环氧丙烯酸酯改性聚醚多元醇及其制备方法与流程
1.本发明涉及一种环氧丙烯酸酯改性聚醚多元醇及其制备方法,属于聚醚多元醇改性技术领域。
背景技术:
2.聚氨酯泡沫应用领域中,尤其喷涂领域,在对建筑行业应用时对泡沫粘结性与强度要求较高,常规聚醚多元醇带来的粘结性与强度不足,造成施工过程需要大量使用界面剂及其他助剂来满足施工要求。而环氧丙烯酸酯固化后涂膜具有良好的附着力、耐化学性和强度,两者的改性对性能提升有明显的影响。
3.环氧丙烯酸酯树脂是用环氧树脂和丙烯酸在催化剂的作用下经开环酯化而制得,如下所示:
[0004][0005]
环氧丙烯酸酯原料来源广,价格低廉,合成工艺简单。从结构上来分,可以分为双酚a型环氧丙烯酸酯、酚醛环氧丙烯酸酯、环氧化油类丙烯酸酯以及改性的环氧丙烯酸酯。当作为一种主体树脂时,具有良好的附着力、耐化学性和强度,但是也存在固化后柔韧性不足,脆性大的缺点。因此,为满足不同领域的需求,对环氧丙烯酸酯进行改性,对聚醚合成进行改性,结合两者优异性能是解决当前研究方向之一。
技术实现要素:
[0006]
本发明要解决的技术问题是,克服现有技术中的不足,提供一种环氧丙烯酸酯改性聚醚多元醇,其具有较高的羟值和较低的粘度,用其制备的聚氨酯泡沫具有较高的粘结性和强度;本发明同时提供了简单易行的制备方法。
[0007]
本发明所述的环氧丙烯酸酯改性聚醚多元醇的制备方法,为环氧丙烯酸酯类化合物与复合起始剂混合,在催化剂的作用下,与环氧烯烃化合物进行反应,制得所述的聚醚多元醇。
[0008]
所述复合起始剂为蔗糖与山梨醇、二乙醇胺、乙醇胺、二乙二醇、甘油、丙二醇、棕榈油或水中的一种或多种的混合物。
[0009]
所述环氧丙烯酸酯类化合物由环氧树脂在催化剂和阻聚剂作用下与丙烯酸或甲基丙烯酸反应得到。
[0010]
优选的,环氧树脂为双酚a型或酚醛型环氧树脂。
[0011]
优选的,丙烯酸或甲基丙烯酸与环氧树脂的摩尔比为1:1.02-1.1。
[0012]
优选的,催化剂为三乙胺、n,n-二甲基苄胺、n,n-二甲基苯胺、三甲基苄基氯化铵、
三苯基膦、三苯基锑、乙酰丙酮铬或四乙基溴化铵。
[0013]
优选的,阻聚剂为对甲氧基苯酚、对苯二酚、2,5-二甲基对苯二酚或2,6-二叔丁基对甲苯酚。
[0014]
优选的,环氧丙烯酸酯类化合物与复合起始剂的混合质量比例为1:2-1:10。
[0015]
所述环氧烯烃化合物为环氧乙烷、环氧丙烷或环氧丁烷中的一种或多种。
[0016]
一种环氧丙烯酸酯改性聚醚多元醇,由上述制备方法制得。
[0017]
优选的,所述的环氧丙烯酸酯改性聚醚多元醇的制备方法,包括以下步骤:
[0018]
1)环氧丙烯酸酯合成:将环氧树脂投入反应釜中,合釜试漏置换后,氮气氛围下升温至80-90℃,然后滴加丙烯酸、催化剂与阻聚剂混合物,控制反应温度100℃,同时取样测定酸值,到反应后期升温至110-120℃,使酸值降至小于2mgkoh/g停止反应;
[0019]
2)在步骤1)反应完后向反应釜中投入复合起始剂和碱性催化剂,搅拌升温至105-115℃,升温至设定温度后保温1小时,开始滴加环氧烯烃化合物进行聚醚合成,进料完毕后内压2-3h;
[0020]
3)步骤2)内压反应完后降温至95-105℃进行氮气鼓泡,脱去产品中残余小分子物质,鼓泡1-2h后降温放料得所述的改性聚醚多元醇。
[0021]
本发明为以聚醚为主进行环氧丙烯酸酯的改性,通过环氧基反应在体系中引入不饱和键,其与异氰酸酯、氨酯键之间的复杂化学交联交联反应能够形成一种自增强互穿网络的微相结构,大大提高了不饱和聚氨酯的粘结、强度等性能。
[0022]
与现有技术相比,本发明具有以下有益效果:
[0023]
(1)本发明所用环氧丙烯酸酯原料来源广,价格低廉,合成工艺简单,具有良好的附着力、耐化学性和强度,促进聚氨酯行业的发展;
[0024]
(2)本发明制备的聚醚多元醇羟值高(380-460mgkoh/g),粘度低(3000-7000mpa
·
s),适用性广,可满足各领域产品应用需求;
[0025]
(3)所述环氧丙烯酸酯反应的温度、压力与工艺均可在常规聚醚生产设备中直接进行。
具体实施方式
[0026]
以下结合实施例对本发明做进一步说明,但本发明的保护范围不仅限于此,该领域专业人员对本发明技术方案所作的改变,均应属于本发明的保护范围内。
[0027]
实施例中用到的所有原料若无特殊说明,均为市购。
[0028]
实施例1
[0029]
在反应釜中加入100g环氧树脂e44,合釜试漏置换后,氮气氛围下升温至85
±
5℃,然后1h内完成滴加丙烯酸15.5g、三乙胺催化剂1.1g与对苯二酚阻聚剂0.15g的混合物,控制反应温度100℃,同时取样测定酸值,滴加完毕后升温至115
±
5℃反应1.5h,使酸值降至小于2mgkoh/g停止反应。
[0030]
内压完毕后取325蔗糖真空抽入反应釜中,同时抽加入80g二乙二醇,2g二乙醇胺,226g棕榈油,12g三乙胺催化剂,搅拌降温至110℃,到温度后开始滴加环氧丙烷,质量在581g,滴加过程中保持温度在112
±
2℃,控制釜内压力在0.2mpa以内。滴加完毕后,充氮气加压至0.25mpa,保持温度110
±
2℃,继续内压反应2h,至釜内压力不再降低。之后降温至80
℃,放料,得到羟值在412mgkoh/g,粘度在6280mpa
·
s的聚醚多元醇a1。
[0031]
实施例2
[0032]
在反应釜中加入100g环氧树脂f51(加热后投料),合釜试漏置换后,氮气氛围下升温至85
±
5℃,然后1h内完成滴加丙烯酸33.4g、n,n-二甲基苄胺催化剂1.3g与对甲氧基苯酚0.2g阻聚剂混合物,控制反应温度100℃,同时取样测定酸值,滴加完毕后升温至115
±
5℃反应2h,使酸值降至小于2mgkoh/g停止反应。
[0033]
内压完毕后取325g蔗糖真空抽入反应釜中,同时抽入165g二乙二醇与3g二乙醇胺,12g二甲胺催化剂,搅拌降温至110℃,到温度后开始滴加环氧丙烷,质量在859g,滴加过程中保持温度在112
±
2℃,控制釜内压力在0.2mpa以内。滴加完毕后,充氮气加压至0.25mpa,保持温度110
±
2℃,继续内压反应2h,至釜内压力不再降低。之后降温至80℃,放料,得到羟值在446mgkoh/g,粘度在4770mpa
·
s的聚醚多元醇a2。
[0034]
实施例3
[0035]
在反应釜中加入100g环氧树脂e55(加热后投料),合釜试漏置换后,氮气氛围下升温至85
±
5℃,然后1h内完成滴加甲基丙烯酸19.9g、n,n-二甲基苄胺催化剂1.2g与对甲氧基苯酚0.17g阻聚剂混合物,控制反应温度100℃,同时取样测定酸值,滴加完毕后升温至115
±
5℃反应3h,使酸值降至小于2mgkoh/g停止反应。
[0036]
内压完毕后取325g蔗糖真空抽入反应釜中,同时抽入165g二乙二醇与2.6g二乙醇胺,12g二甲胺催化剂,搅拌降温至110℃,到温度后开始滴加环氧丙烷,质量在850g,滴加过程中保持温度在112
±
2℃,控制釜内压力在0.2mpa以内。滴加完毕后,充氮气加压至0.25mpa,保持温度110
±
2℃,继续内压反应2h,至釜内压力不再降低。之后降温至80℃,放料,得到羟值在448mgkoh/g,粘度在4325mpa
·
s的聚醚多元醇a3。
[0037]
对比例1
[0038]
在反应釜中加入325g蔗糖,50g固体山梨醇,80g二乙二醇,255g棕榈油,12g脂肪胺催化剂,合釜试漏置换后,负压下升温至105℃,到温度后滴入787g环氧丙烷,同时釜内保持氮气氛围压力在0.13
±
0.02mpa,环氧丙烷滴入完毕后保温反应3h至釜内压力不再降低,鼓泡脱单体2h之后降温至80℃,放料得到聚醚多元醇b1,羟值405mgkoh/g,粘度5830mpa
·
s。
[0039]
对比例2
[0040]
在反应釜中加入325g蔗糖,165g二乙二醇,11.4gdma催化剂,合釜试漏置换后,负压下升温至105℃,到温度后滴入913g环氧丙烷,同时釜内保持氮气氛围压力在0.13
±
0.02mpa,环氧丙烷滴入完毕后保温反应3h至釜内压力不再降低,鼓泡脱单体2h之后降温至80℃,放料得到聚醚多元醇b2,羟值440mgkoh/g,粘度3460mpa
·
s。
[0041]
实施例与对比例指标对比如下表:
[0042]
表1各实施例与对比例制备的产品指标对比
[0043] 羟值(mgkoh/g)粘度(mpa.s)实施例14126280实施例24464770实施例34484325对比例14055830对比例24403460
[0044]
由表1可看出,实施例1与对比例1为含油聚醚对比,实施例2与实施例3同对比例2为不含油聚醚对比,在设计羟值官能度时接近,而加入环氧丙烯酸酯或环氧甲基丙烯酸酯之后对比羟值粘度均比常规聚醚要高。
[0045]
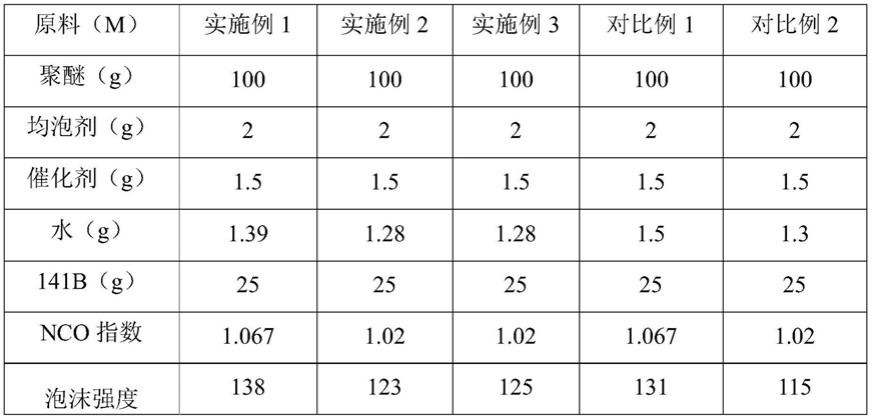
表2各实施例与对比例性能对比
[0046][0047][0048]
通过表2可以看出,实施例1与对比例1活性提高18s,实施例2、3与对比例2相比活性也提高10s左右,强度均提高5%以上。
[0049]
以上所述的仅是本发明的实施例,方案中公知的具体结构及特性等常识在此未作过多描述。应当指出,对于本领域的技术人员来说,在不脱离本发明结构的前提下,还可以作出若干变形和改进,这些也应该视为本发明的保护范围,这些都不会影响本发明实施的效果和专利的实用性。本技术要求的保护范围应当以其权利要求的内容为准,说明书中的具体实施方式等记载可以用于解释权利要求的内容。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1