急性冠脉综合征的风险评估标志物及其应用
1.本发明涉及生物医学技术领域,尤其涉及急性冠脉综合征的风险评估标志物及其相关应用。
背景技术:
2.心血管病主要指冠状动脉粥样硬化性心脏病,简称冠心病(coronary artery disease,cad)。心脏病突发(heart attack)即临床表现的急性冠状动脉综合征(acute coronary syndrome,acs),包括心源性猝死、心肌梗死和不稳定性心绞痛则是冠心病主要致死原因。acs的病理生理机制已公认为冠状动脉粥样斑块炎症性进展至破裂,诱发了血栓形成急性堵塞冠状动脉所致。目前,主流观点认为:包括冠心病在内的心血管疾病是一类免疫代谢性疾病,也是一类全身性、进展性、炎症性疾病。主要病变是动脉粥样硬化斑块形成和炎症性进展,本质特征包括脂质沉积和炎症性细胞聚集所产生的非细菌性炎症反应,即被称为代谢性炎症。因为在粥样硬化斑块和进展过程中,从脂质条纹不断进展到粥样斑块,直至破裂,导致血栓形成的多个环节中,始终都有各种炎症细胞和大量炎症介质参与。由于冠心病的动态性和复杂性,炎性不稳定斑块的形成、进展、破裂的机制仍不清楚,因此,若能阐明冠状动脉斑块炎症不稳定性的启动因素或原因,以及寻找源头干预炎症过程的有效方法,对于有效防范冠状动脉斑块炎症不稳定性的发生、进展和破裂以及acs突发事件,大大降低心血管病的发病率和死亡率;对于保障人民的生命安全和身体健康均具有巨大而深远的社会意义和科学价值。
3.传统认为,总胆固醇(tc)、高血压(hypertension)、年龄等均是与急性冠状动脉综合征有关联的风险因素,但这些因素因个体化差异较大,很难准确用于个体急性冠状动脉综合征的风险评估。
4.另一方面,肠道黏膜是机体最大的具有免疫活性的器官,肠道内寄存的几百亿细菌称为“肠道微生物群”,宿主为肠道菌群提供了适当的环境和必要的营养。反过来,肠道菌群又参与调节人体的各种功能,如向宿主提供代谢营养、参与促进生长和免疫调节、消除致病微生物、保持肠道屏障的完整性和正常的体内平衡。随着新近研究发现,肠道微生物菌群在人类免疫炎症性疾病和代谢性疾病中发挥着源头调节作用,并与存在代谢性炎症和胰岛素抵抗状、动脉粥样硬化、肥胖和糖尿病等疾病密切相关,肠道菌群作为冠心病发生和发展的源头调控影响因素也露出冰山一角。有研究指出,冠心病患者存在肠道菌群失调,表现为大肠杆菌、链球菌和幽门螺杆菌的比例增加。肠道菌群可通过代谢途径、炎性反应等多个途径促进动脉粥样硬化形成。
5.然而,现有技术中并没有通过研究冠心病的肠道菌群特征性预测急性冠脉综合征的发病风险的研究报道。另外,随着宏基因组学等各种测序技术的飞快发展,海量的数据也应运而生。如何从庞杂冗余的生物数据中挖掘出能够对急性冠脉综合征进行风险预测的生物标志物并实现急性冠脉综合征的准确风险预测十分重要。
技术实现要素:
6.本发明的一个目的在于提供一组与急性冠脉综合征发病风险相关的标志物。
7.本发明的另一目的在于提供一种建立急性冠脉综合征发病风险评估模型的方法。
8.本发明的另一目的在于提供一种急性冠脉综合征发病风险评估模型。
9.本发明的另一目的在于提供一种急性冠脉综合征发病风险评估装置。
10.本发明的另一目的在于提供一种急性冠脉综合征发病风险评估方法。
11.本案发明人通过大量的研究与实际检测分析试验,确定了一组与急性冠脉综合征发病风险相关的生物标志物,其包括多个肠道菌群,通过检测来自个体的样本中的这些肠道菌群的相关信息,可以良好地评估个体急性冠脉综合征发病风险。
12.具体而言,一方面,本发明提供了检测个体信息的试剂在制备评估急性冠脉综合征发病风险的检测装置中的应用,其中,所述个体信息包括肠道菌群信息,所述肠道菌群包括:
13.长双歧杆菌(bifidobacterium longum),
14.毛螺旋菌科_5_1_63faa(lachnospiraceae bacterium_5_1_63faa),
15.另枝菌属(alistipes onderdonkii),
16.产气柯林斯菌(collinsella aerofaciens),
17.真杆菌(eubacterium eligens),
18.普氏栖粪杆菌(faecalibacterium prausnitzii),
19.普通拟杆菌(bacteroides vulgatus),
20.未分类颤杆菌(oscillibacter unclassified),
21.卵形拟杆菌(bacteroides ovatus),以及
22.凸腹真桿菌(eubacterium ventriosum)。
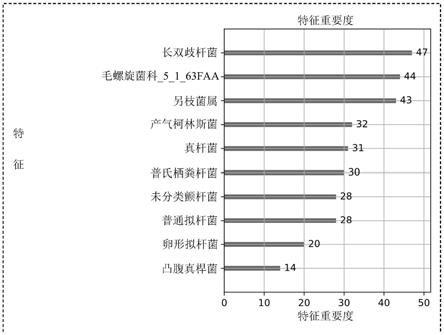
23.根据本发明的具体实施方案,本发明的应用中,所述肠道菌群中各菌在评估急性冠脉综合征发病风险时的特征重要度,长双歧杆菌(bifidobacterium longum)﹥毛螺旋菌科_5_1_63faa(lachnospiraceae bacterium_5_1_63faa)﹥另枝菌属(alistipes onderdonkii)﹥产气柯林斯菌(collinsella aerofaciens)﹥真杆菌(eubacterium eligens)﹥普氏栖粪杆菌(faecalibacterium prausnitzii)﹥普通拟杆菌(bacteroides vulgatus)=未分类颤杆菌(oscillibacter unclassified)﹥卵形拟杆菌(bacteroides ovatus)﹥凸腹真桿菌(eubacterium ventriosum)。
24.根据本发明的具体实施方案,本发明的应用中,所述肠道菌群中各菌在评估急性冠脉综合征发病风险时,按照以下特征重要度确定权重:
25.长双歧杆菌(bifidobacterium longum),47;
26.毛螺旋菌科_5_1_63faa(lachnospiraceae bacterium_5_1_63faa),44;
27.另枝菌属(alistipes onderdonkii),43;
28.产气柯林斯菌(collinsella aerofaciens),32;
29.真杆菌(eubacterium eligens),31;
30.普氏栖粪杆菌(faecalibacterium prausnitzii),30;
31.普通拟杆菌(bacteroides vulgatus),28;
32.未分类颤杆菌(oscillibacter unclassified),28;
33.卵形拟杆菌(bacteroides ovatus),20;
34.凸腹真桿菌(eubacterium ventriosum),14。
35.根据本发明的具体实施方案,本发明的应用中,所述长双歧杆菌(bifidobacterium longum)、毛螺旋菌科_5_1_63faa(lachnospiraceae bacterium_5_1_63faa)、另枝菌属(alistipes onderdonkii)、产气柯林斯菌(collinsella aerofaciens)、真杆菌(eubacterium eligens)、普氏栖粪杆菌(faecalibacterium prausnitzii)、普通拟杆菌(bacteroides vulgatus)、未分类颤杆菌(oscillibacter unclassified)、卵形拟杆菌(bacteroides ovatus)以及凸腹真桿菌(eubacterium ventriosum)均为急性冠脉综合征发病风险因素。各风险因素的异常程度越高(各肠道菌菌相比于健康人的表达丰度差异越大),个体急性冠脉综合征发病风险越高。
36.根据本发明的一些优选具体实施方案,本发明的应用中,所述个体信息还可进一步包括总胆固醇水平、年龄、血压中的一项或多项。
37.根据本发明的具体实施方案,本发明的技术特别适用于对来自东亚人群的个体进行急性冠脉综合征发病风险评估。
38.根据本发明的一些具体实施方案,本发明实施例提供一种建立急性冠脉综合征的风险预测模型的方法,以将所建立的模型用以对急性冠脉综合征进行风险预测,提高预测准确率,该方法包括:
39.获得急性冠脉综合征患者和健康人群的粪便样本dna数据;
40.对所述粪便样本dna数据进行双端测序处理,得到肠道菌群宏基因组数据;
41.利用trimmomatic软件去除肠道菌群宏基因组数据中的接头,并根据预先设定的碱基质量值,对去除接头的肠道菌群宏基因组数据进行修剪;
42.利用fastqc软件对修剪后的肠道菌群宏基因组数据进行质量评估;
43.对质量评估合格的肠道菌群宏基因组数据进行物种注释分析和功能注释分析,得到急性冠脉综合征患者和健康人群的相对丰度信息;
44.根据所述相对丰度信息和预先筛选的急性冠脉综合征的生物标志物,确定肠道菌群特征数据,所述急性冠脉综合征的生物标志物是根据差异菌相对丰度历史信息进行预先筛选的,所述差异菌相对丰度历史信息是对急性冠脉综合征患者和健康人群的相对丰度历史信息进行差异分析得到的;
45.将所述肠道菌群特征数据输入预先建立的机器学习模型中进行训练,得到急性冠脉综合征风险预测模型。
46.根据本发明的一些具体实施方案,本发明还提供了利用所述急性冠脉综合征风险预测模型进行急性冠脉综合征的风险预测的方法。
47.本发明一些实施方案中,还提供一种用于建立急性冠脉综合征的风险预测模型的装置,以将所建立的模型用以对急性冠脉综合征进行风险预测,提高预测准确率,该装置包括:
48.dna数据获得模块,用于获得急性冠脉综合征患者和健康人群的粪便样本dna数据;
49.双端测序处理模块,用于对所述粪便样本dna数据进行双端测序处理,得到肠道菌群宏基因组数据;
50.数据修剪模块,用于利用trimmomatic软件去除肠道菌群宏基因组数据中的接头,并根据预先设定的碱基质量值,对去除接头的肠道菌群宏基因组数据进行修剪;
51.质量评估模块,用于利用fastqc软件对修剪后的肠道菌群宏基因组数据进行质量评估;
52.注释分析模块,用于对质量评估合格的肠道菌群宏基因组数据进行物种注释分析和功能注释分析,得到急性冠脉综合征患者和健康人群的相对丰度信息;
53.特征数据确定模块,用于根据所述相对丰度信息和预先筛选的急性冠脉综合征的生物标志物,确定肠道菌群特征数据,所述急性冠脉综合征的生物标志物是根据差异菌相对丰度历史信息进行预先筛选的,所述差异菌相对丰度历史信息是对急性冠脉综合征患者和健康人群的相对丰度历史信息进行差异分析得到的;
54.模型训练模块,用于将所述肠道菌群特征数据输入预先建立的机器学习模型中进行训练,得到急性冠脉综合征风险预测模型。
55.本发明的另一些实施方案中,还提供了一种急性冠脉综合征的风险预测装置,其包括:风险预测模块,用于利用所述急性冠脉综合征风险预测模型进行急性冠脉综合征的风险预测。
56.本发明实施例还提供一种计算机设备,包括存储器、处理器及存储在存储器上并可在处理器上运行的计算机程序,所述处理器执行所述急性冠脉综合征的风险预测方法。
57.本发明实施例还提供一种计算机可读存储介质,所述计算机可读存储介质存储有执行上述急性冠脉综合征的风险预测方法的计算机程序。
58.另一方面,本发明还提供了另一种急性冠脉综合征发病风险评估装置,其包括检测单元和数据分析单元,其中:
59.所述检测单元用于检测个体信息,获得检测结果;其中,所述个体信息同前述本发明的所述个体信息;
60.所述数据分析单元用于对检测单元的检测结果进行分析处理。
61.根据本发明的具体实施方案,本发明的急性冠脉综合征发病风险评估装置中,所述检测单元包括可获得待测个体肠道菌群中各特征菌(长双歧杆菌、毛螺旋菌科_5_1_63faa、另枝菌属、产气柯林斯菌、真杆菌、普氏栖粪杆菌、普通拟杆菌、未分类颤杆菌、卵形拟杆菌、凸腹真桿菌)信息的任何试剂材料,可以采用现有技术中任何可行的方法检测待测个体肠道菌群中各特征菌的信息。
62.根据本发明的具体实施方案,本发明的急性冠脉综合征发病风险评估装置中,所述检测单元包括检测粪便样本dna数据的试剂材料。优选地,所述检测单元按照以下操作进行检测并获得检测结果:检测粪便样本,获得dna数据;对所获得的粪便样本dna数据进行双端测序处理,得到肠道菌群宏基因组数据;利用trimmomatic软件去除肠道菌群宏基因组数据中的接头,并根据预先设定的碱基质量值,对去除接头的肠道菌群宏基因组数据进行修剪;利用fastqc软件对修剪后的肠道菌群宏基因组数据进行质量评估;对质量评估合格的肠道菌群宏基因组数据进行物种注释分析和功能注释分析,得到肠道菌群中各菌的相对丰度信息。
63.根据本发明的具体实施方案,本发明的急性冠脉综合征发病风险评估装置中,所述数据分析单元对检测单元的检测结果进行分析处理时,包括:将个体信息的检测结果配
以权重系数,以计算所述待测个体的风险评估得分;其中,所述肠道菌群中各菌的特征重要度,长双歧杆菌(bifidobacterium longum)﹥毛螺旋菌科_5_1_63faa(lachnospiraceae bacterium_5_1_63faa)﹥另枝菌属(alistipes onderdonkii)﹥产气柯林斯菌(collinsella aerofaciens)﹥真杆菌(eubacterium eligens)﹥普氏栖粪杆菌(faecalibacterium prausnitzii)﹥普通拟杆菌(bacteroides vulgatus)=未分类颤杆菌(oscillibacter unclassified)﹥卵形拟杆菌(bacteroides ovatus)﹥凸腹真桿菌(eubacterium ventriosum)。
64.根据本发明的具体实施方案,本发明的急性冠脉综合征发病风险评估装置中,所述数据分析单元对检测单元的检测结果进行分析处理时,所述肠道菌群中各菌按照以下特征重要度数值确定权重,或者,所述肠道菌群中各菌的权重比值为:
65.长双歧杆菌(bifidobacterium longum),47;
66.毛螺旋菌科_5_1_63faa(lachnospiraceae bacterium_5_1_63faa),44;
67.另枝菌属(alistipes onderdonkii),43;
68.产气柯林斯菌(collinsella aerofaciens),32;
69.真杆菌(eubacterium eligens),31;
70.普氏栖粪杆菌(faecalibacterium prausnitzii),30;
71.普通拟杆菌(bacteroides vulgatus),28;
72.未分类颤杆菌(oscillibacter unclassified),28;
73.卵形拟杆菌(bacteroides ovatus),20;
74.凸腹真桿菌(eubacterium ventriosum),14。
75.另一方面,本发明还提供了另一种计算机存储介质,所述计算机存储介质存储有计算机程序指令,所述计算机程序指令被执行时实现:基于待测个体信息获得个体急性冠脉综合征发病风险评估结果;
76.其中,所述个体信息同前述本发明的所述个体信息。
77.另一方面,本发明还提供了另一种计算机设备,其包括存储器、处理器及存储在存储器上并可在处理器上运行的计算机程序,其中,所述处理器执行所述计算机程序时实现:基于待测个体信息获得个体急性冠脉综合征发病风险评估结果;
78.其中,所述个体信息同前述本发明的所述个体信息。
79.本发明实施例通过获得急性冠脉综合征患者和健康人群的粪便样本dna数据;对所述粪便样本dna数据进行双端测序处理,得到肠道菌群宏基因组数据;利用trimmomatic软件去除肠道菌群宏基因组数据中的接头,并根据预先设定的碱基质量值,对去除接头的肠道菌群宏基因组数据进行修剪;利用fastqc软件对修剪后的肠道菌群宏基因组数据进行质量评估;对质量评估合格的肠道菌群宏基因组数据进行物种注释分析和功能注释分析,得到急性冠脉综合征患者和健康人群的相对丰度信息;根据所述相对丰度信息和预先筛选的急性冠脉综合征的生物标志物,确定肠道菌群特征数据,所述急性冠脉综合征的生物标志物是根据差异菌相对丰度历史信息进行预先筛选的,所述差异菌相对丰度历史信息是对急性冠脉综合征患者和健康人群的相对丰度历史信息进行差异分析得到的;将所述肠道菌群特征数据输入预先建立的机器学习模型中进行训练,得到急性冠脉综合征风险预测模型;利用所述急性冠脉综合征风险预测模型进行急性冠脉综合征的风险预测。本发明实施
例充分考虑到急性冠脉综合征患者的肠道菌群特征,利用机器学习算法从复杂、繁冗的生物大数据中筛选可用于预测及监测急性冠脉综合征风险的、无创的生物标志物,提高预测准确率,弥补了急性冠脉综合征临床预警的空白。
附图说明
80.为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。在附图中:
81.图1为本发明实施例中急性冠脉综合征的风险预测方法示意图。
82.图2为本发明一具体实施例中训练集中的auroc曲线图。
83.图3为本发明一具体实施例中筛到的对模型起重要作用的急性冠脉综合征的生物标志物示意图。
84.图4为本发明实施例中急性冠脉综合征的风险预测装置结构图。
85.图5为本发明另一具体实施例的急性冠脉综合征风险评估模型的auroc曲线图。
具体实施方式
86.为使本发明实施例的目的、技术方案和优点更加清楚明白,下面结合附图对本发明实施例做进一步详细说明。在此,本发明的示意性实施例及其说明用于解释本发明,但并不作为对本发明的限定。
87.如前所述,随着宏基因组学等各种测序技术的飞快发展,海量的数据也应运而生。如何从庞杂冗余的生物数据中挖掘有用的信息,用于疾病的预测、诊断指标,一直是一项极具挑战的事情。随着大数据时代的来临,科研人员开发了多种算法进行生命科学领域相关数据的挖掘,而对于标志物诊断模型而言,不得不提的就是机器学习算法。机器学习包含了多种方法:线性回归、随机森林等。不同的算法适用的情况和条件不同,易受到生物样本的个体差异,实验方法等影响。
88.为了对急性冠脉综合征进行风险预测,提高预测准确率,本发明实施例提供一种建立急性冠脉综合征的风险预测模型的方法,如图1所示,该方法可以包括:
89.步骤101、获得急性冠脉综合征患者和健康人群的粪便样本dna数据;
90.步骤102、对所述粪便样本dna数据进行双端测序处理,得到肠道菌群宏基因组数据;
91.步骤103、利用trimmomatic软件去除肠道菌群宏基因组数据中的接头,并根据预先设定的碱基质量值,对去除接头的肠道菌群宏基因组数据进行修剪;
92.步骤104、利用fastqc软件对修剪后的肠道菌群宏基因组数据进行质量评估;
93.步骤105、对质量评估合格的肠道菌群宏基因组数据进行物种注释分析和功能注释分析,得到急性冠脉综合征患者和健康人群的相对丰度信息;
94.步骤106、根据所述相对丰度信息和预先筛选的急性冠脉综合征的生物标志物,确定肠道菌群特征数据,所述急性冠脉综合征的生物标志物是根据差异菌相对丰度历史信息进行预先筛选的,所述差异菌相对丰度历史信息是对急性冠脉综合征患者和健康人群的相
对丰度历史信息进行差异分析得到的;
95.步骤107、将所述肠道菌群特征数据输入预先建立的机器学习模型中进行训练,得到急性冠脉综合征风险预测模型。
96.进一步,本发明还提供了一种急性冠脉综合征的风险预测的方法,该方法包括:
97.步骤108、利用所述急性冠脉综合征风险预测模型进行急性冠脉综合征的风险预测。
98.由图1所示可以得知,本发明实施例通过获得急性冠脉综合征患者和健康人群的粪便样本dna数据;对所述粪便样本dna数据进行双端测序处理,得到肠道菌群宏基因组数据;利用trimmomatic软件去除肠道菌群宏基因组数据中的接头,并根据预先设定的碱基质量值,对去除接头的肠道菌群宏基因组数据进行修剪;利用fastqc软件对修剪后的肠道菌群宏基因组数据进行质量评估;对质量评估合格的肠道菌群宏基因组数据进行物种注释分析和功能注释分析,得到急性冠脉综合征患者和健康人群的相对丰度信息;根据所述相对丰度信息和预先筛选的急性冠脉综合征的生物标志物,确定肠道菌群特征数据,所述急性冠脉综合征的生物标志物是根据差异菌相对丰度历史信息进行预先筛选的,所述差异菌相对丰度历史信息是对急性冠脉综合征患者和健康人群的相对丰度历史信息进行差异分析得到的;将所述肠道菌群特征数据输入预先建立的机器学习模型中进行训练,得到急性冠脉综合征风险评估模型;利用所述急性冠脉综合征风险评估模型进行急性冠脉综合征的风险评估。本发明实施例充分考虑到急性冠脉综合征患者的肠道菌群特征,利用机器学习算法从复杂、繁冗的生物大数据中筛选可用于评估及监测急性冠脉综合征风险的、无创的生物标志物,提高评估准确率,弥补了急性冠脉综合征临床预警的空白。
99.实施例中,获得急性冠脉综合征患者和健康人群的粪便样本dna数据。
100.本实施例中,获得急性冠脉综合征患者和健康人群的粪便样本dna数据之后,利用琼脂糖凝胶方法确定所述粪便样本dna数据的总量数据和总浓度数据;将所述总量数据与总浓度数据与预设阈值进行比较;根据比较的结果对所述粪便样本dna数据进行筛选。
101.实施例中,对所述粪便样本dna数据进行双端测序处理,得到肠道菌群宏基因组数据。利用trimmomatic软件去除肠道菌群宏基因组数据中的接头,并根据预先设定的碱基质量值,对去除接头的肠道菌群宏基因组数据进行修剪。利用fastqc软件对修剪后的肠道菌群宏基因组数据进行质量评估。
102.具体实施时,在患者接受项目检测后收集其粪便样本,并在30分钟内放入干冰保存,并尽快储存在-80℃冰箱中待测。提取dna,对提取的核酸物质利用琼脂糖凝胶方法进行质量控制,要求dna总量≥1μg,dna总浓度≥20ng/μl,对质量合格的样本进行建库,然后对粪便样本dna数据进行illumina hiseq4000双端测序,得到每一个样本的双端测序数据,以fastq文件存储。fastq是一种存储了生物序列(通常是核酸序列)以及相应的质量评价的文本格式,它们都是以ascii编码的,几乎是高通量测序的标准格式。
103.具体实施时,用trimmomatic软件对数据进行质量控制,即修剪和去除原始数据中的接头(adapter)和低质量序列。trimmomatic是一个广受欢迎的illumina平台数据过滤工具,其支持多线程,处理数据速度快,主要用来去除fastq序列中的接头,并根据碱基质量值对fastq进行修剪。它包含双端测序和单端测序两种模式同时支持gzip和bzip2压缩文件,也支持phred-33和phred-64格式互相转化。fastqc是一款基于java的软件,它可以快速地
对测序数据进行质量评估。对过滤后的数据,用fastqc软件评价质控后的数据质量。根据fastqc的分析结果,可以判断fastq测序文件的质量。如果fastq测序文件质量合格,则进行后续的数据分析;否则,要重做调整参数,利用trimmomatic软件对双端测序数据进行修剪。需要说明的是,测序出来的序列每个碱基都对应有一个质量值(用字母或符号表示,可转为ascii值减去64来看),这个质量值代表测出的这个碱基的准确性,如果这条序列普遍质量值较低或平均质量值小于20,也或n很多也算低质量序列。
104.实施例中,对质量评估合格的肠道菌群宏基因组数据进行物种注释分析和功能注释分析,得到急性冠脉综合征患者和健康人群的相对丰度信息。
105.本实施例中,对所述肠道菌群宏基因组数据进行物种注释分析和功能注释分析,包括:下载肠道菌群数据库,所述肠道菌群数据库包括多个参考基因组,所述参考基因组包括:细菌,古菌,病毒和真核生物;根据所述肠道菌群数据库,利用metaphian2软件对肠道菌群宏基因组数据进行物种注释分析,利用humann2软件对肠道菌群宏基因组数据进行功能注释分析。
106.本实施例中,对质控后的数据,采用metaphian2软件进行宏基因组物种注释分析。metaphian2整理了17000多个参考基因组,包括13500个细菌和古菌,3500个病毒和110种真核生物。下载对应的数据库后,采用该软件,可以实现精确的分类群分配以及准确的计算物种的相对丰度。其能达到种水平的精度,以及菌株水平的鉴定和追踪。对肠道菌群宏基因组数据进行物种注释和功能注释后,得到肠道菌群的物种丰度信息建立模型进行评估。
107.本实施例中,采用r软件包vegan分析物种多样性,输入文件为肠道菌群物种丰度数据。lefse(lda effect size)有网页运行版本(http://huttenhower.sph.harvard.edu/galaxy/),准备好肠菌物种丰度数据,输入到网页运行版本中,按照默认流程运行,可得到结果,即组间的差异菌群。这里的冠心病肠道菌群特征数据,即从lefse分析得到的差异菌物种丰度数据。
108.实施例中,根据所述相对丰度信息和预先筛选的急性冠脉综合征的生物标志物,确定肠道菌群特征数据,所述急性冠脉综合征的生物标志物是根据差异菌相对丰度历史信息进行预先筛选的,所述差异菌相对丰度历史信息是对急性冠脉综合征患者和健康人群的相对丰度历史信息进行差异分析得到的。
109.本实施例中,差异菌相对丰度历史信息是对急性冠脉综合征患者和健康人群的相对丰度历史信息进行差异分析得到的,包括:所述差异菌相对丰度历史信息是利用lda effect size软件对急性冠脉综合征患者和健康人群的相对丰度历史信息进行差异分析得到的。
110.本实施例中,按如下方式对所述急性冠脉综合征的生物标志物进行预先筛选:利用boruta特征选择包对差异菌相对丰度历史信息进行特征选择,确定急性冠脉综合征的生物标志物。
111.本实施例中,按如下方式利用boruta特征选择包对所述差异菌相对丰度历史信息进行特征选择:根据差异菌相对丰度历史信息,创建阴影特征矩阵;根据所述阴影特征矩阵确定真实特征数据和阴影特征数据;根据所述真实特征数据和阴影特征数据,确定每个差异菌相对丰度历史信息对应的重要度标签;根据所述重要度标签,对差异菌相对丰度历史信息进行特征选择。
112.具体实施时,采用boruta算法进行特征选择。boruta的目标就是选择出所有与因变量相关的特征集合,而不是针对特定模型选择出可以使得模型cost function最小的特征集合。boruta算法的意义在于可以帮助本发明更全面的理解因变量的影响因素,从而更好、更高效地进行特征选择。boruta是python中的一个特征选择包,安装该包后输入差异菌相对丰度历史信息,可以得到适合建模的重要特征。其具体算法步骤为:(1)创建阴影特征(shadow feature):对每个真实特征r,随机打乱顺序,得到阴影特征矩阵s,拼接到真实特征后面,构成新的特征矩阵n=[r,s];(2)用新的特征矩阵n作为输入,训练模型,得到真实特征和阴影特征;(3)取阴影特征的最大值,真实特征中大于该值的,记录一次命中;(4)用(3)中记录的真实特征累计命中,标记特征重要或不重要;(5)删除不重要的特征,重复1-4,直到所有特征都被标记。
[0113]
实施例中,将所述肠道菌群特征数据输入预先建立的机器学习模型中进行训练,得到急性冠脉综合征风险评估模型。
[0114]
本实施例中,将所述肠道菌群特征数据输入预先建立的机器学习模型中进行训练,包括:将所述肠道菌群特征数据输入预先建立的lightgbm机器学习模型进行训练。利用gridsearchcv算法和hyperopt算法对所述lightgbm机器学习模型进行参数调整;利用测试数据对参数调整后的lightgbm机器学习模型进行测试;根据测试的结果,利用auroc指标对lightgbm机器学习模型进行性能评价。
[0115]
本实施例中,gridsearchcv(网格搜索)调整参数,即在指定的参数范围内,按步长依次调整参数,利用调整的参数训练学习器,从所有的参数中找到在验证集上精度最高的参数,这其实是一个循环和比较的过程。lightgbm是比xgboost更强大、速度更快的模型,性能上有很大的提升,与传统算法相比具有的优点:更快的训练效率、低内存使用、更高的准确率、支持并行化学习、可处理大规模数据。采用hyperopt对新模型进一步参数调优,hyperopt是一种通过贝叶斯优化来调整参数的工具,该方法较快的速度,并有较好的效果。此外,hyperopt结合mongodb可以进行分布式调参,快速找到相对较优的参数。
[0116]
本实施例中,采用的是python中的lightgbm包进行lightgbm机器学习构建模型。该模型主要包含两个算法:单边梯度采样(goss)和互斥特征绑定(efb)。goss(从减少样本角度):排除大部分小梯度的样本,仅用剩下的样本计算信息增益。每个数据实例有不同的梯度,根据计算信息增益的定义,梯度大的实例对信息增益有更大的影响,因此在采样时,尽量保留梯度大的样本(预先设定阈值,或者最高百分位间),随机去掉梯度小的样本。此措施在相同的采样率下比随机采样获得更准确的结果,尤其是在信息增益范围较大时。efb(从减少特征角度):捆绑互斥特征,也就是用一个合成特征代替,特别在稀疏特征空间上,许多特征几乎是互斥的(例如许多特征不会同时为非零值)。可以捆绑互斥的特征,将捆绑问题归约到图着色问题,通过贪心算法求得近似解。更具体地,相关参数可以设置如下:
[0117]
params={'boosting_type':'gbdt',
[0118]
'objective':'binary',
[0119]
'metric':'auc',
[0120]
'nthread':4,
[0121]
'learning_rate':0.1,
[0122]
'num_leaves':30,
[0123]
'max_depth':5,
[0124]
'subsample':0.8,
[0125]
'colsample_bytree':0.8,}
[0126]
其中,gbdt即梯度提升树,nthread服务器运行的线程,learning_rate即每个弱学习器的权重缩减系数,num_leaves即每个基学习器输出one-hot向量(长度),max_depth即决策树最大深度,subsample即子采样比例,取值范围为(0,1],colsample_bytree即用来控制每颗树随机采样的列数的占比。
[0127]
本实施例中,gridsearchcv和hyperopt是python中给的包,本发明在python中安装这些包后,进行参数调优。gridsearchcv的名字其实可以拆分为两部分,gridsearch和cv,即网格搜索和交叉验证。网格搜索,搜索的是参数,即在指定的参数范围内,按步长依次调整参数,利用调整的参数训练学习器,从所有的参数中找到在验证集上精度最高的参数,这其实是一个训练和比较的过程。hyperopt是python中的一个用于"分布式异步算法组态/超参数优化"的类库。使用它本发明可以拜托繁杂的超参数优化过程,自动获取最佳的超参数。广泛意义上,可以将带有超参数的模型看作是一个必然的非凸函数,因此hyperopt几乎可以稳定的获取比手工更加合理的调参结果。尤其对于调参比较复杂的模型而言,其更是能以远快于人工调参的速度同样获得远远超过人工调参的最终性能。
[0128]
本实施例中,基于上述方法筛选出的的急性冠脉综合征的生物标志物包括:长双歧杆菌bifidobacterium longum,毛螺旋菌科_5_1_63faa lachnospiraceae bacterium_5_1_63faa,另枝菌属alistipes onderdonkii,产气柯林斯菌collinsella aerofaciens,真杆菌eubacterium eligens,普氏栖粪杆菌faecalibacterium prausnitzii,普通拟杆菌bacteroides vulgatus,未分类颤杆菌oscillibacter unclassified,卵形拟杆菌bacteroides ovatus,凸腹真桿菌eubacterium ventriosum。其相对应的特征重要度见图3。
[0129]
本实施例中,auroc的全称是“接受者操作特征曲线下面积”,往往作为一个评价模型预测能力的指标。在讨论auroc曲线之前,本发明需要理解混淆矩阵(confusion matrix)的概念。一个二元预测可能有4个结果:本发明预测0,而真实类别是0:这被称为真阴性(tn,true negative);本发明预测0,而真实类别是1:这被称为假阴性(fn,false negative);本发明预测1,而真实类别是0:这被称为假阳性(fp,false positive);本发明预测1,而真实类别是1:这被称为真阳性(tp,true positive)。当比较两个不同模型的时候,使用单一指标常常比使用多个指标更方便,下面本发明基于混淆矩阵计算两个指标,之后本发明会将这两个指标组合成一个:
[0130]
真阳性率(tpr),即,灵敏度、命中率、召回,定义为tp/(tp+fn)。这一指标对应被正确识别为阳性的阳性数据点占所有阳性数据点的比例。换句话说,tpr越高,本发明遗漏的阳性数据点就越少。
[0131]
假阳性率(fpr),即,误检率,定义为fp/(fp+tn)。这一指标对应被误认为阳性的阴性数据点占所有阴性数据点的比例。换句话说,fpr越高,本发明错误分类的阴性数据点就越多。
[0132]
为了将fpr和tpr组合成一个指标,本发明首先基于不同的阈值(例如:0.00;0.01,0.02,
…
,1.00)计算前两个指标的逻辑回归,接着将它们绘制为一个图像,其中fpr值为横
轴,tpr值为纵轴。得到的曲线为roc曲线,本发明考虑的指标是该曲线的auc,称为auroc。对角虚线为随机预测器的roc曲线:auroc为0.5。随机预测器通常用作基线,以检验模型是否有用。auroc越高,说明模型的预测能力越好。
[0133]
下面给出一个具体实施例,说明本发明急性冠脉综合征的风险评估方法的具体应用。
[0134]
1、临床入组标准:
[0135]
依据冠状动脉粥样硬化性心脏病的临床特点,将病人分为2组,包括:(1)st段抬高急性心肌梗死(stemi,不稳定斑块破裂组,心肌坏死);非st段抬高急性心肌梗死(nstemi,不稳定斑块部分破裂组,心肌少量坏死)和不稳定心绞痛(uap,斑块濒临破裂或破裂前不稳定组,心肌微量坏死),即acs组,n=212;(2)无动脉粥样硬化斑块的正常对照组,即normal coronary artery组,nca,n=175。在临床信息收集的基础上,采集各组人群新鲜或妥善冷冻的粪便,进行肠道宏基因组测序。
[0136]
研究人群入选标准:稳定性冠心病(陈旧心梗、pci史、稳定性心绞痛或无临床缺血症状的“健康人”,同时冠脉ct/造影发现有冠脉狭窄病变》50%)。
[0137]
排除标准:
[0138]
1)根据国际通用心肌梗死定义诊断为2-5型心肌梗死;
[0139]
2)严重心力衰竭/心源性休克(killip》2级或nyha》2级);
[0140]
3)存在机械并发症(室间隔穿孔、游离壁破裂、乳头肌断裂等);
[0141]
4)发病后曾发生心脏骤停和/或心肺复苏;
[0142]
5)3月内口服或使用静脉任何抗生素≥1周;
[0143]
6)3月内急性冠状动脉综合征(acs)或冠状动脉血管重建(包括pci和cabg);
[0144]
7)3月内创伤或手术;
[0145]
8)3月内脑血管病史(包括脑梗死或脑出血);
[0146]
9)3月内上消化道或下消化道出血;
[0147]
10)3月内明确感染(包括消化道、呼吸道、体表感染等);
[0148]
11)慢性肠道疾病(如克劳恩病、溃疡性结肠炎等等);
[0149]
12)任何肿瘤;
[0150]
13)风湿免疫性疾病;
[0151]
14)慢性肾脏疾病,包括肾脏移植术后。
[0152]
研究对象入选及病例信息收集过程:
[0153]
(1)知情同意书;
[0154]
(2)入选/排除标准;
[0155]
(3)患者生活方式问卷临床资料;
[0156]
(4)在临床信息收集的基础上,采集各组人群的血液、新鲜或妥善冷冻的粪便,进行组学分析。
[0157]
本临床研究遵守《世界医学大会赫尔辛基宣言》和国家相关法规的要求实施。本临床研究方案已获阜外医院的医学伦理委员会批准,所有参与实验的临床患者均已签署本项目《知情同意书》。
[0158]
2、实施方法:
[0159]
共有387名参与者在国家心血管病中心、中国医学科学院阜外医院参加了本次研究。根据诊断指南和排除标准将其分为以下两组:nca组(n=175),acs组(n=212)。
[0160]
在患者入院的第二天上午,空腹时间大于10小时的条件下采集病人的血液样本,由阜外医院完成相关临床常规生化指标检测,所有检测均按照国际标准方法进行。同时收集患者粪便样本,并在30分钟内放入干冰保存,并尽快储存在-80℃冰箱中待测。提取dna,对提取的核酸物质利用琼脂糖凝胶方法进行质量控制。要求dna总量≥1μg,dna总浓度≥20ng/μl。对质量合格的样本进行建库,illumina hiseq4000双端测序。获取原始宏基因组双端测序数据后,用trimmomatic软件对数据进行质量控制,去除低质量序列和接头。并用fastqc软件评价质控后的数据。对质控后的数据,采用metaphian2软件进行宏基因组物种注释分析。获取癌症患者与正常人肠道菌群的物种的丰度信息后,分析物种多样性,并采用lefse(lda effect size)分析组间菌群差异,获得急性冠脉综合征患者肠道菌群的特征,在物种水平建立模型进行评估。采用lightgbm的机器学习方法建模及十乘十交叉验证的方法,将数据随机分成训练集和测试集。首先采用boruta算法进行特征选择。采用gridsearchcv(网格搜索)和hyperopt不断调整参数,选择最优的参数。重新获取一批从未参与建模的外部数据,将构建好的模型用于评估这批数据,通过auroc来判断评估模型的好坏。特征的重要性用其对模型的贡献度表示。所有的分析采用python的scikit-learn包。图2为训练集中的auroc曲线图,图3为筛到的对模型起重要作用的急性冠脉综合征的生物标志物。
[0161]
基于同一发明构思,本发明实施例还提供了一种急性冠脉综合征的风险评估装置,如下面的实施例所述。由于这些解决问题的原理与急性冠脉综合征的风险评估方法相似,因此装置的实施可以参见方法的实施,重复之处不再赘述。
[0162]
图4为本发明实施例中急性冠脉综合征的风险评估装置的结构图,如图4所示,该装置包括:
[0163]
dna数据获得模块401,用于获得急性冠脉综合征患者和健康人群的粪便样本dna数据;
[0164]
双端测序处理模块402,用于对所述粪便样本dna数据进行双端测序处理,得到肠道菌群宏基因组数据;
[0165]
数据修剪模块403,用于利用trimmomatic软件去除肠道菌群宏基因组数据中的接头,并根据预先设定的碱基质量值,对去除接头的肠道菌群宏基因组数据进行修剪;
[0166]
质量评估模块404,用于利用fastqc软件对修剪后的肠道菌群宏基因组数据进行质量评估;
[0167]
注释分析模块405,用于对质量评估合格的肠道菌群宏基因组数据进行物种注释分析和功能注释分析,得到急性冠脉综合征患者和健康人群的相对丰度信息;
[0168]
特征数据确定模块406,用于根据所述相对丰度信息和预先筛选的急性冠脉综合征的生物标志物,确定肠道菌群特征数据,所述急性冠脉综合征的生物标志物是根据差异菌相对丰度历史信息进行预先筛选的,所述差异菌相对丰度历史信息是对急性冠脉综合征患者和健康人群的相对丰度历史信息进行差异分析得到的;
[0169]
模型训练模块407,用于将所述肠道菌群特征数据输入预先建立的机器学习模型中进行训练,得到急性冠脉综合征风险评估模型;
[0170]
风险评估模块408,用于利用所述急性冠脉综合征风险评估模型进行急性冠脉综合征的风险评估。
[0171]
一个实施例中,所述模型训练模块进一步用于:
[0172]
将所述肠道菌群特征数据输入预先建立的lightgbm机器学习模型进行训练。
[0173]
一个实施例中,急性冠脉综合征的风险评估装置还包括:
[0174]
参数调整模块,用于利用gridsearchcv算法和hyperopt算法对所述lightgbm机器学习模型进行参数调整;
[0175]
模型测试模块,用于利用测试数据对参数调整后的lightgbm机器学习模型进行测试;
[0176]
性能评价模块,用于根据测试的结果,利用auroc指标对lightgbm机器学习模型进行性能评价。
[0177]
一个实施例中,本发明的急性冠脉综合征的生物标志物包括:长双歧杆菌bifidobacte rium longum,毛螺旋菌科_5_1_63faa lachnospiraceae bacterium_5_1_63faa,另枝菌属alistipes onderdonkii,产气柯林斯菌collinsella aerofaciens,真杆菌eubacterium eligens,普氏栖粪杆菌faecalibacterium prausnitzii,普通拟杆菌bacteroides vulgatus,未分类颤杆菌os cillibacter unclassified,卵形拟杆菌bacteroides ovatus,凸腹真桿菌eubacterium ventriosum。各生物标志物均为急性冠脉综合征发病风险因素,用于评估急性冠脉综合征发病风险时的特征重要度参见图3。如果某一项或多项生物标志物相比于健康人的表达丰度差异越大,则个体急性冠脉综合征发病风险越高。
[0178]
图5显示了在本发明的肠道菌群特征因素的基础上,进一步整合传统认为与急性冠脉综合征风险密切相关的总胆固醇水平、年龄和高血压因素,所获得的用于对急性冠脉综合征发病风险进行评估的模型的auroc曲线。将其与图2相比,可以看出,进一步整合总胆固醇水平、年龄和血压因素后,与急性冠脉综合征发病风险的关联强度并没有特别显著地提升,可表明本发明的肠道菌群特征因素可独立于传统临床危险因素(总胆固醇水平、年龄和高血压)之外用于评估急性冠脉综合征发病风险。
[0179]
综上所述,本发明实施例通过获得急性冠脉综合征患者和健康人群的粪便样本dna数据;对所述粪便样本dna数据进行双端测序处理,得到肠道菌群宏基因组数据;利用trimmomatic软件去除肠道菌群宏基因组数据中的接头,并根据预先设定的碱基质量值,对去除接头的肠道菌群宏基因组数据进行修剪;利用fastqc软件对修剪后的肠道菌群宏基因组数据进行质量评估;对质量评估合格的肠道菌群宏基因组数据进行物种注释分析和功能注释分析,得到急性冠脉综合征患者和健康人群的相对丰度信息;根据所述相对丰度信息和预先筛选的急性冠脉综合征的生物标志物,确定肠道菌群特征数据,所述急性冠脉综合征的生物标志物是根据差异菌相对丰度历史信息进行预先筛选的,所述差异菌相对丰度历史信息是对急性冠脉综合征患者和健康人群的相对丰度历史信息进行差异分析得到的;将所述肠道菌群特征数据输入预先建立的机器学习模型中进行训练,得到急性冠脉综合征风险评估模型;利用所述急性冠脉综合征风险评估模型进行急性冠脉综合征的风险评估。本发明实施例充分考虑到急性冠脉综合征患者的肠道菌群特征,利用机器学习算法从复杂、繁冗的生物大数据中筛选可用于预测及监测急性冠脉综合征风险的、无创的生物标志物,
提高预测准确率,弥补了急性冠脉综合征临床预警的空白。
[0180]
本领域内的技术人员应明白,本发明的实施例可提供为方法、系统、或计算机程序产品。因此,本发明可采用完全硬件实施例、完全软件实施例、或结合软件和硬件方面的实施例的形式。而且,本发明可采用在一个或多个其中包含有计算机可用程序代码的计算机可用存储介质(包括但不限于磁盘存储器、cd-rom、光学存储器等)上实施的计算机程序产品的形式。
[0181]
本发明是参照根据本发明实施例的方法、设备(系统)、和计算机程序产品的流程图和/或方框图来描述的。应理解可由计算机程序指令实现流程图和/或方框图中的每一流程和/或方框、以及流程图和/或方框图中的流程和/或方框的结合。可提供这些计算机程序指令到通用计算机、专用计算机、嵌入式处理机或其他可编程数据处理设备的处理器以产生一个机器,使得通过计算机或其他可编程数据处理设备的处理器执行的指令产生用于实现在流程图一个流程或多个流程和/或方框图一个方框或多个方框中指定的功能的装置。
[0182]
这些计算机程序指令也可存储在能引导计算机或其他可编程数据处理设备以特定方式工作的计算机可读存储器中,使得存储在该计算机可读存储器中的指令产生包括指令装置的制造品,该指令装置实现在流程图一个流程或多个流程和/或方框图一个方框或多个方框中指定的功能。
[0183]
这些计算机程序指令也可装载到计算机或其他可编程数据处理设备上,使得在计算机或其他可编程设备上执行一系列操作步骤以产生计算机实现的处理,从而在计算机或其他可编程设备上执行的指令提供用于实现在流程图一个流程或多个流程和/或方框图一个方框或多个方框中指定的功能的步骤。
[0184]
最后应说明的是:以上所述实施例,仅为本发明的具体实施方式,用以说明本发明的技术方案,而非对其限制,本发明的保护范围并不局限于此,尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,其依然可以对前述实施例所记载的技术方案进行修改或可轻易想到变化,或者对其中部分技术特征进行等同替换;而这些修改、变化或者替换,并不使相应技术方案的本质脱离本发明实施例技术方案的精神和范围,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应所述以权利要求的保护范围为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1