3h降温2.5℃。当温度为-1℃,过滤,滤饼用120g环己烷淋洗,在75℃,真空度-0.09mpa下真空干燥6h,得85g硫酸氢氯吡格雷i晶型球颗粒。实施例4制备的硫酸氢氯吡格雷i晶型球颗粒的显微镜照片(放大100倍)见图3。
[0066]
实施例5:
[0067]
1、取碳酸钠55g,加入312g水,搅拌,配制成15%的碳酸钠溶液。
[0068]
2、取根据实施例1步骤制备的硫酸氢氯吡格雷210g,加入1000g二氯甲烷,加入上述15%浓度的碳酸钠溶液,直至水层ph在7~9。静置分层,有机层水洗2次,再加入3g活性炭,25g硫酸镁,搅拌后过滤,真空浓缩至氯吡格雷游离碱质量不再变化为止,此时氯吡格雷游离碱质量为158g。
[0069]
3、在上述的氯吡格雷游离碱中加入2-丁醇1200g,搅拌溶清,再加入乙酸160g,搅拌降温至5℃,再滴加46.5g浓硫酸,53min滴毕。滴毕控温在10℃,再滴加混合溶剂220g(其
中乙酸30g,环己烷190g),2.2h滴毕。
[0070]
4、滴加完毕,加入5g硫酸氢氯吡格雷ⅰ晶型晶种,在10℃保温8h后,每隔3h降温1℃。当温度为0℃,过滤,滤饼用150g环己烷淋洗,在55℃,真空度-0.10mpa下真空干燥9h,得152g硫酸氢氯吡格雷i晶型球颗粒。
[0071]
实施例6:
[0072]
1、取碳酸氢钠85g,加入800g水,搅拌,配制成约10%的碳酸氢钠溶液。
[0073]
2、取根据实施例1步骤制备的硫酸氢氯吡格雷210g,加入900g甲苯,加入上述10%浓度的碳酸钠溶液,直至水层ph在7~9。静置分层,有机层水洗2次,再加入6g活性炭,50g硫酸镁,搅拌后过滤,真空浓缩至氯吡格雷游离碱质量不再变化为止,此时氯吡格雷游离碱质量为158g。
[0074]
3、在上述的氯吡格雷游离碱中加入1-丁醇1300g,搅拌溶清,再加入甲酸150g,搅拌降温至10℃,再滴加48g浓硫酸,52min滴毕。滴毕控温在15℃,再滴加混合溶剂200g(其中甲酸25g,正己烷175g),2h滴毕。
[0075]
4、滴加完毕,加入5g硫酸氢氯吡格雷ⅰ晶型晶种,在15℃保温7h后,每隔3h降温1.5℃。当温度为至-2℃,过滤,滤饼用150g正己烷淋洗,在65℃,真空度-0.08mpa下真空干燥8h,得149g硫酸氢氯吡格雷i晶型球颗粒。
[0076]
对比例1:cn2020105148000的实施例2
[0077]
(1)按照实施例3的操作步骤得到氯吡格雷碱,将氯吡格雷碱溶解在乙酸丁酯和正丁醇的混合溶剂中,乙酸丁酯和正丁醇的摩尔比为5:1,得到浓度为0.01g/ml的游离碱溶液,在30℃的条件下,将浓硫酸溶解在同样的乙酸丁酯和正丁醇的混合溶剂中,得到浓度为0.3g/ml的硫酸溶液,将两溶液混合,并保证混合的硫酸与氯吡格雷游离碱的摩尔比为1.4:1;
[0078]
(2)调整温度至35℃,加入硫酸氢氯吡格雷ⅰ晶型晶种,晶种质量为氯吡格雷游离碱的0.1%,以降温速率为0.5℃/min将温度降至0℃,之后保持温度搅拌40min;
[0079]
(3)过滤、洗涤、干燥,得到硫酸氢氯吡格雷i晶型球形晶体。
[0080]
对比例2:cn201410847832的实施例1
[0081]
①
取实施例1制备的氯吡格雷硫酸氢盐760g(纯度大于99.0%)分散于10l二氯甲烷和5l水的混合液中,加入固体碳酸氢钠至水相ph》7。静置分液,取有机相并用水洗涤(1l
×
2),无水硫酸镁除水至溶液澄清。
[0082]
②
将有机相过滤、真空旋蒸至质量不再变化,将剩余物溶于10.5l的2-丁醇中,将溶液于25℃下保温。将100ml浓硫酸(181g)分散于2.5l的2-丁醇中在10分钟内加入体系中,将10g i晶型晶种分散于1l的2-丁醇中一并倒入。25℃下保温2.5h,降温至15℃继续保温3h,抽滤,滤饼用乙酸乙酯洗涤,40℃真空烘干1.0h,得产品610g。
[0083]
对比例3:
[0084]
1、取碳酸钠55g,加入1045g水,搅拌,配制成5%的碳酸钠溶液。
[0085]
2、取根据实施例1步骤制备的硫酸氢氯吡格雷210g,加入900g二氯甲烷,加入上述5%浓度的碳酸钠溶液,直至水层ph在7~9。静置分层,有机层水洗2次,再加入6g活性炭,50g硫酸镁,搅拌后过滤,真空浓缩至氯吡格雷游离碱质量不再变化为止,此时氯吡格雷游离碱质量为158g。
[0086]
3、在上述的氯吡格雷游离碱中加入2-丁醇1300g,搅拌溶清,再加入乙酸180g,搅拌降温至15℃,再滴加48.5g浓硫酸,50min滴毕。滴毕控温在20℃,再滴加环己烷190g,滴加过程中,滴加的环己烷与反应液面接触点变白析料,随后又迅速溶清消失,1.8h滴毕。
[0087]
4、滴加完毕,加入8g硫酸氢氯吡格雷ⅰ晶型晶种,在20℃保温6h后,每隔3h降温2℃,当温度为0℃时,过滤(此时发现过滤缓慢,滤液较难抽滤,整体抽滤时间延长一倍左右,出现此现象主要是本批颗粒度偏小导致。),滤饼用200g环己烷淋洗,在75℃,真空度-0.08mpa下真空干燥6h,得166g硫酸氢氯吡格雷i晶型球颗粒。
[0088]
对比例4:
[0089]
1、取碳酸钠55g,加入1045g水,搅拌,配制成5%的碳酸钠溶液。
[0090]
2、取根据实施例1步骤制备的硫酸氢氯吡格雷210g,加入900g二氯甲烷,加入上述5%浓度的碳酸钠溶液,直至水层ph在7~9。静置分层,有机层水洗2次,再加入6g活性炭,50g硫酸镁,搅拌后过滤,真空浓缩至氯吡格雷游离碱质量不再变化为止,此时氯吡格雷游离碱质量为157g。
[0091]
3、在上述的氯吡格雷游离碱中加入2-丁醇1300g,搅拌溶清降温至15℃,再滴加48.5g浓硫酸,50min滴毕,滴毕发现反应瓶内壁及搅拌桨上油状物析出附着。控温在20℃,再滴加混合溶剂220g(其中乙酸30g,环己烷190g),2h滴毕。滴毕发现油状物溶解了部分,反应瓶内壁及搅拌桨仍少部分附着。
[0092]
4、滴加完毕,加入8g硫酸氢氯吡格雷ⅰ晶型晶种,在20℃保温6h后,每隔3h降温2℃,当温度为0℃时,过滤,滤饼用200g环己烷淋洗,在75℃,真空度-0.08mpa下真空干燥6h,得185g硫酸氢氯吡格雷,经检测为混晶。
[0093]
按中国药典2020版二部硫酸氢氯吡格雷的方法检测实施例3-6和对比例1-4制备的产品质量,结果如表1所示:
[0094]
表1质量对比
[0095][0096]
所得对比例和实施例质量检测如上,可以看出,实施例3~6能保证所有杂质均<0.05%,所得到的产品质量更优。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1