一种苄位氘代的α,α-二氘苄碘、二氘苄胺、二氘药物分子及其合成方法
一种苄位氘代的
α
,
α-二氘苄碘、二氘苄胺、二氘药物分子及其合成方法
技术领域
1.本发明涉及有机化合物及有机合成技术领域,具体涉及一种苄位氘代的α,α-二氘苄碘、二氘苄胺、二氘药物分子及其合成方法。
背景技术:
2.人体对药物的代谢主要依靠氧化自由基反应,因此改变药物代谢主要针对容易被氧化的位点针对性氘代,因为将相对容易氧化的苄位上的c-h键置换成更为稳定的c-d键,增加了药物分子苄位上的抗氧化性和稳定性,能改善药物分子的代谢途径,实现药物分子的改性。2017年,fda批准的第一个氘代药物氘代丁苯那嗪,引起氘代药物研究的热潮。而苄碘作为一种高活性的中间体,广泛运用于有机合成中。鉴于许多药物中含有很多容易被氧化的苄位,开发出一种简易的合成氘代苄碘的方法并将其应用于氘代活性药物分子的合成具有重大意义。
3.现有技术中,关于合成氘代苄碘的方法非常少,关于氘代方法的报道大多涉及贵金属pd,ir或者rh和昂贵的氘源如硼氘化钠或者氘化铝锂的使用,具有成本高、工艺复杂、产率低等缺点;同时,合成的α,α-二氘苄碘种类少。
4.鉴于此,开发一种新的合成工艺解决上述技术问题具有十分重要的意义。
技术实现要素:
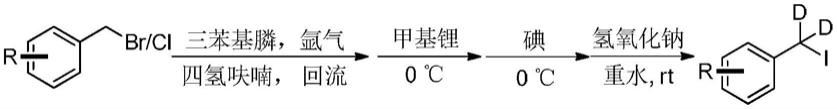
5.本发明要解决的技术问题是提供一种苄位氘代的α,α-二氘苄碘及其合成方法,以苄氯或苄溴为原料,氘水为氘源采用一锅法合成,合成成本低,产率高,可制备得到多种α,α-二氘苄碘产物。
6.为了解决上述问题,本发明的技术方案如下:
7.一种苄位氘代的α,α-二氘苄碘,其结构式如下:
[0008][0009]
其中,r为f、cl、ch3、t-bu、och3、cn、coot-bu、so2net2、ph、2-cnc6h5、喹啉和萘中的一种或两种。
[0010]
所述苄位氘代的α,α-二氘苄碘的合成方法,包括如下步骤:
[0011]
将原料苄氯或苄溴与三苯基膦按一定比例混合,进行氩气保护,在氩气氛围条件下加入干燥四氢呋喃,然后在60-70℃条件下反应10-15h,反应后将反应液转移至冰浴降温;在冰浴条件下加入一定量的甲基锂或者二(三甲基硅烷基)氨基钾并反应10-20min;随后加入一定量的碘并将反应液转移至常温反应6-10h,随即向反应液中加入氢氧化钠的氘水溶液,点板监测反应完毕后,得到反应液;其中原料苄氯或苄溴与三苯基膦在有机溶剂中
的反应温度可以为60℃、65℃、68℃或70℃,也可以为该范围的其他温度值;
[0012]
将反应液过色谱柱进行纯化,得到白色固体,烘干后,即得到苄位氘代的α,α-二氘苄碘。
[0013]
进一步地,苄氯或苄溴、三苯基膦、甲基锂/二(三甲基硅烷基)氨基钾、碘和氢氧化钠的摩尔比为1:1.05-1.2:1.05-1.2:1.05-1.2:3-4;如:摩尔比为1:1.05:1.05:1.05:3、1:1.2:1.2:1.2:4或1:1.08:1.08:1.08:3.5,也可以为该摩尔比范围内的其他比值。
[0014]
进一步地,原料苄氯或苄溴与混合溶剂的摩尔体积比为0.1-0.2mmol/ml,其中混合溶剂包含有机溶剂和氘水溶液;原料苄氯或苄溴与混合溶剂的摩尔体积比可以为0.1mmol/ml、0.2mmol/ml、0.12mmol/ml、0.15mmol/ml、0.17mmol/ml,也可以为该范围内的其他比值。
[0015]
进一步地,色谱柱采用体积比为(0-1)%乙酸乙酯和石油醚作为洗脱剂。
[0016]
基于上述苄位氘代的α,α-二氘苄碘及其合成方法,本发明还提供一种利用苄位氘代的α,α-二氘苄碘作为合成单体,合成苄位氘代的α,α-二氘苄胺的方法。具体的,
[0017]
一种苄位氘代的α,α-二氘苄胺,其结构式如下:
[0018][0019]
其中,r’为氟,苯基,氢中的一种。
[0020]
所述苄位氘代的α,α-二氘苄胺的合成方法,包括如下步骤:
[0021]
将原料苄氯或苄溴与三苯基膦按一定比例混合,进行氩气保护,在氩气氛围条件下加入干燥四氢呋喃,然后在60-70℃条件下反应10-15h,反应后将反应液转移至冰浴降温;在冰浴条件下加入一定量的甲基锂或者二(三甲基硅烷基)氨基钾并反应10-20min;随后加入一定量的碘并将反应液转移至常温反应6-10小时;随即向反应液中加入邻苯二甲酰亚胺钾的氘水溶液,点板监测反应完毕后,得到反应液;其中原料苄氯或苄溴与三苯基膦在有机溶剂中的反应温度可以为60℃、65℃、68℃或70℃,也可以为该范围的其他温度值;
[0022]
将反应液过色谱柱进行纯化,得到白色固体,烘干后,即得所述苄位氘代的α,α-二氘苄胺。
[0023]
进一步地,苄氯或苄溴、三苯基膦、甲基锂/二(三甲基硅烷基)氨基钾、碘和邻苯二甲酰亚胺钾的摩尔比为1:1.05-1.2:1.05-1.2:1.05-1.2:3-4;如:摩尔比为1:1.05:1.05:1.05:3、1:1.2:1.2:1.2:4或1:1.08:1.08:1.08:3.5,也可以为该摩尔比范围内的其他比值。
[0024]
进一步地,苄氯或苄溴与混合溶剂的摩尔体积比为0.1-0.2mmol/ml;其中混合溶剂包括有机溶剂和氘水溶液;原料苄氯或苄溴与混合溶剂的摩尔体积比可以为0.1mmol/ml、0.2mmol/ml、0.12mmol/ml、0.15mmol/ml、0.17mmol/ml,也可以为该范围内的其他比值。
[0025]
进一步地,色谱柱采用体积比为10-20%乙酸乙酯和石油醚作为洗脱剂。
[0026]
基于上述苄位氘代的α,α-二氘苄碘及其合成方法,本发明还提供一种利用苄位氘代的α,α-二氘苄碘作为合成单体,合成苄位氘代的α,α-二氘药物分子的方法。具体的,
[0027]
一种苄位氘代的α,α-二氘药物分子,其结构式如下:
[0028][0029]
其中,r为f、cl、ch3、t-bu、och3、cn、coot-bu、so2net2、ph、2-cnc6h5、喹啉和萘中的一种或两种;r1,r2分别为1-(4-氯二苯甲基)哌嗪,5-(哌嗪-1-基)嘧啶中的一种。
[0030]
所述苄位氘代的α,α-二氘药物分子的合成方法,包括如下步骤:
[0031]
将原料苄氯或苄溴与三苯基膦按一定比例混合,进行氩气保护,在氩气氛围条件下加入干燥四氢呋喃,然后在60-70℃条件下反应10-15h,反应后将反应液转移至冰浴降温;在冰浴条件下加入一定量的甲基锂或者二(三甲基硅烷基)氨基钾并反应10-20min;随后加入一定量的碘并将反应液转移至常温反应6-10小时,随即向反应液中加入氢氧化钠的氘水溶液,反应15-20h后加入一定量的hnr1r2反应3-5h,点板监测反应完毕后,得到反应液;其中原料苄氯或苄溴与三苯基膦在有机溶剂中的反应温度可以为60℃、65℃、68℃或70℃,也可以为该范围的其他温度值;
[0032]
将反应液过色谱柱进行纯化,得到白色固体,烘干后,即得所述苄位氘代的α,α-二氘药物分子。
[0033]
进一步地,苄氯或苄溴、三苯基膦、甲基锂/二(三甲基硅烷基)氨基钾、碘、氢氧化钠和hnr1r2的摩尔比为1:1.05-1.2:1.05-1.2:1.05-1.2:3-4:1.05-1.2;如:摩尔比为1:1.05:1.05:1.05:3:1.05、1:1.2:1.2:1.2:4:1.2或1:1.08:1.08:1.08:3.5:1.08,也可以为该摩尔比范围内的其他比值。
[0034]
进一步地,苄氯或苄溴与混合溶剂的摩尔体积比为0.1-0.2mmol/ml;其中混合溶剂包括有机溶剂和氘水溶液;原料苄氯或苄溴与混合溶剂的摩尔体积比可以为0.1mmol/ml、0.2mmol/ml、0.12mmol/ml、0.15mmol/ml、0.17mmol/ml,也可以为该范围内的其他比值。
[0035]
进一步地,色谱柱采用体积比为10-20%乙酸乙酯和石油醚作为洗脱剂。
[0036]
与现有技术相比,本发明提供的苄位氘代的α,α-二氘苄碘、二氘苄胺、二氘药物分子及其合成方法,有益效果在于:
[0037]
一、本发明的苄位氘代的α,α-二氘苄碘,以苄氯或苄溴为原料,氘水为氘源采用一锅法合成,合成工艺简单成本低,产率和氘代率高,可达到86%的产率和97%的氘代率。
[0038]
二、本发明的苄位氘代的α,α-二氘苄碘,作为重要的合成单体,通过向反应体系中加入邻苯二甲酰亚胺钾或二级胺,可在一锅法条件下进一步合成苄位氘代的α,α-二氘苄胺和苄位氘代的α,α-二氘药物分子,充分说明了该合成方法的实用价值。
附图说明
[0039]
为了更清楚地说明本发明实施例中的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0040]
图1为实施例1的产物的质谱图。
[0041]
图2为实施例2的产物的质谱图。
[0042]
图3为实施例3的产物的质谱图。
[0043]
图4为实施例4的产物的质谱图。
[0044]
图5为实施例5的产物的质谱图。
[0045]
图6为实施例6的产物的质谱图。
[0046]
图7为实施例7的产物的质谱图。
[0047]
图8为实施例8的产物的质谱图。
[0048]
图9为实施例9的产物的质谱图。
[0049]
图10为实施例10的产物的质谱图。
[0050]
图11为实施例11的产物的质谱图。
[0051]
图12为实施例12的产物的质谱图。
[0052]
图13为实施例13的产物的质谱图。
[0053]
图14为实施例14的产物的质谱图。
[0054]
图15为实施例15的产物的质谱图。
[0055]
图16为实施例16的产物的质谱图。
[0056]
图17为实施例17的产物的质谱图。
[0057]
图18为实施例18的产物的质谱图。
[0058]
图19为实施例19的产物的质谱图。
[0059]
图20为实施例20的产物的质谱图。
[0060]
图21为实施例21的产物的质谱图。
[0061]
图22为实施例22的产物的质谱图。
[0062]
图23为实施例23的产物的质谱图。
[0063]
图24为实施例24的产物的质谱图。
具体实施方式
[0064]
为了使本技术领域的人员更好地理解本发明实施例中的技术方案,并使本发明的上述目的、特征和优点能够更加明显易懂,下面对本发明的具体实施方式作进一步的说明。
[0065]
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应该被视为在本文中具体公开。
[0066]
本发明的苄位氘代的α,α-二氘苄碘,合成方法包括如下步骤:
[0067]
将原料苄氯或苄溴与三苯基膦按一定比例混合,进行氩气保护,在氩气氛围条件下加入干燥四氢呋喃,然后在60-70℃条件下反应10-15h,反应后将反应液转移至冰浴降温;在冰浴条件下加入一定量的甲基锂或者二(三甲基硅烷基)氨基钾并反应10-20min;随后加入一定量的碘并将反应液转移至常温反应6-10h,随即向反应液中加入氢氧化钠的氘水溶液,点板监测反应完毕后,得到反应液;
[0068]
将反应液过色谱柱进行纯化,得到白色固体,烘干后,即得到苄位氘代的α,α-二氘苄碘。
[0069]
其合成线路如下:
[0070][0071]
制备得到的苄位氘代的α,α-二氘苄碘的结构式如下:
[0072][0073]
其中,r为f、cl、ch3、t-bu、och3、cn、coot-bu、so2net2、ph、2-cnc6h5、喹啉和萘中的一种或两种。
[0074]
以本发明的苄位氘代的α,α-二氘苄碘为合成单体,通过向反应体系中加入邻苯二甲酰亚胺钾或二级胺,可在一锅法条件下进一步合成苄位氘代的α,α-二氘苄胺和苄位氘代的α,α-二氘药物分子,具体的合成工艺如下:
[0075]
一种苄位氘代的α,α-二氘苄胺的合成方法,包括如下步骤:
[0076]
将原料苄氯或苄溴与三苯基膦按一定比例混合,进行氩气保护,在氩气氛围条件下加入干燥四氢呋喃,然后在60-70℃条件下反应10-15h,反应后将反应液转移至冰浴降温;在冰浴条件下加入一定量的甲基锂或者二(三甲基硅烷基)氨基钾并反应10-20min;随后加入一定量的碘并将反应液转移至常温反应6-10小时;随即向反应液中加入邻苯二甲酰亚胺钾的氘水溶液,点板监测反应完毕后,得到反应液;
[0077]
将反应液过色谱柱进行纯化,得到白色固体,烘干后,即得所述苄位氘代的α,α-二氘苄胺。
[0078]
其合成线路如下:
[0079][0080]
制备得到的苄位氘代的α,α-二氘苄胺结构式如下:
[0081][0082]
其中,r’为氟,苯基,氢中的一种。
[0083]
一种苄位氘代的α,α-二氘药物分子的合成方法,包括如下步骤:
[0084]
将原料苄氯或苄溴与三苯基膦按一定比例混合,进行氩气保护,在氩气氛围条件下加入干燥四氢呋喃,然后在60-70℃条件下反应10-15h,反应后将反应液转移至冰浴降温;在冰浴条件下加入一定量的甲基锂或者二(三甲基硅烷基)氨基钾并反应10-20min;随后加入一定量的碘并将反应液转移至常温反应6-10小时,随即向反应液中加入氢氧化钠的氘水溶液,反应15-20h后加入一定量的hnr1r2反应3-5h,点板监测反应完毕后,得到反应液;
[0085]
将反应液过色谱柱进行纯化,得到白色固体,烘干后,即得所述苄位氘代的α,α-二氘药物分子。
[0086]
其合成线路如下:
[0087][0088]
制备得到的苄位氘代的α,α-二氘药物分子的结构式如下:
[0089][0090]
其中,r为f、cl、ch3、t-bu、och3、cn、coot-bu、so2net2、ph、2-cnc6h5、喹啉和萘中的一种或两种;r1,r2分别为1-(4-氯二苯甲基)哌嗪,5-(哌嗪-1-基)嘧啶中的一种。
[0091]
以下通过具体的实施例对本发明提供的苄位氘代的α,α-二氘苄碘、苄位氘代的α,α-二氘苄胺、苄位氘代的α,α-二氘药物分子及其合成工艺进行详细阐述。
[0092]
实施例1
[0093]
将0.8mmol原料1(结构式如下)与0.84mmol三苯基膦混合,随后对反应体系进行氩气保护。在氩气氛围条件下加入4ml干燥四氢呋喃,然后在65℃条件下反应。12h后,将反应液转移至0度冰浴降温。在此温度下加入0.84mmol的甲基锂并反应15分钟。加入0.84mmol碘并将反应液转移至常温反应8小时。待到反应液变成固态,在此温度下向反应液体中加入含有2.4mmol的氢氧化钠1ml氘水溶液反应18h。待到溶液澄清时,进行点板,确认反应完毕后,将反应液过色谱柱,并使用体积比为(0-1)%的乙酸乙酯和石油醚进行洗脱,得到白色固体183mg,产率77%,将固体进行核磁、红外和质谱测试。
[0094][0095]
核磁测试结果为:1h nmr(400mhz,chloroform-d)δ7.58
–
7.54(m,2h),7.52
–
7.48(m,2h),7.46
–
7.39(m,4h),7.36
–
7.31(m,1h),4.48(d,j=4.8hz,0.07h);
[0096]
13
c nmr(101mhz,chloroform-d)δ140.77,140.43,138.15,129.14,128.79,127.50(d,j=5.5hz),127.00,6.12
–
4.44(m);
[0097]
质谱测试结果为:hrms(esi)m/z[m
–
i-]
+
calcd for c
13
h9d
2+
:169.0981,found 169.0981.(结构如图1所示);
[0098]
红外测试结果为:ir(neat)ν3027,2269,1793,1483,1117,895,754,687cm
–1;
[0099]
结合核磁、红外和质谱的测试结果,可得本实施例产物1的结构如下:
[0100][0101]
实施例2
[0102]
将0.8mmol原料2(结构式如下)与0.84mmol三苯基膦混合,随后对反应体系进行氩
气保护。在氩气氛围条件下加入4ml干燥四氢呋喃,然后在65℃条件下反应。12h后,将反应液转移至0度冰浴降温。在此温度下加入0.84mmol的甲基锂并反应15分钟。加入0.84mmol碘并将反应液转移至常温反应8小时。待到反应液变成固态,在此温度下向反应液体中加入含有2.4mmol的氢氧化钠1ml氘水溶液反应18h。待到溶液澄清时,进行点板,确认反应完毕后,将反应液过色谱柱,并使用体积比为(0-1)%的乙酸乙酯和石油醚进行洗脱,得到白色固体147mg,产率57%,氘代率93%,将固体进行核磁、红外和质谱测试。
[0103][0104]
核磁测试结果为:1h nmr(400mhz,chloroform-d)δ7.76(ddd,j=7.8,1.4,0.6hz,1h),7.64(td,j=7.7,1.4hz,1h),7.52
–
7.51(m,1h),7.50(s,4h),7.45(td,j=7.6,1.2hz,1h),4.50(d,j=4.8hz,0.15h);
[0105]
13
c nmr(101mhz,chloroform-d)δ144.73,139.82,137.63,133.84,132.93,130.00,129.20(d,j=8.8hz),127.79,118.64,111.15,5.34
–
3.87(m);
[0106]
质谱测试结果为:hrms(esi)m/z[m
–
i-]
+
calcd for c
14
h8d2n
+
194.0933,found 194.0932.(结构如图2所示);
[0107]
红外测试结果为:ir(neat)ν2923,2219,1913,1593,1401,1111,902,756cm
–1;
[0108]
结合核磁、红外和质谱的测试结果,可得本实施例的产物2的结构如下:
[0109][0110]
实施例3
[0111]
将0.8mmol原料3(结构式如下)与0.84mmol三苯基膦混合,随后对反应体系进行氩气保护。在氩气氛围条件下加入4ml干燥四氢呋喃,然后在65℃条件下反应。12h后,将反应液转移至0度冰浴降温。在此温度下加入0.84mmol的甲基锂并反应15分钟。加入0.84mmol碘并将反应液转移至常温反应8小时。待到反应液变成固态,在此温度下向反应液体中加入含有2.4mmol的氢氧化钠1ml氘水溶液反应18h。待到溶液澄清时,进行点板,确认反应完毕后,将反应液过色谱柱,并使用体积比为(0-1)%的乙酸乙酯和石油醚进行洗脱,得到白色固体118mg,产率62%,氘代率96%,将固体进行核磁、红外和质谱测试。
[0112][0113]
核磁测试结果为:1h nmr(400mhz,chloroform-d)δ7.33(td,j=7.6,1.8hz,1h),7.23(tdd,j=7.4,5.2,1.8hz,1h),7.06(td,j=7.6,1.2hz,1h),6.99(ddd,j=9.7,8.2,
1.2hz,1h),4.41(d,j=5.3hz,0.09h);
[0114]
13
c nmr(101mhz,chloroform-d)δ161.63,159.15,130.78,129.93(d,j=8.2hz),124.53(d,j=3.7hz),116.03,115.82,-3.31
–‑
4.27(m);
[0115]
质谱测试结果为:hrms(esi)m/z[m
–
i-]
+
calcd for c7h4d2f
+
111.0574,found 111.0567.(结构如图3所示);
[0116]
红外测试结果为:ir(neat)ν2917,2849,1583,1492,1454,1236,899,753cm
–1;
[0117]
结合核磁、红外和质谱的测试结果,可得本实施例产物3的结构如下:
[0118][0119]
实施例4
[0120]
将0.8mmol原料4(结构式如下)与0.84mmol三苯基膦混合,随后对反应体系进行氩气保护。在氩气氛围条件下加入4ml干燥四氢呋喃,然后在65℃条件下反应。12h后,将反应液转移至0度冰浴降温。在此温度下加入0.84mmol的甲基锂并反应15分钟。加入0.84mmol碘并将反应液转移至常温反应8小时。待到反应液变成固态,在此温度下向反应液体中加入含有2.4mmol的氢氧化钠1ml氘水溶液反应18h。待到溶液澄清时,进行点板,确认反应完毕后,将反应液过色谱柱,并使用体积比为(0-1)%的乙酸乙酯和石油醚进行洗脱,得到白色固体163mg,产率80%,氘代率91%,将固体进行核磁、红外和质谱测试。
[0121][0122]
核磁测试结果为:1h nmr(400mhz,chloroform-d)δ7.37(dt,j=7.4,3.7hz,1h),7.33
–
7.28(m,1h),7.20
–
7.14(m,2h),4.48(d,j=5.5hz,0.18h);
[0123]
13
c nmr(101mhz,chloroform-d)δ134.27(d,j=5.2hz),131.41,128.20,127.76,127.04,124.95,1.00
–‑
1.24(m);
[0124]
质谱测试结果为:hrms(esi)m/z[m-i-+oh-+na]
+
calcd for c7h5d2clnao
+
167.0203,found 167.0215.(结构如图4所示);
[0125]
红外测试结果为:ir(neat)ν2916,1593,1419,1411,1217,1047,846,751cm
–1;
[0126]
结合核磁、红外和质谱的测试结果,可得本实施例产物3的结构如下:
[0127][0128]
实施例5
[0129]
将0.8mmol原料5(结构式如下)与0.84mmol三苯基膦混合,随后对反应体系进行氩
气保护。在氩气氛围条件下加入4ml干燥四氢呋喃,然后在65℃条件下反应。12h后,将反应液转移至0度冰浴降温。在此温度下加入0.84mmol的甲基锂并反应15分钟。加入0.84mmol碘并将反应液转移至常温反应8小时。待到反应液变成固态,在此温度下向反应液体中加入含有2.4mmol的氢氧化钠1ml氘水溶液反应18h。待到溶液澄清时,进行点板,确认反应完毕后,将反应液过色谱柱,并使用体积比为(0-1)%的乙酸乙酯和石油醚进行洗脱,得到白色固体128mg,产率68%,氘代率95%,将固体进行核磁、红外和质谱测试。
[0130][0131]
核磁测试结果为:1h nmr(400mhz,chloroform-d)δ7.20
–
7.12(m,3h),7.02(s,1h),4.38(s,0.10h),2.30(s,3h);
[0132]
13
c nmr(101mhz,chloroform-d)δ139.14(d,j=5.9hz),138.53,129.50,128.81,125.88,21.41,6.61
–
5.34(m);
[0133]
质谱测试结果为:hrms(esi)m/z[m-i-+oh-+na]
+
calcd for c8h8d2ko
+
163.0489,found 163.0467.(结构如图5所示);
[0134]
红外测试结果为:ir(neat)ν1998,1746,1530,1343,1206,937,773cm
–1;
[0135]
结合核磁、红外和质谱的测试结果,可得本实施例产物5的结构如下:
[0136][0137]
实施例6
[0138]
将0.8mmol原料6(结构式如下)与0.84mmol三苯基膦混合,随后对反应体系进行氩气保护。在氩气氛围条件下加入4ml干燥四氢呋喃,然后在65℃条件下反应。12h后,将反应液转移至0度冰浴降温。在此温度下加入0.84mmol的甲基锂并反应15分钟。加入0.84mmol碘并将反应液转移至常温反应8小时。待到反应液变成固态,在此温度下向反应液体中加入含有2.4mmol的氢氧化钠1ml氘水溶液反应18h。待到溶液澄清时,进行点板,确认反应完毕后,将反应液过色谱柱,并使用体积比为(0-1)%的乙酸乙酯和石油醚进行洗脱,得到白色固体125mg,产率66%,氘代率95%,将固体进行核磁、红外和质谱测试。
[0139][0140]
核磁测试结果为:1h nmr(400mhz,chloroform-d)δ7.24(td,j=8.0,5.8hz,1h),7.13(dt,j=7.6,1.3hz,1h),7.06(dt,j=9.5,2.1hz,1h),6.92(tdd,j=8.4,2.6,1.0hz,1h),4.38(dd,j=3.8,2.4hz,0.10h);
[0141]
13
c nmr(101mhz,chloroform-d)δ163.88,161.43,141.49,130.32,124.40,115.74(d,j=22.0hz),114.95(d,j=21.1hz),4.50
–
3.09(m);
[0142]
质谱测试结果为:hrms(esi)m/z[m-i-]
+
calcd for c7h4d2f
+
111.0574,found 111.0568.(结构如图6所示);
[0143]
红外测试结果为:ir(neat)ν2251,1684,1439,1265,1151,858,779,684cm
–1;
[0144]
结合核磁、红外和质谱的测试结果,可得本实施例产物6的结构如下:
[0145][0146]
实施例7
[0147]
将0.8mmol原料7(结构式如下)与0.84mmol三苯基膦混合,随后对反应体系进行氩气保护。在氩气氛围条件下加入4ml干燥四氢呋喃,然后在65℃条件下反应。12h后,将反应液转移至0度冰浴降温。在此温度下加入0.84mmol的甲基锂并反应15分钟。加入0.84mmol碘并将反应液转移至常温反应8小时。待到反应液变成固态,在此温度下向反应液体中加入含有2.4mmol的氢氧化钠1ml氘水溶液反应18h。待到溶液澄清时,进行点板,确认反应完毕后,将反应液过色谱柱,并使用体积比为(0-1)%的乙酸乙酯和石油醚进行洗脱,得到白色固体164mg,产率82%,氘代率95%,将固体进行核磁、红外和质谱测试。
[0148][0149]
核磁测试结果为:1h nmr(400mhz,chloroform-d)δ7.13
–
7.06(m,1h),6.88
–
6.83(m,1h),6.82
–
6.77(m,1h),6.70
–
6.65(m,1h),4.30(d,j=4.8hz,0.10h),3.68(s,3h).
[0150]
13
c nmr(101mhz,chloroform-d)δ159.71,140.58,129.89,121.10,114.23,113.72,55.34,6.61
–
4.50(m);
[0151]
质谱测试结果为:hrms(esi)m/z[m-i-]
+
calcd for c8h
17
d2o
+
123.0773;found 123.0773.(结构如图7所示);
[0152]
红外测试结果为:ir(neat)ν2932,2357,1597,1485,1263,1036,773,687cm
–1;
[0153]
结合核磁、红外和质谱的测试结果,可得本实施例产物7的结构如下:
[0154][0155]
实施例8
[0156]
将0.8mmol原料8(结构式如下)与0.84mmol三苯基膦混合,随后对反应体系进行氩气保护。在氩气氛围条件下加入4ml干燥四氢呋喃,然后在65℃条件下反应。12h后,将反应液转移至0度冰浴降温。在此温度下加入0.84mmol的甲基锂并反应15分钟。加入0.84mmol碘并将反应液转移至常温反应8小时。待到反应液变成固态,在此温度下向反应液体中加入含有2.4mmol的氢氧化钠1ml氘水溶液反应18h。待到溶液澄清时,进行点板,确认反应完毕后,将反应液过色谱柱,并使用体积比为(0-1)%的乙酸乙酯和石油醚进行洗脱,得到白色固体
125mg,产率66%,氘代率96%,将固体进行核磁、红外和质谱测试。
[0157][0158]
核磁测试结果为:1h nmr(400mhz,chloroform-d)δ7.37
–
7.30(m,2h),6.96(t,j=8.6hz,2h),4.41(d,j=5.1hz,0.09h);
[0159]
13
c nmr(101mhz,chloroform-d)δ163.36,160.90,135.12,130.42,115.91,115.70,4.92
–
3.65(m);
[0160]
质谱测试结果为:hrms(esi)m/z[m-i-]
+
calcd for c7h4d2f
+
111.0574,found 111.0555.(结构如图8所示);
[0161]
红外测试结果为:ir(neat)ν2921,2851,1603,1508,1230,816,543cm
–1;
[0162]
结合核磁、红外和质谱的测试结果,可得本实施例产物8的结构如下:
[0163][0164]
实施例9
[0165]
将0.8mmol原料9(结构式如下)与0.84mmol三苯基膦混合,随后对反应体系进行氩气保护。在氩气氛围条件下加入4ml干燥四氢呋喃,然后在65℃条件下反应。12h后,将反应液转移至0度冰浴降温。在此温度下加入0.84mmol的甲基锂并反应15分钟。加入0.84mmol碘并将反应液转移至常温反应8小时。待到反应液变成固态,在此温度下向反应液体中加入含有2.4mmol的氢氧化钠1ml氘水溶液反应18h。待到溶液澄清时,进行点板,确认反应完毕后,将反应液过色谱柱,并使用体积比为(0-1)%的乙酸乙酯和石油醚进行洗脱,得到白色固体175mg,产率86%,氘代率97%,将固体进行核磁、红外和质谱测试。
[0166][0167]
核磁测试结果为:1h nmr(400mhz,chloroform-d)δ7.25(q,j=8.5hz,4h),4.37(d,j=4.6hz,0.06h).
[0168]
13
c nmr(101mhz,chloroform-d)δ137.78,133.62,130.10,129.06,4.92
–
3.44(m);
[0169]
质谱测试结果为:hrms(esi)m/z[m-i-]
+
calcd for c7h4d2cl
+
127.0278,found 127.0278.(结构如图9所示);
[0170]
红外测试结果为:ir(neat)ν2916,2274,1905,1592,1490,1224,1087,818cm
–1;
[0171]
结合核磁、红外和质谱的测试结果,可得本实施例产物9的结构如下:
[0172][0173]
实施例10
[0174]
将0.8mmol原料10(结构式如下)与0.84mmol三苯基膦混合,随后对反应体系进行氩气保护。在氩气氛围条件下加入4ml干燥四氢呋喃,然后在65℃条件下反应。12h后,将反应液转移至0度冰浴降温。在此温度下加入0.84mmol的甲基锂并反应15分钟。加入0.84mmol碘并将反应液转移至常温反应8小时。待到反应液变成固态,在此温度下向反应液体中加入含有2.4mmol的氢氧化钠1ml氘水溶液反应18h。待到溶液澄清时,进行点板,确认反应完毕后,将反应液过色谱柱,并使用体积比为(0-1)%的乙酸乙酯和石油醚进行洗脱,得到白色固体175mg,产率86%,氘代率97%,将固体进行核磁、红外和质谱测试。
[0175][0176]
核磁测试结果为:1h nmr(400mhz,chloroform-d)δ7.61
–
7.56(m,2h),7.49
–
7.44(m,2h),4.43(d,j=4.6hz,0.13h);
[0177]
13
c nmr(101mhz,chloroform-d)δ144.61,132.62,129.44,118.50,111.55,3.41
–
2.48(m);
[0178]
质谱测试结果为:hrms(esi)m/z[m-i-+oh-+h]
+
calcd for c8h6d2no
+
136.0726,found 136.0725.(结构如图10所示);
[0179]
红外测试结果为:ir(neat)ν2917,2222,1932,1605,1503,833,812,736cm
–1;
[0180]
结合核磁、红外和质谱的测试结果,可得本实施例产物10的结构如下:
[0181][0182]
实施例11
[0183]
将0.8mmol原料11(结构式如下)与0.84mmol三苯基膦混合,随后对反应体系进行氩气保护。在氩气氛围条件下加入4ml干燥四氢呋喃,然后在65℃条件下反应。12h后,将反应液转移至0度冰浴降温。在此温度下加入0.84mmol的甲基锂并反应15分钟。加入0.84mmol碘并将反应液转移至常温反应8小时。待到反应液变成固态,在此温度下向反应液体中加入含有2.4mmol的氢氧化钠1ml氘水溶液反应18h。待到溶液澄清时,进行点板,确认反应完毕后,将反应液过色谱柱,并使用体积比为(0-1)%的乙酸乙酯和石油醚进行洗脱,得到白色固体188mg,产率85%,氘代率95%,将固体进行核磁、红外和质谱测试。
[0184][0185]
核磁测试结果为:1h nmr(400mhz,chloroform-d)δ7.30(s,4h),4.44(d,j=5.0hz,0.11h),1.30(s,9h).
[0186]
13
c nmr(101mhz,chloroform-d)δ151.03,136.06,128.47,125.82,34.67,31.28,6.18
–
5.60(m);
[0187]
质谱测试结果为:hrms(esi)m/z[m-i-]
+
calcd for c
11h13d2+
149.1294,found 149.1291.(结构如图11所示);
[0188]
红外测试结果为:ir(neat)ν3049,2958,2066,1599,1505,1191,799,473cm
–1;
[0189]
结合核磁、红外和质谱的测试结果,可得本实施例产物11的结构如下:
[0190][0191]
实施例12
[0192]
将0.8mmol原料12(结构式如下)与0.84mmol三苯基膦混合,随后对反应体系进行氩气保护。在氩气氛围条件下加入4ml干燥四氢呋喃,然后在65℃条件下反应。12h后,将反应液转移至0度冰浴降温。在此温度下加入0.84mmol的甲基锂并反应15分钟。加入0.84mmol碘并将反应液转移至常温反应8小时。待到反应液变成固态,在此温度下向反应液体中加入含有2.4mmol的氢氧化钠1ml氘水溶液反应18h。待到溶液澄清时,进行点板,确认反应完毕后,将反应液过色谱柱,并使用体积比为(0-1)%的乙酸乙酯和石油醚进行洗脱,得到白色固体156mg,产率61%,氘代率91%,将固体进行核磁、红外和质谱测试。
[0193][0194]
核磁测试结果为:1h nmr(400mhz,chloroform-d)δ7.83
–
7.79(m,2h),7.31
–
7.28(m,2h),4.33(d,j=4.8hz,0.18h),1.49(s,9h);
[0195]
13
c nmr(101mhz,chloroform-d)δ165.15,143.74,131.45,129.95,128.56,81.11,28.23,4.50
–
3.65(m);
[0196]
质谱测试结果为:hrms(esi)m/z[m-i-]
+
calcd for c
12h13
d2o
2+
193.1192,found 193.1169.(结构如图12所示);
[0197]
红外测试结果为:ir(neat)ν2813,2533,1670,1420,1280,1016,849,695cm
–1;
[0198]
结合核磁、红外和质谱的测试结果,可得本实施例产物的结构如下:
[0199][0200]
实施例13
[0201]
将0.8mmol原料13(结构式如下)与0.84mmol三苯基膦混合,随后对反应体系进行氩气保护。在氩气氛围条件下加入4ml干燥四氢呋喃,然后在65℃条件下反应。12h后,将反应液转移至0度冰浴降温。在此温度下加入0.84mmol的甲基锂并反应15分钟。加入0.84mmol碘并将反应液转移至常温反应8小时。待到反应液变成固态,在此温度下向反应液体中加入含有2.4mmol的氢氧化钠1ml氘水溶液反应18h。待到溶液澄清时,进行点板,确认反应完毕后,将反应液过色谱柱,并使用体积比为(0-1)%的乙酸乙酯和石油醚进行洗脱,得到白色固体241mg,产率85%,氘代率96%,将固体进行核磁、红外和质谱测试。
[0202][0203]
核磁测试结果为:1h nmr(400mhz,chloroform-d)δ7.71(d,j=8.3hz,2h),7.49(d,j=8.4hz,2h),4.45(d,j=4.7hz,0.09h),3.23(q,j=7.1hz,4h),1.12(t,j=7.2hz,6h).
[0204]
13
c nmr(101mhz,chloroform-d)δ143.84,139.52,129.38,127.44,42.17,14.29,4.08
–
2.67(m);
[0205]
质谱测试结果为:hrms(esi)m/z[m-i-]
+
calcd for c
11h14
d2no2s
+
228.1022,found 228.1025.(结构如图13所示);
[0206]
红外测试结果为:ir(neat)ν2972,2928,1932,1464,1326,1145,1087,941cm
–1;
[0207]
红外测试结果为:ir(kbr)ν3062,2920,1642,1466,1260,1209,1172,832,763,696,525cm-1
;
[0208]
结合核磁、红外和质谱的测试结果,可得本实施例产物13的结构如下:
[0209][0210]
实施例14
[0211]
将0.8mmol原料14(结构式如下)与0.84mmol三苯基膦混合,随后对反应体系进行氩气保护。在氩气氛围条件下加入4ml干燥四氢呋喃,然后在65℃条件下反应。12h后,将反应液转移至0度冰浴降温。在此温度下加入0.84mmol的甲基锂并反应15分钟。加入0.84mmol碘并将反应液转移至常温反应8小时。待到反应液变成固态,在此温度下向反应液体中加入含有2.4mmol的氢氧化钠1ml氘水溶液反应18h。待到溶液澄清时,进行点板,确认反应完毕后,将反应液过色谱柱,并使用体积比为(0-1)%的乙酸乙酯和石油醚进行洗脱,得到白色
固体106mg,产率50%,氘代率95%,将固体进行核磁、红外和质谱测试。
[0212][0213]
核磁测试结果为:1h nmr(400mhz,chloroform-d)δ6.84(dt,j=4.5,2.0hz,2h),6.69(d,j=8.4hz,2h),5.92(s,2h),4.41(d,j=5.0hz,0.10h);
[0214]
13
c nmr(101mhz,chloroform-d)δ147.85,147.39,132.88,122.19,109.26,108.41,101.35,7.45
–
5.76(m);
[0215]
质谱测试结果为:hrms(esi)m/z[m-i-]
+
calcd for c8h5d2o
2+
137.0566,found 137.0557.(结构如图14所示);
[0216]
红外测试结果为:ir(neat)ν2192,1980,1529,1343,1206,1148,937cm
–1;
[0217]
结合核磁、红外和质谱的测试结果,可得本实施例产物14的结构如下:
[0218][0219]
实施例15
[0220]
将0.8mmol原料15(结构式如下)与0.84mmol三苯基膦混合,随后对反应体系进行氩气保护。在氩气氛围条件下加入4ml干燥四氢呋喃,然后在65℃条件下反应。12h后,将反应液转移至0度冰浴降温。在此温度下加入0.84mmol的甲基锂并反应15分钟。加入0.84mmol碘并将反应液转移至常温反应8小时。待到反应液变成固态,在此温度下向反应液体中加入含有2.4mmol的氢氧化钠1ml氘水溶液反应18h。待到溶液澄清时,进行点板,确认反应完毕后,将反应液过色谱柱,并使用体积比为(0-1)%的乙酸乙酯和石油醚进行洗脱,得到白色固体197mg,产率91%,氘代率96%,将固体进行核磁、红外和质谱测试。
[0221][0222]
核磁测试结果为:1h nmr(400mhz,chloroform-d)δ7.82
–
7.73(m,4h),7.49
–
7.40(m,3h),4.59(d,j=5.0hz,0.08h);
[0223]
13
c nmr(101mhz,chloroform-d)δ136.50,133.32,132.84,128.78,127.80(d,j=6.6hz),127.06,126.93,126.50,126.41,6.74
–
5.96(m);
[0224]
质谱测试结果为:hrms(esi)m/z[m-i-+oh-+na]
+
calcd for c
11
h8d2nao
+
183.0749,found 183.0772.(结构如图15所示);
[0225]
红外测试结果为:ir(neat)ν2174,1980,1734,1528,1344,1145,918cm
–1;
[0226]
结合核磁、红外和质谱的测试结果,可得本实施例产物15的结构如下:
1)%的乙酸乙酯和石油醚进行洗脱,得到白色固体241mg,产率85%,氘代率96%,将固体进行核磁、红外和质谱测试。
[0240][0241]
核磁测试结果为:1h nmr(cdcl3,400mhz)δ7.83
–
7.77(m,1h),7.71(d,j=7.6hz,1h),7.59(t,j=7.7hz,1h),7.52
–
7.37(m,6h),7.34
–
7.23(m,3h),7.16(d,j=7.9hz,2h),5.42(s,0.11h),3.72(s,3h),2.92(t,j=7.9hz,2h),2.77(s,3h),1.87(h,j=7.5hz,2h),1.05(t,j=7.4hz,3h);
[0242]
13
c{1h}nmr(cdcl3,101mhz)δ156.41,154.62,144.54,143.20,142.90,137.88,136.70,136.41(d,j=6.1hz),134.90,133.74,132.9,129.96,129.49(d,j=13.4hz),127.80,126.53,123.98,122.48,122.27,119.49,118.53,111.14,109.58,108.81,47.13
–
45.66(m),31.81,29.81,21.85,16.89,14.09;
[0243]
质谱测试结果为:hrms(esi)m/z:[m+na]
+
calcd for c
33h27
d2n5na
+
520.2441;found 520.2449.(结果如图17所示);
[0244]
红外测试结果为:ir(neat)ν2921,2223,1595,1282,863,763,562cm
–1;
[0245]
结合核磁、红外和质谱的测试结果,可得本实施例产物17的结构如下:
[0246][0247]
实施例18
[0248]
将0.8mmol原料4(结构式如下)与0.84mmol三苯基膦混合,随后对反应体系进行氩气保护。在氩气氛围条件下加入4ml干燥四氢呋喃,然后在65℃条件下反应。12h后,将反应液转移至0度冰浴降温。在此温度下加入0.84mmol的甲基锂并反应15分钟。加入0.84mmol碘并将反应液转移至常温反应8小时。待到反应液变成固态,在此温度下向反应液体中加入含有2.4mmol的氢氧化钠1ml氘水溶液反应18h。待到溶液澄清后,加入0.84mmol的原料胺2(结构式如下)反应4h后,进行点板,确认反应完毕后,将反应液过色谱柱,并使用体积比为(0-1)%的乙酸乙酯和石油醚进行洗脱,得到白色固体241mg,产率85%,氘代率96%,将固体进行核磁、红外和质谱测试。
[0249][0250]
核磁测试结果为:1h nmr(cdcl3,400mhz)δ7.52(dd,j=7.6,1.9hz,1h),7.33(dd,j=7.7,1.5hz,1h),7.18(dtd,j=22.4,7.4,1.7hz,2h),7.02(d,j=5.2hz,1h),6.67(d,j=5.1hz,1h),3.78(d,j=9.3hz,0.18h),3.60(s,2h),2.86(d,j=4.6hz,2h),2.82(dd,j=6.2,3.6hz,2h);
[0251]
13
c{1h}nmr(cdcl3,101mhz)δ136.18(d,j=5.5hz),134.36,134.01,133.50,130.74,129.53,128.31,126.81,125.35,122.70,58.58
–
57.11(m),53.18,50.78,25.66;
[0252]
质谱测试结果为:hrms(esi)m/z:[m+na]
+
calcd for c
14h12
d2clnnas
+
288.0553;found 288.0557.(结果如图18所示);
[0253]
红外测试结果为:ir(neat)ν2917,1650,1436,1242,1025,748,697cm
–1;
[0254]
结合核磁、红外和质谱的测试结果,可得本实施例产物18的结构如下:
[0255][0256]
实施例19
[0257]
将0.8mmol原料5(结构式如下)与0.84mmol三苯基膦混合,随后对反应体系进行氩气保护。在氩气氛围条件下加入4ml干燥四氢呋喃,然后在65℃条件下反应。12h后,将反应液转移至0度冰浴降温。在此温度下加入0.84mmol的甲基锂并反应15分钟。加入0.84mmol碘并将反应液转移至常温反应8小时。待到反应液变成固态,在此温度下向反应液体中加入含有2.4mmol的氢氧化钠1ml氘水溶液反应18h。待到溶液澄清后,加入0.84mmol的原料胺3(结构式如下)反应4h后,进行点板,确认反应完毕后,将反应液过色谱柱,并使用体积比为(0-1)%的乙酸乙酯和石油醚进行洗脱,得到白色固体241mg,产率85%,氘代率96%,将固体进行核磁、红外和质谱测试。
[0258][0259]
核磁测试结果为:1h nmr(cdcl3,400mhz)δ7.32(t,j=7.7hz,4h),7.20(q,j=8.5,7.9hz,4h),7.14(q,j=7.7hz,2h),7.10
–
7.04(m,2h),7.01(d,j=7.4hz,1h),3.42(d,j=5.1hz,0.10h),2.42(d,j=22.9hz,8h),2.29(s,3h);
[0260]
13
c{1h}nmr(cdcl3,101mhz)δ142.31,141.53,137.83(d,j=5.8hz),132.58,130.11,129.34,128.68(d,j=6.3hz),128.16,127.95(d,j=10.3hz),127.20,126.50,75.55,63.15
–
61.82(m),53.41,51.91,21.53;
[0261]
质谱测试结果为:hrms(esi)m/z:[m+h]
+
calcd for c
25h26
d2cln
2+
393.2061;found 393.2077.(结果如图19所示);
[0262]
红外测试结果为:ir(neat)ν3024,2803,2042,1679,1486,1139,756cm
–1;
[0263]
结合核磁、红外和质谱的测试结果,可得本实施例产物19的结构如下:
[0264][0265]
实施例20
[0266]
将0.8mmol原料11(结构式如下)与0.84mmol三苯基膦混合,随后对反应体系进行氩气保护。在氩气氛围条件下加入4ml干燥四氢呋喃,然后在65℃条件下反应。12h后,将反应液转移至0度冰浴降温。在此温度下加入0.84mmol的甲基锂并反应15分钟。加入0.84mmol碘并将反应液转移至常温反应8小时。待到反应液变成固态,在此温度下向反应液体中加入含有2.4mmol的氢氧化钠1ml氘水溶液反应18h。待到溶液澄清后,加入0.84mmol的原料胺3(结构式如下)反应4h后,进行点板,确认反应完毕后,将反应液过色谱柱,并使用体积比为(0-1)%的乙酸乙酯和石油醚进行洗脱,得到白色固体241mg,产率85%,氘代率96%,将固体进行核磁、红外和质谱测试。
[0267][0268]
核磁测试结果为:1h nmr(cdcl3,400mhz)δ7.34
–
7.27(m,6h),7.23
–
7.15(m,6h),7.11(t,j=7.3hz,1h),4.18(s,1h),3.44(d,j=12.1hz,0.11h),2.41(d,j=24.8hz,8h),1.27(s,9h);
[0269]
13
c{1h}nmr(cdcl3,101mhz)δ149.97,142.34,141.57,135.03(d,j=7.1hz),132.63,129.40,129.17,128.73(d,j=6.7hz),128.06,127.25,125.20,75.62,62.88
–
61.51(m),53.40,52.01,34.60,31.63;
[0270]
质谱测试结果为:hrms(esi)m/z:[m+na]
+
calcd for c
28h31
d2cln2na
+
457.2350;found 457.2319.(结果如图20所示);
[0271]
红外测试结果为:ir(neat)ν2960,2803,1738,1487,1139,906,731,543cm
–1;
[0272]
结合核磁、红外和质谱的测试结果,可得本实施例产物20的结构如下:
[0273][0274]
实施例21
[0275]
将0.8mmol原料14(结构式如下)与0.84mmol三苯基膦混合,随后对反应体系进行氩气保护。在氩气氛围条件下加入4ml干燥四氢呋喃,然后在65℃条件下反应。12h后,将反应液转移至0度冰浴降温。在此温度下加入0.84mmol的甲基锂并反应15分钟。加入0.84mmol碘并将反应液转移至常温反应8小时。待到反应液变成固态,在此温度下向反应液体中加入含有2.4mmol的氢氧化钠1ml氘水溶液反应18h。待到溶液澄清后,加入0.84mmol的原料胺4(结构式如下)反应4h后,进行点板,确认反应完毕后,将反应液过色谱柱,并使用体积比为(0-1)%的乙酸乙酯和石油醚进行洗脱,得到白色固体241mg,产率85%,氘代率96%,将固体进行核磁、红外和质谱测试。
[0276][0277]
核磁测试结果为:1h nmr(cdcl3,400mhz)δ8.28(d,j=4.7hz,2h),6.88(s,1h),6.75(s,2h),6.44(t,j=4.7hz,1h),5.93(s,2h),3.86
–
3.77(m,4h),3.42(d,j=12.3hz,0.09h),2.52
–
2.42(m,4h).
[0278]
13
c{1h}nmr(cdcl3,101mhz)δ161.66,157.65,147.67,146.67,131.73,122.21,109.68,109.48,107.88,100.88,62.18(dq,j=40.9,20.4hz),52.76,43.69;
[0279]
质谱测试结果为:hrms(esi)m/z:[m+k]
+
calcd for c
16h16
d2kn4o
2+
339.1187;found 339.1189.(结果如图21所示);
[0280]
红外测试结果为:ir(neat)ν2923,2056,1578,1480,1248,1037,886,638cm
–1;
[0281]
结合核磁、红外和质谱的测试结果,可得本实施例产物21的结构如下:
[0282][0283]
实施例22
[0284]
将0.8mmol原料1(结构式如下)与0.84mmol三苯基膦混合,随后对反应体系进行氩气保护。在氩气氛围条件下加入4ml干燥四氢呋喃,然后在65℃条件下反应。12h后,将反应液转移至0度冰浴降温。在此温度下加入0.84mmol的甲基锂并反应15分钟。加入0.84mmol碘
并将反应液转移至常温反应8小时。待到反应液变成固态,在此温度下向反应液体中加入含有2.4mmol的邻苯二甲酰亚胺钾的1ml氘水溶液反应18h。待到溶液澄清后进行点板,确认反应完毕后,将反应液过色谱柱,并使用体积比为(10-15)%的乙酸乙酯和石油醚进行洗脱,得到白色固体202mg,产率80%,氘代率98%,将固体进行核磁、红外和质谱测试。
[0285][0286]
核磁测试结果为:1h nmr(cdcl3,400mhz)δ7.77(tt,j=5.1,2.5hz,1h),7.63(td,j=5.3,2.1hz,2h),7.48
–
7.41(m,5h),7.33(t,j=7.5hz,2h),7.27
–
7.22(m,1h),4.80(d,j=5.9hz,0.04h);
[0287]
13
c{1h}nmr(cdcl3,100mhz)δ168.09,140.85,135.26,134.01,132.17,129.12,128.76,127.40(d,j=9.7hz),127.11,123.38,40.85(dd,j=24.6,18.9hz);
[0288]
质谱测试结果为:hrms(esi)m/z:[m+na]
+
calcd for c
21h13
d2nnao
2+
338.1121;found 338.1120.(结果如图22所示);
[0289]
红外测试结果为:ir(neat)ν2165,1769,1700,1485,1185,918,729,525cm
–1;
[0290]
结合核磁、红外和质谱的测试结果,可得本实施例产物22的结构如下:
[0291][0292]
实施例23
[0293]
将0.8mmol原料8(结构式如下)与0.84mmol三苯基膦混合,随后对反应体系进行氩气保护。在氩气氛围条件下加入4ml干燥四氢呋喃,然后在65℃条件下反应。12h后,将反应液转移至0度冰浴降温。在此温度下加入0.84mmol的甲基锂并反应15分钟。加入0.84mmol碘并将反应液转移至常温反应8小时。待到反应液变成固态,在此温度下向反应液体中加入含有2.4mmol的邻苯二甲酰亚胺钾的1ml氘水溶液反应18h。待到溶液澄清后进行点板,确认反应完毕后,将反应液过色谱柱,并使用体积比为(10-15)%的乙酸乙酯和石油醚进行洗脱,得到白色固体101mg,产率50%,氘代率98%,将固体进行核磁、红外和质谱测试。
[0294][0295]
核磁测试结果为:1h nmr(cdcl3,400mhz)δ7.82(dd,j=5.4,3.1hz,2h),7.68(dd,j=5.5,3.1hz,2h),7.45
–
7.39(m,2h),7.02
–
6.94(m,2h),4.78(s,0.04h);
[0296]
13
c{1h}nmr(cdcl3,101mhz)δ167.90,163.57,161.12,134.00,132.06,130.56(d,j=8.2hz),123.32,115.60,115.39,41.02
–
39.81(m);
[0297]
质谱测试结果为:hrms(esi)m/z:[m+na]
+
calcd for c
15
h8d2fnnao
2+
280.0713;found 280.0689.(结果如图23所示);
[0298]
红外测试结果为:ir(neat)ν3071,1762,1709,1506,1388,1186,917,716cm
–1;
[0299]
结合核磁、红外和质谱的测试结果,可得本实施例产物23的结构如下:
[0300][0301]
实施例24
[0302]
将0.8mmol原料17(结构式如下)与0.84mmol三苯基膦混合,随后对反应体系进行氩气保护。在氩气氛围条件下加入4ml干燥四氢呋喃,然后在65℃条件下反应。12h后,将反应液转移至0度冰浴降温。在此温度下加入0.84mmol的甲基锂并反应15分钟。加入0.84mmol碘并将反应液转移至常温反应8小时。待到反应液变成固态,在此温度下向反应液体中加入含有2.4mmol的邻苯二甲酰亚胺钾的1ml氘水溶液反应18h。待到溶液澄清后进行点板,确认反应完毕后,将反应液过色谱柱,并使用体积比为(10-15)%的乙酸乙酯和石油醚进行洗脱,得到白色固体72mg,产率38%,氘代率98%,将固体进行核磁、红外和质谱测试。
[0303][0304]
核磁测试结果为:1h nmr(cdcl3,400mhz)δ7.80(dt,j=7.4,3.7hz,2h),7.71
–
7.61(m,2h),7.46
–
7.39(m,2h),7.35
–
7.21(m,3h),4.82(d,j=5.5hz,0.04h);
[0305]
13
c{1h}nmr(cdcl3,100mhz)δ168.00,136.30,133.95,132.14,128.66(d,j=3.5hz),127.84,123.31,41.14(dt,j=42.2,21.4hz);
[0306]
质谱测试结果为:hrms(esi)m/z:[m+na]
+
calcd for c
15
h8d2fnnao
2+
280.0713;found 280.0689.(结果如图24所示);
[0307]
红外测试结果为:ir(neat)ν3071,1762,1709,1506,1388,1186,917,716cm
–1;
[0308]
结合核磁、红外和质谱的测试结果,可得本实施例产物24的结构如下:
[0309][0310]
与现有技术相比,本发明提供的苄位氘代的α,α-二氘苄碘、二氘苄胺、二氘药物分子及其合成方法,有益效果在于:
[0311]
一、本发明的苄位氘代的α,α-二氘苄碘,以苄氯或苄溴为原料,氘水为氘源采用一锅法合成,合成工艺简单成本低,产率和氘代率高,可达到86%的产率和97%的氘代率。
[0312]
二、本发明的苄位氘代的α,α-二氘苄碘,作为重要的合成单体,通过向反应体系中
加入邻苯二甲酰亚胺钾或二级胺,可在一锅法条件下进一步合成苄位氘代的α,α-二氘苄胺和苄位氘代的α,α-二氘药物分子,充分说明了该合成方法的实用价值。
[0313]
以上对本发明的实施方式作出详细说明,但本发明不局限于所描述的实施方式。对本领域的技术人员而言,在不脱离本发明的原理和精神的情况下对这些实施例进行的多种变化、修改、替换和变型均仍落入在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1