一种酰基化海洋生物多糖衍生物及其制备方法和应用
1.本发明属于海洋化工工程技术,具体涉及一种酰基化海洋生物多糖衍生物及其制备方法和应用。
背景技术:
2.化学农药为减少农业病虫害,作物增产做出了重要的贡献。然而,化学农药的不合理使用,也带来了农药残留、耐药性、环境污染和生态平衡破坏等一系列问题,严重威胁我国农产品安全和农业生态环境安全。因此,需要大力发展绿色、高效、低毒的生物农药,推动农业病害防治的绿色发展。
3.壳聚糖(chitosan),是目前发现的唯一的一种天然阳离子聚合物,它由虾蟹壳中提取的甲壳素(chitin)经脱乙酰处理制得,具有优异的生物活性和物理化学性质。壳寡糖(chitooligosaccharide)是壳聚糖经降解后得到的小分子多糖,分子量一般低于2000da,安全无毒且具有良好的生物兼容性,与人体细胞有良好的亲和性,无免疫原性。壳寡糖及其衍生物因其特有生物活性对多种细菌、真菌具有广谱抗菌的功能。
4.吡啶酰氯作为天然维生素b3烟酸的类似物,其合成方法和生物活性受到科学家的广泛关注,而其多变的结构类型又为不断合成新化合物奠定了基础。为开发新型生物农药的开辟了新途径。
技术实现要素:
5.本发明就是针对上述问题,提供了一种酰基化海洋生物多糖衍生物及其制备方法和应用。
6.为了实现本发明的上述目的,本发明采用如下技术方案为:
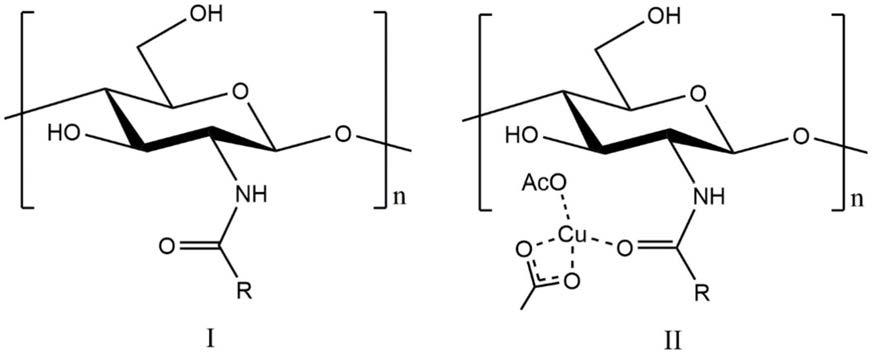
7.一种酰基化海洋生物多糖衍生物,衍生物如式i或式ⅱ所示,
[0008][0009]
式i或式ⅱ中,r为吡啶-3-基,吡啶-4-基或2-氯吡啶-3-基;n=2-20。
[0010]
一种酰基化海洋生物多糖衍生物的制备方法:
[0011]
1)将壳寡糖溶于过量的溶剂a中,而后再加入三甲基氯硅烷和咪唑,在室温条件下进行反应生成o-三甲基硅基壳寡糖;其中,壳寡糖与三甲基氯硅烷的摩尔比为1:1-3;
[0012]
所述壳寡糖分子量为1000-1500。
[0013]
2)o-三甲基硅基壳寡糖溶于过量的溶剂b中,而后再加入吡啶酰氯与缚酸剂,在室温条件下进行反应,得到o-三甲基硅基-n-吡啶甲酰基壳寡糖;其中,o-三甲基硅基壳寡糖与吡啶酰氯的摩尔比为1:1-3;
[0014]
3)将上述得到的o-三甲基硅基-n-吡啶甲酰基壳寡糖溶于过量的溶剂c中,而后再加入四丁基氟化铵的四氢呋喃溶液在室温条件下进行反应,得到式i所示n-吡啶甲酰基壳寡糖;
[0015]
4)将上述式i所示n-吡啶甲酰基壳寡糖溶于过量溶剂d中,再加入一水乙酸铜在室温搅拌下反应即得式ⅱ所示配合物吡啶甲酰基壳寡糖铜配合物;其中,n-吡啶甲酰基壳寡糖与一水乙酸铜的质量比为1:1-2。
[0016]
所述步骤1)壳寡糖溶于溶剂a中,而后再加入三甲基氯硅烷和咪唑在室温下反应12-16小时生成o-三甲基硅基壳寡糖;其中,溶剂a的体积与壳寡糖质量比为10-20:1;壳寡糖与咪唑的质量比为1:1;溶剂a为n,n-二甲基甲酰胺和/或n-甲基吡咯烷酮。
[0017]
所述步骤2)o-三甲基硅基壳寡糖溶于过量的溶剂b中,而后再加入吡啶酰氯在室温条件下反应12-24小时,即得o-三甲基硅基-n-吡啶甲酰基壳寡糖;其中,o-三甲基硅基壳寡糖与吡啶酰氯的摩尔比为1:1-3;溶剂b的体积与壳寡糖质量比为10-20:1;所述吡啶酰氯为烟酰氯盐酸盐、异烟酰氯盐酸盐或2-氯烟酰氯;所述的缚酸剂为吡啶和/或三乙胺;溶剂为n,n-二甲基甲酰胺和/或n-甲基吡咯烷酮。
[0018]
所述步骤3)将上述得到的o-三甲基硅基-n-吡啶甲酰基壳寡糖溶于过量的溶剂c中,而后再加入四丁基氟化铵的四氢呋喃溶液在室温条件下反应6-12小时,得到式i所示n-吡啶甲酰基壳寡糖;其中,溶剂c的体积与o-三甲基硅基-n-吡啶甲酰基壳寡糖质量比为10-20:1;四丁基氟化铵的四氢呋喃溶液与o-三甲基硅基-n-吡啶甲酰基壳寡糖质量比为10-20:1;溶剂c为水和/或四氢呋喃。
[0019]
所述步骤4)将获得式i所示n-吡啶甲酰基壳寡糖溶于过量溶剂d中,再加入一水乙酸铜在室温搅拌下反应6-12小时,反应后过滤,滤饼用溶剂洗涤,在50-80℃下进行干燥,即得式ⅱ所示配合物吡啶甲酰基壳寡糖铜配合物;其中,溶剂的体积与n-吡啶甲酰基壳寡糖质量比为40-100:1。
[0020]
一种酰基化海洋生物多糖衍生物的应用,所述式i所示衍生物用于制备农用杀菌剂中的应用。
[0021]
所述式i所示衍生物用于制备农用真菌杀菌剂中的应用。
[0022]
原理:壳寡糖结构中含有-nh2,可与吡啶酰氯发生亲核取代反应在壳寡糖结构中引入吡啶甲酰基,进而与壳寡糖产生协同作用,显著提高衍生物的生物活性;同时再用吡啶甲酰基壳寡糖中电子密度高的羰基配位铜离子,进而与壳寡糖产生协同作用,显著提高衍生物的生物活性。
[0023]
本发明的优点:
[0024]
1.本发明在壳寡糖结构中引入吡啶甲酰基,二者产生协同增效作用,显著提高了壳寡糖的抑菌活性;同时进一步在壳寡糖结构中引入吡啶甲酰基与铜离子,三者产生协同增效作用,显著提高了壳寡糖的抑菌活性。
[0025]
2.本发明制备的吡啶甲酰基壳寡糖衍生物具有良好的水溶性,在提高抑菌效果的同时还避免了重金属离子在土壤中残留,扩大了其应用领域,在农药领域有着潜在的应用
价值。
附图说明
[0026]
图1为壳寡糖的红外光谱图,其红外特征吸收(cm-1
):3259,2884,1520,1410,1068。
[0027]
图2为本发明实施例提供的壳寡糖衍生物1的红外光谱图,其红外特征吸收(cm-1
):3357,1725,1652,1555,1421,1288。
[0028]
图3为本发明实施例提供的壳寡糖衍生物2的红外光谱图,其红外特征吸收(cm-1
):3294,1732,1651,1541,1410,1282。
[0029]
图4为本发明实施例提供的壳寡糖衍生物3的红外光谱图,其红外特征吸收(cm-1
):3364,1733,1653,1562,1404,1297。
[0030]
图5为本发明实施例提供的壳寡糖衍生物4的红外光谱图,其红外特征吸收(cm-1
):3275,1728,1647,1558,1421,1286。
[0031]
图6为本发明实施例提供的壳寡糖衍生物5的红外光谱图,其红外特征吸收(cm-1
):3272,1734,1649,1558,1412,1284。
[0032]
图7为本发明实施例提供的壳寡糖衍生物6的红外光谱图,其红外特征吸收(cm-1
):3290,1735,1655,1561,1403,1296。
具体实施方式
[0033]
下面结合说明书附图对本发明作进一步说明,并且本发明的保护范围不仅局限于以下实施例。
[0034]
本发明中首先采用三甲基氯硅烷保护壳寡糖的6位羟基,再将吡啶酰氯与壳寡糖c2位上的-nh2发生反应生成o-三甲基硅基-n-吡啶甲酰基壳寡糖,之后在四丁基氟化铵作用下脱去三甲基硅基生成n-吡啶甲酰基壳寡糖衍生物,该壳寡糖衍生物上羰基中的氧与乙酸铜的铜离子配位生成吡啶甲酰基壳寡糖铜配合物衍生物,所得的衍生物经红外光谱分析确定其结构,壳寡糖与接入的基团有效地结合生成吡啶甲酰基壳寡糖衍生物,同时在经铜配合获得衍生物。
[0035]
实施例1衍生物1的制备
[0036]
将15克分子量为1000的壳寡糖(参见图1)、12.1克三甲基氯硅烷和15克咪唑加入到100ml n,n-二甲基甲酰胺中,35℃反应12小时,反应物加入三倍体积乙酸乙酯,抽滤得到沉淀,用无水乙醇洗涤,50摄氏度下干燥,即得o-三甲基硅基壳寡糖。
[0037]
将4克o-三甲基硅基壳寡糖加入到30mln,n-二甲基甲酰胺和30ml吡啶中,搅拌下向其加入6.12g烟酰氯盐酸盐,35℃反应24小时,加入丙酮沉淀,沉淀抽滤,丙酮洗涤,50℃下干燥,得黄色粉末,即为o-三甲基硅基-n-烟酰基壳寡糖。
[0038]
将1.5克o-三甲基硅基-n-烟酰基壳寡糖加入到15ml水中,搅拌下向其15ml四丁基氟化铵的四氢呋喃溶液(1mol/l),25℃反应4小时,加入乙醇沉淀,沉淀抽滤,无水乙醇洗涤,50℃下干燥,得黄色粉末,即为吡啶甲酰基壳寡糖衍生物1,结构见通式i(r为吡啶-3-基;n=2-20)。
[0039]
红外光谱表明:吡啶甲酰基壳寡糖衍生物1的红外谱图(图2)与全壳寡糖的红外谱图(图1)相比,出现了位于1725cm-1
的羰基特征吸收峰,以及1652cm-1
、1555cm-1
以及1288cm-1
的仲酰胺特征吸收峰;1594cm-1
为吡啶环的特征吸收;证明目衍生物1合成成功。
[0040]
实施例2衍生物2的制备
[0041]
将15克分子量为1000的壳寡糖、12.1克三甲基氯硅烷和15克咪唑加入到100ml n,n-二甲基甲酰胺中,35℃反应12小时,反应物加入三倍体积乙酸乙酯,抽滤得到沉淀,用无水乙醇洗涤,50摄氏度下干燥,即得o-三甲基硅基壳寡糖。
[0042]
将4克o-三甲基硅基壳寡糖加入到30mln,n-二甲基甲酰胺和30ml吡啶中,搅拌下向其加入6.12g异烟酰氯盐酸盐,35℃反应24小时,加入丙酮沉淀,沉淀抽滤,丙酮洗涤,50℃下干燥,得黄色粉末,即为o-三甲基硅基-n-异烟酰基壳寡糖。
[0043]
将1.5克o-三甲基硅基-n-异烟酰基壳寡糖加入到15ml水中,搅拌下向其15ml四丁基氟化铵的四氢呋喃溶液(1mol/l),25℃反应4小时,加入乙醇沉淀,沉淀抽滤,无水乙醇洗涤,50℃下干燥,得黄色粉末,即为吡啶甲酰基壳寡糖衍生物2,结构见通式i(r为吡啶-4-基;n=2-20)。
[0044]
红外光谱表明:吡啶甲酰基壳寡糖衍生物2的红外谱图(图3)与全壳寡糖的红外谱图(图1)相比,出现了位于1732cm-1
的羰基特征吸收峰,以及1651cm-1
、1541cm-1
以及1282cm-1
的仲酰胺特征吸收峰;1603cm-1
为吡啶环的特征吸收;证明目衍生物2合成成功。
[0045]
实施例3衍生物3的制备
[0046]
将15克分子量为1000的壳寡糖、12.1克三甲基氯硅烷和15克咪唑加入到100ml n,n-二甲基甲酰胺中,35℃反应12小时,反应物加入三倍体积乙酸乙酯,抽滤得到沉淀,用无水乙醇洗涤,50摄氏度下干燥,即得o-三甲基硅基壳寡糖。
[0047]
将4克o-三甲基硅基壳寡糖加入到30mln,n-二甲基甲酰胺和30ml吡啶中,搅拌下向其加入6.04g 2-氯烟酰氯,35℃反应24小时,加入丙酮沉淀,沉淀抽滤,丙酮洗涤,50℃下干燥,得黄色粉末,即为o-三甲基硅基-n-(2-氯烟酰基)壳寡糖。
[0048]
将1.5克o-三甲基硅基-n-(2-氯烟酰基)壳寡糖加入到15ml水中,搅拌下向其15ml四丁基氟化铵的四氢呋喃溶液(1mol/l),25℃反应4小时,加入乙醇沉淀,沉淀抽滤,无水乙醇洗涤,50℃下干燥,得黄色粉末,即为吡啶甲酰基壳寡糖衍生物3,结构见通式i(r为2-氯吡啶-3-基;n=2-20)。
[0049]
红外光谱表明:吡啶甲酰基壳寡糖衍生物3的红外谱图(图4)与全壳寡糖的红外谱图(图1)相比,出现了位于1733cm-1
的羰基特征吸收峰,以及1653cm-1
、1562cm-1
以及1297cm-1
的仲酰胺特征吸收峰;1579cm-1
为吡啶环的特征吸收;证明目衍生物3合成成功。
[0050]
实施例4衍生物4的制备
[0051]
将15克分子量为1500的壳寡糖(参见图1)、12.1克三甲基氯硅烷和15克咪唑加入到100ml n,n-二甲基甲酰胺中,35℃反应12小时,反应物加入三倍体积乙酸乙酯,抽滤得到沉淀,用无水乙醇洗涤,50摄氏度下干燥,即得o-三甲基硅基壳寡糖。
[0052]
将4克o-三甲基硅基壳寡糖加入到30mln,n-二甲基甲酰胺和30ml吡啶中,搅拌下向其加入6.12g烟酰氯盐酸盐,35℃反应24小时,加入丙酮沉淀,沉淀抽滤,丙酮洗涤,50℃下干燥,得黄色粉末,即为o-三甲基硅基-n-烟酰基壳寡糖。
[0053]
将1.5克o-三甲基硅基-n-烟酰基壳寡糖加入到15ml水中,搅拌下向其15ml四丁基氟化铵的四氢呋喃溶液(1mol/l),25℃反应4小时,加入乙醇沉淀,沉淀抽滤,无水乙醇洗涤,50℃下干燥,得黄色粉末,即为n-烟酰基壳寡糖。
[0054]
将0.5克n-烟酰基壳寡糖溶于20ml水中,0.38克cu(oac)2·
h2o溶于5ml水中,室温搅拌下将乙酸铜水溶液滴加到n-烟酰基壳寡糖水溶液中,反应6小时,加入无水乙醇沉淀,离心,无水乙醇洗涤洗,50℃干燥,得深绿色粉末,即为吡啶甲酰基壳寡糖铜配合物衍生物1,结构见通式i(r为吡啶-3-基;n=2-20)。
[0055]
红外光谱表明:吡啶甲酰基壳寡糖铜配合物衍生物4的红外谱图(图5)与全壳寡糖的红外谱图(图1)相比,出现了位于1728cm-1
的羰基特征吸收峰,以及1647cm-1
、1558cm-1
以及1286cm-1
的仲酰胺特征吸收峰;1590cm-1
为吡啶环的特征吸收;证明目衍生物1合成成功。
[0056]
实施例2衍生物5的制备
[0057]
将15克分子量为1500的壳寡糖、12.1克三甲基氯硅烷和15克咪唑加入到100ml n,n-二甲基甲酰胺中,35℃反应12小时,反应物加入三倍体积乙酸乙酯,抽滤得到沉淀,用无水乙醇洗涤,50摄氏度下干燥,即得o-三甲基硅基壳寡糖。
[0058]
将4克o-三甲基硅基壳寡糖加入到30mln,n-二甲基甲酰胺和30ml吡啶中,搅拌下向其加入6.12g异烟酰氯盐酸盐,35℃反应24小时,加入丙酮沉淀,沉淀抽滤,丙酮洗涤,50℃下干燥,得黄色粉末,即为o-三甲基硅基-n-异烟酰基壳寡糖。
[0059]
将1.5克o-三甲基硅基-n-异烟酰基壳寡糖加入到15ml水中,搅拌下向其15ml四丁基氟化铵的四氢呋喃溶液(1mol/l),25℃反应4小时,加入乙醇沉淀,沉淀抽滤,无水乙醇洗涤,50℃下干燥,得黄色粉末,即为n-异烟酰基壳寡糖。
[0060]
将0.5克n-异烟酰基壳寡糖溶于20ml水中,0.38克cu(oac)2·
h2o溶于5ml水中,室温搅拌下将乙酸铜水溶液滴加到n-异烟酰基壳寡糖水溶液中,反应6小时,加入无水乙醇沉淀,离心,无水乙醇洗涤洗,50℃干燥,得深绿色粉末,即为吡啶甲酰基壳寡糖铜配合物衍生物2,结构见通式i(r为吡啶-4-基;n=2-20)。
[0061]
红外光谱表明:吡啶甲酰基壳寡糖铜配合物衍生物5的红外谱图(图6)与全壳寡糖的红外谱图(图1)相比,出现了位于1734cm-1
的羰基特征吸收峰,以及1649cm-1
、1558cm-1
以及1284cm-1
的仲酰胺特征吸收峰;1590cm-1
为吡啶环的特征吸收;证明目衍生物2合成成功。
[0062]
实施例3衍生物6的制备
[0063]
将15克分子量为1500的壳寡糖、12.1克三甲基氯硅烷和15克咪唑加入到100ml n,n-二甲基甲酰胺中,35℃反应12小时,反应物加入三倍体积乙酸乙酯,抽滤得到沉淀,用无水乙醇洗涤,50摄氏度下干燥,即得o-三甲基硅基壳寡糖。
[0064]
将4克o-三甲基硅基壳寡糖加入到30mln,n-二甲基甲酰胺和30ml吡啶中,搅拌下向其加入6.04g 2-氯烟酰氯,35℃反应24小时,加入丙酮沉淀,沉淀抽滤,丙酮洗涤,50℃下干燥,得黄色粉末,即为o-三甲基硅基-n-(2-氯烟酰基)壳寡糖。
[0065]
将1.5克o-三甲基硅基-n-(2-氯烟酰基)壳寡糖加入到15ml水中,搅拌下向其15ml四丁基氟化铵的四氢呋喃溶液(1mol/l),25℃反应4小时,加入乙醇沉淀,沉淀抽滤,无水乙醇洗涤,50℃下干燥,得黄色粉末,即为n-(2-氯烟酰基)基壳寡糖。
[0066]
将0.5克n-(2-氯烟酰基)壳寡糖溶于20ml水中,0.38克cu(oac)2·
h2o溶于5ml水中,室温搅拌下将乙酸铜水溶液滴加到n-(2-氯烟酰基)壳寡糖水溶液中,反应6小时,加入无水乙醇沉淀,离心,无水乙醇洗涤洗,50℃干燥,得深绿色粉末,即为吡啶甲酰基壳寡糖铜配合物衍生物3,结构见通式i(r为2-氯吡啶-3-基;n=2-20)。
[0067]
红外光谱表明:吡啶甲酰基壳寡糖铜配合物衍生物6的红外谱图(图7)与全壳寡糖
的红外谱图(图1)相比,出现了位于1735cm-1
的羰基特征吸收峰,以及1655cm-1
、1561cm-1
以及1296cm-1
的仲酰胺特征吸收峰;1578cm-1
为吡啶环的特征吸收;证明目衍生物3合成成功。
[0068]
抑菌活性测定
[0069]
采用生长速率法测定样品对辣椒疫霉病原菌的抑菌活性。测试在2个样品浓度下即:0.04mg/ml,0.08mg/ml对辣椒疫霉的抑制效果。
[0070]
实验以相同浓度的噻菌铜药剂(市售为20%的悬浮剂)为阳性对照,以蒸馏水为阴性对照。将培养基均匀倒入1个直径为9cm的培养皿中,待完全凝固后,在每个培养皿中接种3块直径为5mm的菌饼。在27℃下培养72小时后,测量菌落直径,计算样品的抑菌率。每次处理设置1个培养皿,每皿接种3个菌落,对每个菌落测定最大直径(d
max
)和最小直径(d
min
),取平均值为样品抑菌圈直径d
样品
,全部试验重复一次。根据下式计算抑菌率(参见表1)。
[0071]
抑菌率(%)=(d
空白-d样品
)/(d
空白-
5)
×
100
[0072]
表1部分通式1壳寡糖衍生物对辣椒疫霉的抑制活性
[0073][0074]
由上述表1中实验结果表明吡啶甲酰基壳寡糖衍生物和吡啶甲酰基壳寡糖铜配合物衍生物高浓度下对辣椒疫霉具有良好的抑菌效果,好于现在已有的噻菌铜产品。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1