一种氟雷拉纳的合成方法与流程
1.本发明属于医药化工领域,特别涉及一种氟雷拉纳的合成方法。
背景技术:
2.氟雷拉纳是异噁唑啉类广谱杀虫剂,目前作为兽药已登记上市,商品名称为 bravecto
tm
[1]。氟雷拉纳的cas号为864731-61-3,cas名称为4-[5-(3,5-二氣苯基)-4,5-二氢-5-(三氟甲基)-3-异噁唑基]-2-甲基-义[2-氧代-2-[(2,2,2-三氟乙基)氧基]乙基] 苯甲酰胺,中文通用名为氟雷拉纳。其化学结构式如下所示:
[0003][0004]
氟雷拉纳主要用于动物寄生虫的治疗,与苯基吡唑类、环戊二烯类以及大环内酯类等杀虫剂的作用靶标类似,属于y-氨基丁酸门控氯离子通道干扰剂,其作用机制主要是通过干扰y-氨基丁酸门控氯离子通道从而达到杀虫效果+51。据相关文献报道,氟雷拉纳作为一种广谱型杀虫剂,除动物寄生虫外,对大多数农业害虫同样具有较好的杀虫活性。与市场上其他杀虫剂相比,氟雷拉纳具有与之相当或更高的杀虫活性,尤其是对虱目、蛋目、半翅目、双翅目以及鳞翅目等害虫都具有良好的杀虫效果。因此,氟雷拉纳有望作为农药进行研制开发,用于农业害虫的防治。
[0005]
目前关于氟雷拉纳的合成有多种方法,《精细化工》第37卷第5期讲述了三种氟雷拉纳的合成路线,第一条是以4-乙酰基-2-甲基苯甲酸为起始原料,首先制成4-乙酰基-2
‑ꢀ
甲基苯酰氯,然后与2-氨基-;v-(2,2,2-三氟乙基)乙酰胺进行酰胺化反应得到4-乙酰基
ꢀ‑
2-甲基-#-[2-氧代-2-[(2,2,2-三氟乙基)氨基]乙基]苯甲酰胺,再与3',5'-二氯-2,2,2-三氟苯乙酮反应制得4-[(2£)-3-(3,5-二氯苯基)-4,4,4-三氟-1-氧代-2-丁烯-1-基]-2-甲基界[2-氧代
ꢀ‑
2-[(2,2,2-三氟乙基)氨基]乙基]苯甲酰胺,最后进行合环得到目标产物氟雷拉纳,第二条是以4-溴-2-甲基苯甲酸为起始原料,经过酯化、酰胺化反应得到2-甲基-4-甲醛肟基苯甲酸叔丁酯,然后与1,3-二氯-5-(1-三氟甲基-乙烯基)苯进行1,3-偶极加成环合反应得到4-[5-(3,5-二氯苯基)-4,5-二氢-5-(三氟甲基)-3-异噁唑基]-2-甲基苯甲酸叔丁酯,再经过碘取代及水解反应最后得到4-[5-(3,5-二氯苯基)-4,5-二氢-5-三氟甲基-3-异噁唑基]-2-碘
ꢀ‑
6-甲基苯甲酸中间体,得到的中间体再通过与2-氨基-n(2,2,2-三氟乙基)乙酰胺盐酸盐反应以及去碘化反应,最终得到目标产物氟雷拉纳,第三种那个路线为3,5-二氯苯甲酸为起始原料,首先与甲醇进行酯化得到3,5-二氯苯甲酸甲酯,然后与三甲基(三氟甲基)硅烷反应得到[1-(3,5-二氯苯基)-2,2,2-三氟-1-甲氧基乙氧基]三甲基硅烷,然后与ak4-乙酰基-2-甲基苯基)乙酰胺反应得到4-[5-(3,5-二氯苯基)-5-三氟甲基-4,5-二氢异嗯唑-3-基]-2
‑ꢀ
甲基苯胺,再经过重氮化反应得到3-(4-溴-3-甲基苯基)-5-(3,5-二氯苯基)-5-三氟甲基-4,5
‑ꢀ
二氢异噁唑,再与一氧化碳反应,得到4-[5-(3,5-二
氯苯基)-5-三氟甲基-4,5-二氢异嚷唑
ꢀ‑
3-基]-2-甲基苯甲酸,最后与2-氨基-ak2,2,2-三氟乙基)乙酰胺反应得到最终产物氟雷拉纳以上三条路线中第一条存在制备价格高的问题,第二条路线制备价格昂贵且关键步骤偶极加成收率较低,第三条需要气体co参与,反应条件苛刻。以上路线均不适合工业化生产。
[0006]
cn109879826、cn111675667、cn112457267均提到通过4-甲酰基-2-甲基苯甲酸为原料,通过肟化,取代,缩合,关环反应得到氟雷拉纳,但在关键步骤,关环反应中,存在收率低的问题。
[0007]
此外,现有的合成工艺还包括以4-溴-2-甲基苯甲酸为原料,经suzuki偶联反应得到4-乙酰基-2-甲基苯甲酸,再经过缩合,脱水环合制备氟雷拉纳,但是存在经济成本高,反应时间长等问题。
技术实现要素:
[0008]
发明目的:为了解决现有技术的问题,本发明提供了一种氟雷拉纳的合成方法,该合成方法降低了反应成本,提高了收率以及缩短了反应周期。
[0009]
技术方案:本发明所述的氟雷拉纳的合成方法,包括以下步骤:
[0010]
(s1):4-溴-2-甲基-苯甲酸经suzuki偶联反应得到4-乙酰基-2-甲基苯甲酸,所用的催化剂为摩尔比为1:2~4的醋酸钯与1,3-双(二苯基膦)丙烷;
[0011][0012]
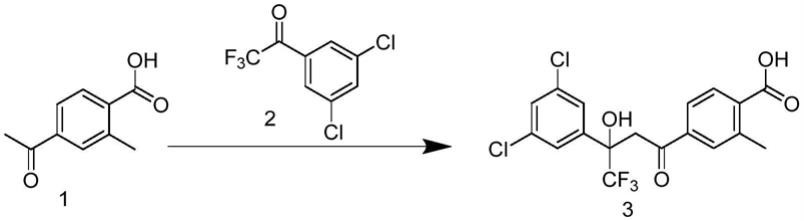
(s2):4-乙酰基-2-甲基苯甲酸(中间体1)与3,5-二氯-三氟苯乙酮(化合物2)在碱性条件下加热缩合制备4-(3-(3,5-二氯苯基)-4,4,4-三氟-3-羟基丁基)-苯甲酸(中间体3);
[0013][0014]
(s3):4-(3-(3,5-二氯苯基)-4,4,4-三氟-3-羟基丁基)-苯甲酸(中间体3)与三乙胺形成三乙胺盐,再在dmap催化下脱水形成4-(3-(3,5-二氯苯基)-4,4,4-三氟-2-烯醇)-苯甲酸(中间体4);
[0015]
[0016]
(s4):4-(3-(3,5-二氯苯基)-4,4,4-三氟-2-烯醇)-苯甲酸(中间体4)与四丁基溴化铵、氢氧化钠与盐酸羟胺制备4-(5-(3,5-二氯苯基)-5-(三氟甲基)-3-(4,5-二氢异噻唑基))-苯甲酸(中间体5);
[0017][0018]
(s5):4-(5-(3,5-二氯苯基)-5-(三氟甲基)-3-异噁唑基)-苯甲酸(中间体5) 与氯化亚砜形成酰氯后,无氧条件下,在碱性条件下,与2-氨基-n-(2,2,2-三氟乙基)乙酰胺(化合物6)合成氟雷拉纳(化合物7)。
[0019][0020]
作为本发明的一种优选实施方式,步骤(s1)以及步骤(s2)为连投步骤,以及步骤(s3)、(s4)连投缩短整个流程反应时间,减少后处理的时间成本及目标化合物的中间损失,提高了收率,适合工业化生产。
[0021]
作为本发明的一种优选实施方式,步骤(s1)中,所述4-溴-2-甲基-苯甲酸与钯催化剂的摩尔比为1:0.007~0.0085。
[0022]
作为本发明的一种优选实施方式,所述4-溴-2-甲基-苯甲酸与钯催化剂的摩尔比为 1:0.0075~0.008。
[0023]
作为本发明的一种优选实施方式,所述4-溴-2-甲基-苯甲酸与钯催化剂的摩尔比为 1:0.0076~0.0078。
[0024]
作为本发明的一种优选实施方式,所述酸钯与1,3-双(二苯基膦)丙烷的摩尔比为 1:4。
[0025]
作为本发明的一种优选实施方式,步骤(s1)中,将摩尔比为1:4.8~5:0.03~0.032: 0.0075~0.008:1.65~1.7的4-溴-2-甲基-苯甲酸、正丁基乙烯基醚、1,3-双(二苯基膦) 丙烷、醋酸钯以及碳酸钾加入正丁醇,氮气置换2~3次,加热85~95℃回流8~10h,停止加热,冷至室温,将反应产物提纯。
[0026]
步骤(s1)中,所述反应产物提纯的方法为:向反应产物中加入水和浓盐酸调节 ph到1-2,乙酸乙酯萃取,有机相依次用水洗,饱和氯化钠水溶液洗涤,用硅藻土过滤后,无水硫酸钠干燥,旋干。
[0027]
作为本发明的一种优选实施方式,步骤(s2)中,所述4-乙酰基-2-甲基苯甲酸、 3,5-二氯-三氟苯乙酮、月硅酸钠、碳酸钾以及混合均匀,在55~65℃温度下搅拌反应 20~
24h,所述4-乙酰基-2-甲基苯甲酸与水的摩尔比为1:0.2~0.5。
[0028]
作为本发明的一种优选实施方式,步骤(s2)中,将4-乙酰基-2-甲基苯甲酸、3,5
‑ꢀ
二氯-三氟苯乙酮、月硅酸钠、碳酸钾以及水的摩尔比为1:1:0.06~0.07:1.5~1.6:0.2~0.5,在55~65℃温度下搅拌反应20~24h,反应物成微白色泥状,将所得产物纯化,得微黄色固体。
[0029]
作为本发明的一种优选实施方式,步骤(s2)中,4-乙酰基-2-甲基苯甲酸、3,5-二氯-三氟苯乙酮、月硅酸钠、碳酸钾以及水的摩尔比为1:1:0.06~0.07:1.5~1.6:0.2~0.3。
[0030]
作为本发明的一种优选实施方式,步骤(s2)中,所述纯化的方式为:向微白色泥状的反应物中加入水,浓盐酸调ph至1-2,用乙酸乙酯萃取,无水硫酸钠干燥,柱层析纯化。
[0031]
作为本发明的一种优选实施方式,步骤(s3)中,4-(3-(3,5-二氯苯基)-4,4,4
‑ꢀ
三氟-3-羟基丁基)-苯甲酸与三乙胺的摩尔比为1:5~6。
[0032]
作为本发明的一种优选实施方式,步骤(s3)中,所述脱水的反应条件为三乙胺盐溶于有机溶剂中,加入4-二甲氨基吡啶,加热至55~65℃,逐滴加入乙酸酐,滴加完毕后,升温到75~85℃,搅拌反应5.5~6.5h。
[0033]
作为本发明的一种优选实施方式,步骤(s3)中,所述4-(3-(3,5-二氯苯基)-4,4,4
‑ꢀ
三氟-3-羟基丁基)-苯甲酸、4-二甲氨基吡啶以及乙酸酐的摩尔比为1~1.2:0.2~0.3:4~4.5。
[0034]
作为本发明的一种优选实施方式,步骤(s3)中,将4-(3-(3,5-二氯苯基)-4,4,4
‑ꢀ
三氟-3-羟基丁基)-苯甲酸溶于二氯甲烷中,搅拌下加入三乙胺,室温反应0.5~1h,后将反应液旋干,得到三乙胺盐,向三乙胺盐中加入甲苯,和4-二甲氨基吡啶加热到 55~65℃,逐滴加入乙酸酐,滴加完毕后,升温到75~85℃,搅拌5.5~6.5h,监测反应完全。
[0035]
作为本发明的一种优选实施方式,步骤(s4)中,4-(3-(3,5-二氯苯基)-4,4,4
‑ꢀ
三氟-2-烯醇)-苯甲酸、四丁基溴化铵、氢氧化钠与盐酸羟胺的摩尔比为 1:0.3~0.4:4.5~5.5:2~3。
[0036]
作为本发明的一种优选实施方式,步骤(s4)中,反应条件为室温搅拌反应12~24h。
[0037]
作为本发明的一种优选实施方式,步骤(s5)中,反应时间为5~6小时。
[0038]
作为本发明的一种优选实施方式,步骤(s5)中,将4-(5-(3,5-二氯苯基)-5-(三氟甲基)-3-异噁唑基)-苯甲酸溶于苯中,加入氯化亚砜,回流反应4~6h,反应完全后,旋干,得到反应产物中间体0;在氮气保护情况下,将2-氨基-n-(2,2,2-三氟乙基)乙酰胺溶于二氯甲烷中,0℃下加入三乙胺,搅拌5~10min,滴加溶于二氯甲烷的中间体0,滴加完毕,保持5~10min,撤去冰浴,室温反应5~6h,监测反应完全后,旋干,柱层析。
[0039]
有益效果:(1)本发明调整催化剂的用量,在保证产率以及生产效率的同时,降低了生产成本;(2)本发明在步骤s3中提高加热温度,从而缩短反应时间;(3)本发明步骤s1以及s2可以连投,步骤s3以及s4可以连投,缩短整个流程反应时间,减少后处理的时间成本及目标化合物的中间损失,提高了收率,适合工业化生产;(4)本发明的合成方法提高了经济效益,减少试剂用量,增加反应安全性。
具体实施方式
[0040]
现通过以下具体实施方式说明本发明的有益效果,本领域普通人员根据本发明做出的显而易见的改变和修饰也在本发明范围内。
[0041]
实施例1:4-乙酰基-2-甲基苯甲酸(中间体1)的制备
[0042][0043]
将4-溴-2-甲基-苯甲酸(化合物0)(0.277g,1.29mmol),正丁基乙烯基醚(0.83 ml,6.42mmol),1,3-双(二苯基膦)丙烷(0.17g,0.04mmol),醋酸钯(0.0023g, 0.01mmol),碳酸钾(0.3g,2.17mmol)加入到瓶中,加入正丁醇10ml,氮气置换3 次,加热控制温度为90℃回流9~10小时,原料反应完全,停止加热,冷至室温,加入水和浓盐酸调节ph到1-2,乙酸乙酯萃取,有机相依次用水洗,饱和氯化钠水溶液洗涤,用硅藻土过滤后,无水硫酸钠干燥,旋干。得到0.263g化合物1,黄色固体,产率为95%。1hnmr(300mhz,cdcl3)δ8.17(d,j=7.9hz,1h),7.88(d,j=8.4hz,2h),2.72(d, j=19.4hz,6h).
[0044]
实施例2:4-(3-(3,5-二氯苯基)-4,4,4-三氟-3-羟基丁基)-苯甲酸(中间体3)的制备
[0045][0046]
将化合物1(0.5g,2.8mmol),化合物2(0.68g,2.8mmol),月硅酸钠(0.04g,0.18 mmol),碳酸钾(0.6g,4.34mmol),水(12ml,0.66mol)混合均匀,60℃搅拌24h,此时反应物成微白色泥状,向反应液中加入水,浓盐酸调ph至1-2,用乙酸乙酯萃取,无水硫酸钠干燥,柱层析纯化,流动相为石油醚(pe)与乙酸乙酯(ea),(pe:乙酸乙酯ea=2:1,v:v),得到0.915g微黄色固体,产率为77.42%。
[0047]1hnmr(300mhz,cdcl3)δ8.21(d,j=8.3hz,1h),7.87(d,j=7.5hz,2h),7.55(d,j=1.8hz,2h),7.41(d,j=1.8hz,1h),5.64(s,1h),3.92(d,j=17.6hz,1h),3.76(d,j=17.6 hz,1h),2.78(s,3h).
[0048]
实施例3:(1)4-(3-(3,5-二氯苯基)-4,4,4-三氟-2-烯醇)-苯甲酸(中间体4)的制备
[0049]
[0050]
将化合物3(0.5g,1.19mmol)溶于20ml二氯甲烷中,搅拌下加入三乙胺(0.62g, 6.12mmol)室温反应1h,后将反应液旋干,得到三乙胺盐,向三乙胺盐中加入甲苯20ml,和4-二甲氨基吡啶(0.03g,0.24mmol,)加热到60℃,逐滴加入乙酸酐(0.4ml,4.2mmol),滴加完毕后,升温到80℃,搅拌6h,监测反应完全,冷却至室温,加水,浓盐酸调节 ph到1-2,ea萃取,水洗,盐洗,无水硫酸钠干燥,得到0.405g黄色固体。产率84.6%,进行下一步制备。
[0051]
实施例4:4-(5-(3,5-二氯苯基)-5-(三氟甲基)-3-(4,5-二氢异噻唑基))-苯甲酸(中间体5)的制备
[0052][0053]
步骤(1):取氢氧化钠0.2g,盐酸羟胺0.17g分别配制成质量浓度为50%的溶液,冷却后,混合,冷至室温备用。
[0054]
步骤(2):二颈瓶中加入化合物4(0.405g,1mmol),四丁基溴化铵(0.12g,0.37mmol),氮气保护,注射甲苯20ml,0℃搅拌10min,滴加步骤(1)配制好的溶液,滴加完毕后,室温搅拌过夜,反应完毕后,加水,浓盐酸调节ph1~2,ea萃取,水洗,干燥,柱层析(pe:ea=2:1)得到0.42g黄色固体,产率为84.6%。
[0055]1hnmr(300mhz,cdcl3)δ7.97(d,j=8.1hz,1h),7.59(d,j=18.9hz,2h),7.45(d,j= 18.7hz,1h),7.37(s,1h),7.18~7.12(m,1h),4.14(dd,j=15.6,7.1hz,1h),3.76(d,j=17.4 hz,1h),2.72(s,1h),2.63(s,2h).
[0056]
为了缩短反应周期,本发明的中间体4不进行提纯操作,中间体3-中间体5的反应进行连投操作:具体步骤如下:
[0057][0058]
步骤(1):取氢氧化钠0.2g,盐酸羟胺0.17g分别配制成质量浓度为50%的溶液,冷却后,混合,冷至室温备用。
[0059]
步骤(2):将化合物3(0.5g,1.19mmol)溶于20ml二氯甲烷中,搅拌下加入三乙胺(0.62g,6.12mmol)室温反应1h,后将反应液蒸干,得到三乙胺盐,氮气保护,向三乙胺盐中加入甲苯20ml,和4-二甲氨基吡啶(0.03g,0.24mmol)加热到60℃,逐滴加入乙酸酐(0.4ml,4.2mmol),滴加完毕后,升温到80℃,搅拌6h,监测反应完全,冷却至室温,0℃加入四丁基溴化铵(0.12g,0.37mmol),搅拌10min,滴加步骤(1) 配制好的溶液,滴加完毕后,室温搅拌过夜,反应完毕后,加水,浓盐酸调节ph至1~2, ea萃取,水洗,干燥,柱层析(pe:ea=2:1,v:v)得到黄色固体,产物经核磁图谱验证为中间体5,产率与实施例4得到的产率相当,表明本发明中间体3-中间5可以直接连投。
[0060]
实施例5:氟雷拉纳(化合物7)的制备
[0061][0062]
将化合物5(0.42g,1mmol)溶于苯20ml中,加入氯化亚砜(0.33g,2.8mmol),85℃加热回流反应5h,反应完全后,旋干,得到中间体0。
[0063]
氮气保护,将化合物6(0.18g,1.2mmol)溶于10ml二氯甲烷中,0℃下加入三乙胺 (0.13g,1.3mmol),搅拌10min,滴加溶于5ml二氯甲烷的步骤(1)得到的中间体0(0.47g),滴加完毕,保持10min,撤去冰浴,室温反应5h,监测反应完全后,旋干,柱层析, (pe:ea =2.5:1)得到0.465g微黄色固体。产率为85.73%。
[0064]1hnmr(300mhz,dmso~d6)δ8.65(dt,j=6.5,3.6hz,2h),7.84(d,j=1.9hz,1h), 7.70~7.56(m,4h),7.53(d,j=8.4hz,1h),4.43(d,j=18.4hz,1h),4.33(d,j=18.5hz,1h), 4.11
–
3.87(m,4h),2.43(s,3h) 。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1