一种恩格列净中间体的合成方法与流程
1.本发明涉及一种恩格列净中间体(3s)-3-[4-[(5-碘-2-氯苯基)甲基]苯氧基]四氢呋喃的合成方法,属于有机合成技术领域。
背景技术:
[0002]
恩格列净(empagliflozin)是勃林格殷格翰和礼来联合开发的一种钠-葡萄糖协同转运蛋白(sglt2)抑制剂,于2014年5月首先获得欧洲药物管理局批准上市,2014年8月获得美国fda批准上市,2014年12月获日本药品与医疗器械管理局批准上市,2017年9月获准在中国上市,可用于治疗2型糖尿病。作为一种高选择性的sglt2抑制剂,恩格列净拥有独特的不依赖胰岛素的降糖途径,即通过减少葡萄糖在肾脏的重吸收而从尿中直接排糖,除具有明确的降糖效果外,还能带来减轻体重、降低血压、降低尿酸的益处。化学名称:(1s)-1,5-脱水-1-c-[4-氯-3-[[4-[[(3s)-四氢-3-呋喃基]氧基]苯基]甲基]苯基]-d-葡萄糖醇,恩格列净分子式:c23h27clo7,分子量:450.91。化学结构式如下:
[0003][0004]
原研专利cn102574829a公开了一种恩格列净的合成方法,其合成路线如下所示。
[0005]
[0006][0007]
其中,化合物v.1是(3s)-3-[4-[(5-碘-2-氯苯基)甲基]苯氧基]四氢呋喃,作为恩格列净的重要中间体,其合成方法主要有以下几种:
[0008]
1)中国专利cn107311962a公开了一种(s)-3-(4-(5-溴-2-氯苄基)苯氧基)四氢呋喃的合成方法,合成路线如下所示。
[0009][0010]
该合成方法采用了简单易得的4-氟甲苯作为起始原料,合成路线较短,但是该合成路线的第1-2步反应及后处理步骤较为繁琐,有待进一步改善。
[0011]
2)中国专利cn108178751 a公开了一种恩格列净中间体的合成方法,该合成方法以4-羟基苄基氯为起始原料,依次与甲基磺酰氯、(s)-3-羟基四氢呋喃进行反应后得到化合物ⅲ,然后再与4-碘苯胺进行反应得到化合物ⅳ,最后经重氮化反应后再与氯化亚铜反
应得到(s)-3-(4-(5-碘-2-氯苄基)苯氧基)四氢呋喃。
[0012][0013]
该合成路线较短,但产率较低,不利于工业生产。
技术实现要素:
[0014]
针对现有技术中的上述技术问题,提供了一种新的恩格列净中间体的合成方法。
[0015]
本发明提供一种技术方案:一种恩格列净中间体-(s)-3-(4-(5-碘-2-氯苄基)苯氧基)四氢呋喃的合成方法,包括以下步骤:
[0016][0017][0018]
(1)、由(s)-3-羟基四氢呋喃与苯酚在binap和mtad作用下生成(s)-3-苯氧基四氢呋喃;
[0019]
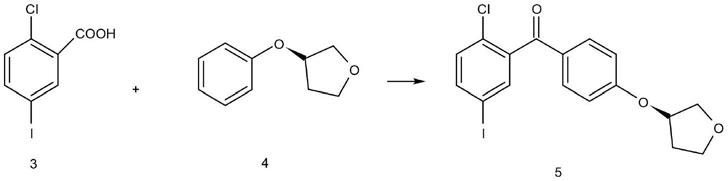
(2)、(s)-3-苯氧基四氢呋喃与2-氯-5-碘苯甲酸酰化反应得到2-3(5-碘-2-氯苯基)[4-[[(3s)-四氢-3-呋喃基]氧基]苯基]甲酮;
[0020]
(3)、化合物2-3(5-碘-2-氯苯基)[4-[[(3s)-四氢-3-呋喃基]氧基]苯基]甲酮还
原反应得到(s)-3-(4-(5-碘-2-氯苄基)苯氧基)四氢呋喃。
[0021]
其中,所述步骤(1)的反应溶剂为四氢呋喃、乙腈中的一种或多种的混合溶剂,优选为四氢呋喃。
[0022]
其中,所述步骤(2)的反应溶剂为四氢呋喃、二甲亚砜、dmf中的一种或多种的混合溶剂,优选为四氢呋喃和二甲亚砜的混合溶剂。
[0023]
进一步的,所述步骤(2)的中酰化剂为tscl(对甲苯磺酰氯),反应温度为室温。
[0024]
进一步的,所述步骤(3)的反应溶剂为二氧六环、tbme(甲基叔丁基醚)、乙二醇二乙醚(dme)中的一种或多种的混合溶剂,优选为二氧六环。
[0025]
进一步的,所述步骤(3)的催化剂为三乙胺和edta(n,n-二异丙基乙胺),优选为edta。
[0026]
具体包括以下步骤:
[0027]
1)在thf(四氢呋喃)中加入苯酚、binap(联萘二苯膦)和(s)-3-羟基四氢呋喃,逐滴添加mtad(n-甲基-1,2,4-三唑啉-3,5-二酮),室温下将溶液搅拌过夜。反应结束后,过滤、浓缩、萃取和纯化后得到(s)-3-苯氧基四氢呋喃;
[0028]
2)在四氢呋喃和二甲亚砜的混合溶液中,依次加入2-氯-5-碘苯甲酸、(s)-3-苯氧基四氢呋喃和tscl,室温搅拌,缓慢地滴加edta,tlc显示反应1h后完成,在室温下用稀酸调ph为中性,减压除去溶剂,抽滤,水洗后干燥得白色固体。
[0029]
3)将步骤2)的产物加入到干燥的二氧六环中,加入氯化锌,再向该混合液中加入硼氢化钠,室温下搅拌2h。经hplc分析测定其转化情况,待反应完全后,使用乙酸乙酯萃取,减压蒸馏,水洗至中性,无水硫酸钠干燥,过滤,滤液减压回收溶剂得目标产物。
[0030]
本发明的有益效果:
[0031]
本技术合成方法以(s)-3-羟基四氢呋喃为起始原料,依次与2-氯-5-碘苯甲酸进行反应后得到化合物2-3(5-碘-2-氯苯基)[4-[[(3s)-四氢-3-呋喃基]氧基]苯基]甲酮,最后经羰基还原反应得到(s)-3-(4-(5-碘-2-氯苄基)苯氧基)四氢呋喃。本技术通过摸索更合适的反应试剂和溶剂,与现有技术相比,反应步骤短,条件温和,后处理简单,产品的收率高。
具体实施方式
[0032]
以下实施例是对本发明的进一步说明,但是本发明并不局限于此。
[0033]
本技术的原料购于上海易汇生物科技有限公司,试剂购于百灵威化学技术有限公司。
[0034]
核磁共振由德国bruker公司的mercury-plus300/bruker500型核磁共振仪进行测定,质谱由美国waters公司的waters uplc ms测定。
[0035]
实施例1:
[0036]
步骤1)(s)-3-苯氧基四氢呋喃的制备
[0037]
在thf(200ml)中加入苯酚(4.70g,50mmol)、binap(32.4g,52mmol)和(r)-3-羟基四氢呋喃(4.58g,52mmol),逐滴添加mtad(5.65g,50mmol)。室温下将溶液搅拌过夜(hplc监控反应是否完成)。反应结束后,过滤、浓缩、萃取和纯化后得到(s)-3-苯氧基四氢呋喃6.90g,产率为84.6%,纯度为99.5%。
[0038]
步骤2)2-3(5-碘-2-氯苯基)[4-[[(3s)-四氢-3-呋喃基]氧基]苯基]甲酮的制备
[0039]
在150ml的四氢呋喃和二甲亚砜的混合溶液(比例2:1)中,依次加入2-氯-5-碘苯甲酸(11.30g,40mmol)和(s)-3-苯氧基四氢呋喃(6.57g,40mmol),然后加入tscl(0.3g,1.6mmol),室温搅拌,缓慢地滴加8ml edta,tlc显示反应1h后完成,在室温下用稀酸调ph为中性,减压除去溶剂,抽滤,水洗后干燥得白色固体2-3(5-碘-2-氯苯基)[4-[[(3s)-四氢-3-呋喃基]氧基]苯基]甲酮15.75g。收率91.9%,纯度98.3%。
[0040]
步骤3)(s)-3-(4-(5-碘-2-氯苄基)苯氧基)四氢呋喃的制备
[0041]
将步骤2)的产物加入到200ml干燥的二氧六环中,加入氯化锌(6.8g,50mmol),再向该混合液中加入硼氢化钠(1.89g,50mmol),室温下搅拌2h。经hplc分析测定其转化情况,待反应完全后,使用乙酸乙酯150ml萃取两次,减压蒸馏,水洗至中性,无水硫酸钠干燥,过滤,滤液减压回收溶剂得目标产物13.1g。收率为86.1%,纯度为99.61%。1h nmr(500mhz,cdcl3)δ7.53-7.42(m,2h),7.15-7.09(m,3h),6.81(d,j=8.6hz,2h),4.95(dd,j=5.6,4.2hz,1h),4.05-3.96(m,5h),3.91(td,j=8.2,4.5hz,1h),2.27-2.13(m,2h).esi-ms m/z:414.0(m+1)
+
。
[0042]
实施例2:
[0043]
步骤1)(s)-3-苯氧基四氢呋喃的制备
[0044]
在thf(300ml)中加入苯酚(7.05g,75mmol)、binap(46.73g,75mmol)和(r)-3-羟基四氢呋喃(6.61g,75mmol),逐滴添加mtad(8.48g,75mmol)。室温下将溶液搅拌过夜(hplc监控反应是否完成)。反应结束后,过滤、浓缩、萃取和纯化后得到(s)-3-苯氧基四氢呋喃10.53g,产率为84.6%,纯度为99.5%。
[0045]
步骤2)2-3(5-碘-2-氯苯基)[4-[[(3s)-四氢-3-呋喃基]氧基]苯基]甲酮的制备
[0046]
在240ml的四氢呋喃和二甲亚砜的混合溶液(比例2:1)中,依次加入2-氯-5-碘苯甲酸(16.95g,60mmol)和(s)-3-苯氧基四氢呋喃(9.86g,60mmol),然后加入tscl(0.4g,2.1mmol),室温搅拌,缓慢地滴加10ml edta,tlc显示反应1h后完成,在室温下用稀酸调ph为中性,减压除去溶剂,抽滤,水洗后干燥得白色固体2-3(5-碘-2-氯苯基)[4-[[(3s)-四氢-3-呋喃基]氧基]苯基]甲酮23.86g。收率92.8%,纯度98.5%。
[0047]
步骤3)(s)-3-(4-(5-碘-2-氯苄基)苯氧基)四氢呋喃的制备
[0048]
将步骤2)的产物加入到250ml干燥的二氧六环中,加入氯化锌(8.2g,60mmol),再向该混合液中加入硼氢化钠(2.27g,60mmol),室温下搅拌2h。经hplc分析测定其转化情况,待反应完全后,使用乙酸乙酯150ml萃取两次,减压蒸馏,水洗至中性,无水硫酸钠干燥,过滤,滤液减压回收溶剂得目标产物16.0g。收率为87.3%,纯度为99.50%。
[0049]
以上显示和描述了本发明的基本原理和主要特征以及本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1