一种两性霉素B的杂质A或杂质B的分离纯化方法与流程
一种两性霉素b的杂质a或杂质b的分离纯化方法
技术领域
1.本发明属于药物分离纯化领域,具体地涉及两性霉素b的杂质a和杂质b分离制备方法。
背景技术:
2.随着器官移植、免疫抑制药物、放疗和抗癌化疗等在先进的医学治疗中的应用以及人类免疫缺陷病毒(hiv)感染,使真菌病患者的人数有所增加。两性霉素b(amphotericin b)为多烯类抗真菌药物,其主要作用机制为通过选择性与细胞膜的麦角固醇结合,增加膜的通透性,使重要物质外漏、有毒物质内渗,从而降低真菌生命力,达到抗真菌作用。两性霉素b因其抗菌谱广、抗真菌作用强而在临床治疗上发挥着重要的作用。
[0003][0004]
两性霉素b作为临床应用药物,其含量必须满足药品质量控制标准。因此,对其杂质控制即杂质谱的研究,即掌握两性霉素b杂质种类、含量、来源及结构信息是非常必要的。截至目前,国内外相关期刊文献和专利著作等对两性霉素b的研究主要集中在临床应用及其毒副作用和含量测定这几个方面,对两性霉素b杂质分离纯化相关研究少有报道,尤其是两性霉素b杂质a和杂质b的分离纯化方法。
[0005]
如有学者对两性霉素b杂质进行了系统的研究,包括两性霉素b的碱性条件降解产物的考察研究、两性霉素b的酸性条件降解产物的考察研究以及两性霉素b的dmf溶液条件下水浴加热降解产物的研究。
[0006]
cn102115484a发明了一种两性霉素b降解产物、其制备方法及其应用,该发明通过制备液相色谱仪分离纯化降解产物,再经凝胶过滤纯化,冷冻干燥后得两性霉素b降解产物(amb(h)),产率仅9%。
[0007]
目前对两性霉素b杂质的考察研究资料较少,两性霉素b杂质稳定性较差,分离具有一定难度,如果能开发通用的适用于两性霉素b杂质的分离制备方法,得到两性霉素b杂质纯品,即能完善两性霉素b的杂质谱,对于两性霉素b杂质的研究具有重大意义。
技术实现要素:
[0008]
本发明的目的在于提供两性霉素b杂质的分离制备方法,该方法操作简单,分离效
率高,能够得到纯度较高的杂质a和杂质b。
[0009]
本发明的目的通过以下技术方案实现:
[0010]
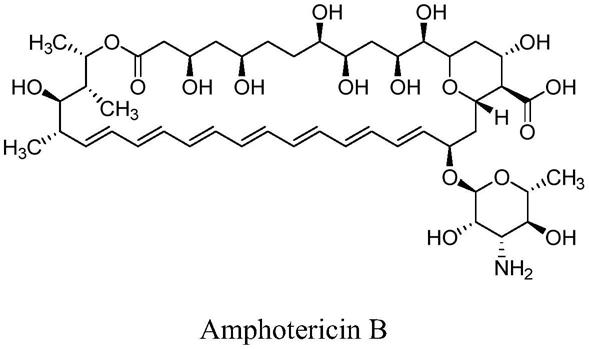
本发明所述两性霉素b杂质a、杂质b的结构式如下所示:
[0011][0012]
本发明具体地涉及两性霉素b杂质a或杂质b分离制备方法,包括如下操作步骤:
[0013]
(1)对两性霉素b进行预处理,得到两性霉素b的杂质溶液;
[0014]
(2)在动态轴向压缩制备色谱中,以柠檬酸水溶液/乙腈为流动相进行洗脱,收集目标杂质馏分,然后经过减压浓缩,得到两性霉素b杂质浓缩液;
[0015]
(3)以水/乙腈为流动相平衡色谱柱,改变流动相比例,将步骤(2)中得到的两性霉素b杂质浓缩液进行再洗脱除盐,收集目标馏分;
[0016]
(4)将步骤(3)收集的馏分进行减压蒸馏,除去大部分溶剂后,然后进行冷冻干燥,得到两性霉素b的杂质a或杂质b。
[0017]
当两性霉素b的杂质为杂质a时,优选的,步骤(1)中,前处理方式为:50mg两性霉素b,加入5ml的nmp、35ml乙腈以及0.1ml浓盐酸。超声搅拌至完全溶解,再加入5ml的10g/l的乙酸铵溶液。
[0018]
当两性霉素b的杂质为杂质a时,作为优选,步骤(2)中,流动相比例:0.01~0.03m柠檬酸水溶液与乙腈体积比为65~75:25~35;检测波长:383nm和303nm;流速:45~55ml/min;进样体积:200ml;采用50dac制备液相色谱进行制备。作为进一步的优选,步骤(2)中,液相色谱条件为:0.02m柠檬酸水溶液/乙腈(67:33),检测波长383nm以及303nm,流速50ml/min,进样体积200ml,采用50dac制备液相色谱制备(图1)。
[0019]
当两性霉素b的杂质为杂质b时,优选的,步骤(1)中,前处理方式为:50mg两性霉素b,加入5ml的nmp、35ml甲醇以及0.1ml浓盐酸。超声搅拌至完全溶解,避光破坏30min后,再加入5ml的10g/l的乙酸铵溶液。
[0020]
当两性霉素b的杂质为杂质b时,作为优选,步骤(2)中,流动相比例:0.01~0.03m柠檬酸水溶液与乙腈的体积比为60~70:30~40;检测波长:383nm;流速:45~55ml/min;进样体积200ml;采用50dac制备液相色谱制备;作为进一步的优选,步骤(2)中,制备液相色谱条件为:0.02m柠檬酸水溶液/乙腈(65:35),检测波长383nm,流速50ml/min,进样体积200ml,50dac制备液相色谱制备(图2)。
[0021]
优选的,步骤(2)中,所述两性霉素b杂质a、杂质b纯度均可达90%以上(图3~图4)。
[0022]
作为优选,步骤(3)中,平衡色谱柱时,水和乙腈的体积比为85~95:5~15;洗脱时,水和乙腈的体积比为60~70:30~40;作为进一步的优选,所述两性霉素b杂质a、杂质b除盐方法为:以水/乙腈(90:10)平衡色谱柱,用泵抽入两性霉素b杂质浓缩液250ml,再以
水/乙腈(90:10)平衡5min,改变流动相水/乙腈(65:35)进行洗脱,收集目标馏分。
[0023]
本发明所采用前处理方式、制备液相色谱条件、除盐方式均可根据具体情况进行适当调整。
[0024]
因此,本发明方法能纯化两性霉素b杂质,对两性霉素b杂质具有较好的普适性及通用性。两性霉素b杂质样品不稳定,分离制备难度大,本发明所述的两性霉素b杂质a、杂质b的分离制备方法分离效率高、分离速度快、操作简便。
附图说明
[0025]
图1为两性霉素b杂质a制备液相色谱图;
[0026]
图2为两性霉素b杂质b制备液相色谱图;
[0027]
图3为两性霉素b杂质a纯品液相色谱图;
[0028]
图4为两性霉素b杂质b纯品液相色谱图。
具体实施方式
[0029]
下面结合具体实施例对本发明的技术方案作进一步的描述,但本发明并不限于这些实施例。以下实施例中所用试剂和原料,除制备方法外,其余均为市售。除非另有定义,否则本文中所有科技术语具有的含义与权利要求主题所属技术领域人员通常理解的含义相同。
[0030]
实施例1:两性霉素b杂质a分离纯化
[0031]
(1)称取50mg两性霉素b,加入5ml的nmp以及35ml乙腈以及0.1ml浓盐酸。超声搅拌至完全溶解,再加入5ml的10g/l的乙酸铵溶液混合均匀后进样。
[0032]
(2)动态轴向压缩制备色谱条件:流动相a相为0.02m柠檬酸水溶液(氨水调节ph至4.7),b相为乙腈;流动相a:流动相b=67:33;检测波长为383nm以及303nm;流速为50ml/min;进样体积为200ml,50dac动态轴向压缩柱为ods柱,粒径为10um。
[0033]
按上述色谱条件运行洗脱方法,收集目标杂质(rt=166.47min)馏分,用分析型液相色谱法测定各馏分纯度,纯度可达90%以上。将收集的馏分30℃减压蒸馏,除去有机溶剂后,得两性霉素b杂质a浓缩液,待用。
[0034]
(3)除盐:以水:乙腈=90:10作流动相平衡色谱柱,用泵抽入两性霉素b杂质a浓缩液250ml,再以水:乙腈=90:10作流动相平衡5min,改变流动相为水:乙腈=65:35进行洗脱,收集目标馏分。
[0035]
(4)将收集的馏分30℃减压蒸馏,除去绝大部分溶剂后,将样品转移至培养皿中,冷冻完全后放入冷冻干燥机干燥,最终得到30mg黄色粉末,即为两性霉素b杂质a纯品,用分析型液相色谱法测定其纯度,纯度为99%,结果见图3。
[0036]
实施例2:两性霉素b杂质b分离纯化
[0037]
(1)称取50mg两性霉素b,加入5ml的nmp以及35ml甲醇以及0.1ml浓盐酸。超声搅拌至完全溶解,避光破坏30min后,再加入5ml的10g/l的乙酸铵溶液,混合均匀后即可进样。
[0038]
(2)动态轴向压缩制备色谱条件:流动相a相为0.02m柠檬酸水溶液(氨水调节ph至4.7),b相为乙腈;流动相a:流动相b=65:35;检测波长为383nm;流速为50ml/min;进样体积为200ml,50dac动态轴向压缩柱为ods柱,粒径为10um。
[0039]
按上述制备液相色谱条件运行洗脱方法,收集目标杂质(rt=42.75min)馏分,用液相色谱法测定各馏分纯度,纯度可达90%以上。将收集的馏分30℃减压蒸馏,除去有机溶剂后,得两性霉素b杂质b浓缩液,待用。
[0040]
(3)除盐:以水:乙腈=90:10作流动相平衡色谱柱,用泵抽入两性霉素b杂质b浓缩液250ml,再以水:乙腈=90:10作流动相平衡5min,改变流动相为水:乙腈=65:35进行洗脱,收集目标馏分。
[0041]
(4)将收集的馏分30℃减压蒸馏,除去绝大部分溶剂后,将样品转移至培养皿中,冷冻完全后放入冷冻干燥机干燥,最终得到40mg黄色粉末,即为两性霉素b杂质b纯品,用分析型液相色谱法测定其纯度,纯度为95%,结果见图4。
[0042]
实施例3:两性霉素b杂质a和杂质b的预处理
[0043]
(1)称取2000mg两性霉素b,加入150ml的nmp以及700ml乙腈以及0.1ml浓盐酸。超声搅拌至完全溶解,再加入150ml的10g/l的乙酸铵溶液混合均匀后进样,按实施例1中的(2)动态轴向压缩制备色谱条件、(3)除盐和(4)30℃减压蒸馏等步骤处理,发现增大样品处理量,且放置时间比较长,其会产生其他的杂质,而目标杂质a和杂质b的含量更少,更不利于分离,故优选实施例中的比例进行处理。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1