一种无对称元素的羧酸配体及其合成方法和应用与流程
1.本发明涉及mof配体的制备技术领域,具体涉及一种无对称元素的羧酸配体及其合成方法和应用。
背景技术:
2.金属-有机框架材料(mofs)是近十年来发展迅速的一种配位聚合物,具有三维的孔结构,一般以金属离子为连接点,有机配位体支撑构成空间3d延伸,是沸石和碳纳米管之外的又一类重要的新型多孔材料,在催化、储能和分离中都有广泛应用。mof已成为无机化学、有机化学等多个化学分支的重要研究方向。
3.mof结构通常高度规整,主要原因也是众多的mof配体都是规则对称的,要么是中心对称,抑或是轴对称。在mof中构筑“缺陷”,也需要从配体入手,一个基本的思路,就是引入“不对称”,但是含量不宜过多,否则,mof难以结晶。而不对称的配体设计与合成,也需要在合理的范围内。
技术实现要素:
4.本发明的目的在于提供一种无对称元素的羧酸配体及其合成方法和应用,以解决现有技术中构建“不对称”配体时mof难以结晶的缺陷。
5.以下通过三个方面进一步阐述本发明的方案:
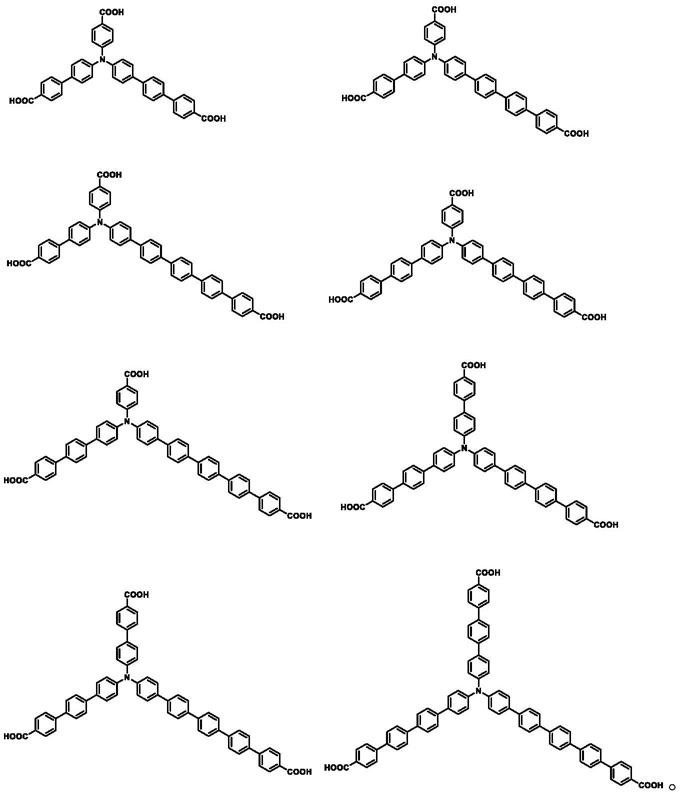
6.第一方面,提供了一种无对称元素的羧酸配体,包括如下化学式:
[0007][0008]
其中,x、y、z代表连接苯环的个数;x、y、z取值范围为1~5,且x≠y≠z;苯环连接的方式为对位单键连接。
[0009]
结合第一方面,所述配体包括如下化学式:
[0010][0011]
第二方面,提供了一种无对称元素羧酸配体的合成方法,所述方法包括如下步骤:
[0012]
将含有x个苯环的化合物a与含有y个苯环的化合物b通过偶联反应连接得到化合物o1;
[0013]
将化合物o1与含有z个苯环的化合物c进行偶联反应得到化合物o2;
[0014]
反应结束后通过重结晶提纯,即得到产物。
[0015]
结合第二方面,所述化合物a上的苯环“一”字形连接,其中一端的苯环上连接有氨基,另一端苯环上连接有甲酯或乙酯。
[0016]
结合第二方面,所述化合物b上的苯环“一”字形连接,其中一端的苯环上连接有溴或碘,另一端苯环上连接有甲酯或乙酯。
[0017]
结合第二方面,所述化合物c上的苯环“一”字形连接,其中一端的苯环上连接有溴或碘,另一端苯环上连接有甲酯或乙酯。
[0018]
结合第二方面,所述偶联反应包括采用钯催化剂催化的buchwald-hartwig反应和采用铜催化剂催化的ullmann反应中的一种或多种。
[0019]
第三方面,提供了一种无对称元素羧酸配体的应用,将上述制备获得的无对称元素羧酸作为mof的配体,使mof的晶格产生缺陷,有助于负载或修饰。
[0020]
本发明的优点在于:该种无对称元素的羧酸配体,利用简便的合成方法,可以得到不含对称元素(包括对称轴、对称中心、对称面)的羧酸配体,有潜力在mof中形成可控的晶
格缺陷,提升mof的部分性质,并开拓mof的应用领域。
具体实施方式
[0021]
为使本发明实现的技术手段、创作特征、达成目的与功效易于明白了解,下面结合具体实施方式,进一步阐述本发明。
[0022]
一种无对称元素的羧酸配体,包括如下化学式:
[0023][0024]
其中,x、y、z代表连接苯环的个数;x、y、z取值范围为1~5,且x≠y≠z;苯环连接的方式为对位单键连接;
[0025]
所述配体包括如下化学式:
[0026][0027]
一种无对称元素羧酸配体的合成方法,所述方法包括如下步骤:
[0028]
将含有x个苯环的化合物a与含有y个苯环的化合物b通过偶联反应连接得到化合物o1;
[0029]
其中,化合物a上的苯环“一”字形连接,其中一端的苯环上连接有氨基,另一端苯环上连接有甲酯或乙酯,包括但不仅限于如下结构式:
[0030][0031]
化合物b上的苯环“一”字形连接,其中一端的苯环上连接有溴或碘,另一端苯环上连接有甲酯或乙酯,包括但不仅限于如下结构式:
[0032][0033]
将化合物o1与含有z个苯环的化合物c进行偶联反应得到化合物o2;
[0034]
其中,化合物c上的苯环“一”字形连接,其中一端的苯环上连接有溴或碘,另一端苯环上连接有甲酯或乙酯,包括但不仅限于如下结构式:
[0035][0036]
反应结束后通过重结晶提纯,即得到产物;
[0037]
上述偶联反应包括采用钯催化剂催化的buchwald-hartwig反应和采用铜催化剂催化的ullmann反应。
[0038]
一种无对称元素羧酸配体的应用,将上述制备获得的无对称元素羧酸作为mof的配体,使mof的晶格产生缺陷,有助于负载或修饰。
[0039]
以下通过实施例进一步对本发明的技术方案进行解释:
[0040]
实施例1
[0041]
x=1,y=2,z=3配体的合成路线为:
(dba)3(三二亚苄基丙酮二钯)与10mmol三叔丁基膦,氮气保护下,100℃反应8小时;
[0055]
将上述反应体系降温,随后将含有4个苯环的20mmol的化合物c溶于100ml的无水甲苯中,通过注射器加入到混合液中,再次升温至100℃下继续反应24小时。反应结束后冷却,混合液旋干,以二氯甲烷:正己烷(2:1)作为洗脱剂,分离得到8.6g纯品m134;
[0056]
将上述得到的8.6g m134溶于100ml四氢呋喃,再加入100ml的5mol/l的氢氧化钾溶液,70℃搅拌48小时,旋蒸除去有机相,用二氯甲烷清洗水相三次,用1n稀盐酸将ph调至中性,得到固体沉淀,过滤后水洗,烘干得到5.9g白色固体。
[0057]
其中,hplc(洗脱剂为甲醇);纯度98.59%。
[0058]
元素分析:c
51h35
no6理论值:c:80.83,h:4.66,n:1.85;
[0059]
实测值:c:80.68,h:4.63,n:1.89;
[0060]
质谱结果:[c
39h26
no6]-理论值:756.2392;
[0061]
实测值:756.2382。
[0062]
实施例3
[0063]
x=2,y=3,z=4配体的合成路线为:
[0064][0065]
将含有两个苯环的22mmol的化合物a(见上述反应路线)与含有3个苯环的20mmol的化合物b溶于200ml无水甲苯中,常温搅拌,先加入60mmol的叔丁醇钠,再加入2mmol的pd2(dba)3(三二亚苄基丙酮二钯)与10mmol三叔丁基膦,氮气保护下,100℃反应8小时;
[0066]
将上述反应体系降温,随后将含有4个苯环的20mmol的化合物c溶于100ml的无水甲苯中,通过注射器加入到混合液中,再次升温至100℃下继续反应24小时。反应结束后冷却,混合液旋干,以二氯甲烷:正己烷(2:1)作为洗脱剂,分离得到8.5g纯品m234;
[0067]
将上述得到的8.5g m234溶于100ml四氢呋喃,再加入100ml的5mol/l的氢氧化钾溶液,70℃搅拌48小时,旋蒸除去有机相,用二氯甲烷清洗水相三次,用1n稀盐酸将ph调至中性,得到固体沉淀,过滤后水洗,烘干得到5.2g白色固体。
[0068]
其中,hplc(洗脱剂为甲醇);纯度99.05%。
[0069]
元素分析:c
57h39
no6理论值:c:82.10,h:4.71,n:1.68;
[0070]
实测值:c:82.03,h:4.60,n:1.73;
[0071]
质谱结果:[c
39h26
no6]-理论值:832.1705;
[0072]
实测值:832.1708。
[0073]
实施例4
[0074]
x=3,y=4,z=5配体的合成路线为:
[0075][0076]
将含有三个苯环的22mmol的化合物a(见上述反应路线)与含有4个苯环的20mmol的化合物b溶于200ml无水甲苯中,常温搅拌,先加入60mmol的叔丁醇钠,再加入2mmol的pd2(dba)3(三二亚苄基丙酮二钯)与10mmol三叔丁基膦,氮气保护下,100℃反应8小时;
[0077]
将上述反应体系降温,随后将含有5个苯环的20mmol的化合物c溶于100ml的无水甲苯中,通过注射器加入到混合液中,再次升温至100℃下继续反应24小时。反应结束后冷却,混合液旋干,以二氯甲烷:正己烷(2:1)作为洗脱剂,分离得到9.2g纯品m345;
[0078]
将上述得到的9.2g m345溶于100ml四氢呋喃,再加入100ml的5mol/l的氢氧化钾溶液,70℃搅拌48小时,旋蒸除去有机相,用二氯甲烷清洗水相三次,用1n稀盐酸将ph调至中性,得到固体沉淀,过滤后水洗,烘干得到6.6g白色固体。
[0079]
其中,hplc(洗脱剂为甲醇);纯度97.89%。
[0080]
元素分析:c
75h50
no6理论值:c:84.80,h:4.84,n:1.32;
[0081]
实测值:c:84.68,h:4.69,n:1.33;
[0082]
质谱结果:[c
75h50
no6]-理论值:1060.3644;
[0083]
实测值:1060.363。
[0084]
由技术常识可知,本发明可以通过其它的不脱离其精神实质或必要特征的实施方
案来实现。因此,上述公开的实施方案,就各方面而言,都只是举例说明,并不是仅有的。所有在本发明范围内或在等同于本发明的范围内的改变均被本发明包含。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1