用于治疗COVID-19的化合物和方法与流程
用于治疗covid-19的化合物和方法
1.发明背景
2.本发明涉及抑制病毒复制活性的化合物和方法,其包括使 sars-cov-2相关的3c样(“3cl”)蛋白酶与治疗有效量的 sars-cov-2相关的3c样蛋白酶抑制剂接触。本发明也涉及通过给有 此需要的患者施用治疗有效量的sars-cov-2相关的3c样蛋白酶抑 制剂来治疗患者中的冠状病毒疾病2019(“covid-19”)的方法。本发 明进一步涉及治疗患者中的covid-19的方法,所述方法包括给有此 需要的患者施用药物组合物,其包含治疗有效量的sars-cov-2相关 的3c样蛋白酶抑制剂。
3.到2020年4月上旬,covid-19的爆发已蔓延到全球许多国家, 已确认超过100万人被感染,并导致5,0000多人死亡,到2021年3 月,全球已经有超过150万人死亡。covid-19的病原体已被确定为 一种新型冠状病毒,已被命名为严重急性呼吸综合征冠状病毒2 (“sars-cov-2”)。sars-cov-2的基因组序列是从中国武汉的9 名患者身上获得的分离物中测序的,并被发现属于β冠状病毒属的 sarbecovirus亚属。lu,r.等人.the lancet,2020年1月29日; http://doi.org/10.1016/s0140-6736(20)。发现sars-cov-2的序列与 2018年在中国东部舟山采集的两种蝙蝠来源的sars样冠状病毒 bat-sl-covzc45和bat-sl-covzxc21具有88%的同源性。还发现 sars-cov-2与严重急性呼吸综合征冠状病毒(“sars-cov”) (2002-2003年sars爆发的病原体)有约79%的同源性,与中东呼 吸综合征冠状病毒(“mers-cov”)(2012年起源于中东的呼吸道病 毒爆发的病原体)有约50%的同源性。根据最近对sars-cov-2的 103个测序基因组的分析,已经提出sars-cov-2可分为两大类型(l 型和s型),s型是祖先型,l型已经从s型演变。lu,j.;cui,j.et al. on the origin and continuing evolution of sars-cov-2; http://doi.org/10.1093/nsr/nwaa036。s和l型可以仅由位于8,782 (orf1ab:t8517c,同义)和28,144(orf8:c251t,s84l)的两 个紧密连接的snp明确定义。在分析的103个基因组中,大约70% 属于l型,大约30%属于s型。尚不清楚l型从s型的演变是发生 在人类身上还是通过人畜共患病中间体发生的,但似乎l型比s型更 具侵略性,在sars-cov-2爆发后不久人类试图控制疫情的干预可能 使l型和s型的相对丰度发生了变化。所提出的sars-cov-2的s和 l亚型的发现增加了个体可能依次感染单个亚型或同时感染两种亚型 的可能性。鉴于这种不断演变的威胁,本领域迫切需要有效治疗 covid-19和抑制sars-cov-2冠状病毒复制的方法。
4.最近的证据清楚地表明,新出现的冠状病毒sars-cov-2,即 covid-19(疾病控制中心,cdc)的病原体,已经获得了人际传播 的能力,导致病毒在社区传播。sars-cov-2受体结合结构域(“rbd”) 的序列,包括其直接接触血管紧张素2受体ace2的受体结合基序 (rbm),与sars-cov的rbd和rbm相似,强烈表明sars-cov-2 使用ace2作为其受体。sars-cov-2rbm中的几个关键残基(特别 是gln
493
)提供了与人类ace2的良好相互作用,这与sars-cov-2 感染人类细胞的能力一致。sars-cov-2的rbm中的其它几个关键 残基(特别是asn
501
)与结合人类ace2兼容,但并不理想,这表明 sars-cov-2在一定程度上利用ace2结合进行人际传播。
5.冠状病毒的复制和转录功能由所谓的“复制酶”基因编码 (ziebuhr,j.,snijder,
e.j.,和gorbaleya,a.e.;virus-encodedproteinases and proteolytic processing in nidovirales.j.gen.virol. 2000,81,853-879;和fehr,a.r.;perlman,s.;coronaviruses:anoverview of their replication and pathogenesis methods mol biol. 2015;1282:1
–
23.doi:10.1007/978-1-4939-2438-7_1),它由两个重叠的 多蛋白组成,这些多蛋白被病毒蛋白酶广泛加工。冠状病毒主要或“3c 样”蛋白酶在11个保守的域间连接处处理c-近端区域(ziebuhr、 snijder、gorbalya,2000和fehr、perlman等人,2015)。“3c样
”ꢀ
蛋白酶的名称来源于冠状病毒酶与众所周知的小核糖核酸病毒3c蛋 白酶之间存在某些相似之处。这些包括底物偏好、使用半胱氨酸作为 催化中的活性位点亲核物质,以及它们推定的整体多肽折叠的相似性。 与sars-cov 3cl蛋白酶(登记号yp_009725301.1)相比,发现 sars-cov-2 3cl蛋白酶序列(登记号yp_009725301.1)具有96.08% 的同源性。xu,j.;zhao,s.;teng,t.;abdalla,a.e.;zhu,w.;xie,l.; wang,y.;guo,x.;systematic comparison of two animal-to-humantransmitted human coronaviruses:sars-cov-2and sars-cov; viruses 2020,12,244;doi:10.3390/v12020244。最近hilgenfeld及其同 事发表了sars-cov-2冠状病毒主蛋白酶(3cl)的高分辨率x射线结 构zhang,l.;lin,d.;sun,x.;rox,k.;hilgenfeld,r.;x-raystructure of main protease of the novel coronavirus sars-cov-2 enables design ofα-ketoamide inhibitors;biorxiv preprint doi: https://doi.org/10.1101/2020.02.17.952879。结构表明,比较 sars-cov-2和sars-cov的3cl蛋白酶时存在差异。在sars-cov 而非sars-cov-2 3cl蛋白酶二聚体中,两个结构域iii之间存在极 性相互作用,涉及每个原聚体残基thr
285
的侧链羟基之间的氢 键,并由ile
286
和thr
285
cγ2侧链之间的疏水性接触支持。与sars-cov3cl中的相同残基相比,在sars-cov-2 3cl中,苏氨酸被丙氨酸取 代,异亮氨酸被亮氨酸取代。在sars-cov-2 3cl蛋白酶中观察到的 thr285ala置换使两个结构域iii彼此靠近一些(分子a和b中残基 285的cα原子之间的距离在sars-cov 3cl蛋白酶中为在 sars-cov-2 3cl蛋白酶中为两个结构域iii的质心之间的距 离从缩小到)。在sars-cov-2 3cl的活性位点,cys
145
和his
41
形成一个催化二联体,当它与与his
41
氢键结合的埋藏水分子 结合在一起时,可以认为构成了sars-cov-2 3cl蛋白酶的催化三联 体。鉴于sars-cov-2的持续传播已导致当前全球范围内的 covid-19爆发,因此需要有新的方法来抑制sars-cov-2病毒复制 和治疗患者的covid-19。
技术实现要素:
6.本发明提供了新的化合物,其在抑制或预防sars-cov-2病毒复 制中起作用,且因而可用于治疗covid-19。本发明也提供了包含所 述化合物的药物组合物和通过施用本发明的化合物或包含本发明的化 合物的药物组合物来治疗covid-19和抑制sars-cov-2病毒复制的 方法。
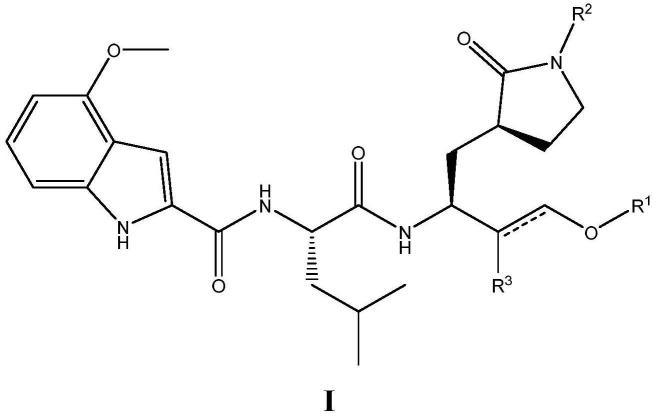
7.本发明的第一方面的第一个实施方案是式i的化合物
[0008][0009]
其中
‑‑‑‑‑‑
不存在或是键;r1选自-ch(r
4a
)-oc(o)r4、-c(o)or4、
ꢀ‑
ch(r
4a
)-oc(o)or4、-p(o)(or5)2、-p(o)(c
1-c6烷基)(or5)和
ꢀ‑
c(o)n(r6)2;r2选自氢、-c(o)r7、-co2r7和-c
1-c6烷基-oc(o)or7; 且当r2是-c(o)r7、-co2r7或-c
1-c6烷基-oc(o)or7时;则r1选自 氢、-ch(r
4a
)-oc(o)r4、-c(o)or4、-ch(r
4a
)-oc(o)or4、-p(o)(or5)2、
ꢀ‑
p(o)(c
1-c6烷基)(or5)和-c(o)n(r6)2;当
‑‑‑‑‑
不存在时r3是氧代, 或当
‑‑‑‑‑
是键时,r3与r1以及r1所连接的氧一起是-oc(o)o-;r4和r7各自独立地选自未被取代的或被1-3个r8取代的c
1-c6烷基、未 被取代的或被1-3个r8取代的c
3-c7环烷基、未被取代的或被1-3个 r8取代的c
5-c
12
二环烷基、包含1-3个独立地选自n、o和s的杂原 子且未被取代的或被1-3个r8取代的4-7元杂环烷基、未被取代的或 被1-3个r8取代的c
6-c
10
芳基、和包含1-4个独立地选自n、o和s 的杂原子且未被取代的或被1-3个r8取代的5-10元杂芳基;r
4a
是氢 或c
1-c6烷基;r5在每次出现时独立地是氢或c
1-c6烷基;或两个r5基团一起为任选地被苯基取代的c
2-c4亚烷基;r6在每次出现时独立 地选自氢和未被取代的或被1-3个r8取代的c
1-c6烷基;或两个r6基团与它们所连接的氮一起为4-7元杂环烷基,其任选地包含另外1-3 个独立地选自n、o和s的杂原子;其中所述杂环烷基是未被取代的 或被1-3个r8取代;且r8在每次出现时独立地选自卤代(halo)、 羟基、氰基、c
1-c3烷基、c
1-c3烷氧基、c
1-c3烷氧基c
1-c3烷基、 c
3-c6环烷基、c
3-c6环烷氧基、二(c
1-c3烷基)氨基、(c
1-c3烷基)氨 基、氨基、二(c
1-c3烷基)氨基-c
1-c3烷基、(c
1-c3烷基)氨基-c
1-c3烷基、氨基-c
1-c3烷基和包含1-3个独立地选自n、o和s的杂原子 的4-7元杂环烷基;或其药学上可接受的盐。
[0010]
本发明的第一方面的第二个实施方案是式ia的第一方面的第一 个实施方案的化合物
[0011][0012]
或其药学上可接受的盐。
[0013]
本发明的第一方面的第三个实施方案是第一方面的第二个实施方 案的化合物,其中r2选自氢、-c(o)och3、-c(o)oc(ch3)3、
ꢀ‑
ch(ch3)oc(o)och3;和-ch2oc(o)och3;和r4选自甲基、乙基、 异丙基和叔丁基;或其药学上可接受的盐。本发明的第一方面的第四 个实施方案是第一方面的第三个实施方案的化合物,其中r2是氢;或 其药学上可接受的盐。本发明的第一方面的第五个实施方案是第一方 面的第三个实施方案的化合物,其选自:碳酸(3s)-3-({n-[(4-甲氧基
ꢀ‑
1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷
ꢀ‑
3-基]丁基甲酯;碳酸(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基丙烷-2-基酯;碳 酸(3s)-4-[(3s)-1-乙酰基-2-氧代吡咯烷-3-基]-3-({n-[(4-甲氧基-1h-吲 哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代丁基甲酯;碳酸 (3s)-4-[(3s)-1-{(1s)-1-[(甲氧基羰基)氧基]乙基}-2-氧代吡咯烷-3
‑ꢀ
基]-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代丁 基甲酯;碳酸乙基(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰 基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯;(3s)-3-[(2s)-4-[(甲 氧基羰基)氧基]-2-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨 基)-3-氧代丁基]-2-氧代吡咯烷-1-甲酸甲酯;碳酸叔丁基(3s)-3-({n-[(4
‑ꢀ
甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代 吡咯烷-3-基]丁酯;和(3s)-3-[(2s)-4-[(叔-丁氧基羰基)氧基]-2-({n-[(4
‑ꢀ
甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-3-氧代丁基]-2-氧代吡 咯烷-1-甲酸叔丁酯;或其药学上可接受的盐。
[0014]
本发明的第一方面的第六个实施方案是式ib的第一方面的第一 个实施方案的化合物
[0015][0016]
或其药学上可接受的盐。
[0017]
本发明的第一方面的第七个实施方案是第一方面的第六个实施方 案的化合物,其中r2选自氢、-c(o)ch3、-co2ch3、-ch2oc(o)och3和-ch(ch3)oc(o)och3;r4选自甲基、乙基、异丙基和叔丁基;和 r
4a
选自氢、甲基和乙基;或其药学上可接受的盐。
[0018]
本发明的第一方面的第八个实施方案是第一方面的第六个实施方 案的化合物,其选自:碳酸(1r)-1-({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2
‑ꢀ
基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基} 氧基)乙基甲酯;碳酸(1r)-1-({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基) 乙基丙烷-2-基酯;碳酸(1r)-1-({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基) 羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基) 丙基甲酯;碳酸(1r)-1-({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l
‑ꢀ
亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基)丙基丙 烷-2-基酯;碳酸({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰 基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基)甲基甲基酯; 碳酸({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2
‑ꢀ
氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基)甲基丙烷-2-基酯;碳酸乙 基(1r)-1-({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨 基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基)乙酯;碳酸乙基(1r)-1-({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨 基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基)丙酯;碳酸乙基 ({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基)甲酯;(3s)-3-[(2s)-4-{[(甲氧基 羰基)氧基]甲氧基}-2-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-3-氧代丁基]-2-氧代吡咯烷-1-甲酸甲酯;碳酸叔丁基 (1r)-1-({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)
ꢀ‑
2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基)乙酯;碳酸叔丁基 (1r)-1-({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)
ꢀ‑
2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基)丙酯;碳酸叔丁基 ({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基)甲酯;碳酸{(3s)-3-[(2s)-4-{[(甲 氧基羰基)氧基]甲氧
4-[(3s)-2-氧代吡咯烷-3-基]丁酯;哌啶-1-甲酸 (3s)-4-[(3s)-1-(甲氧基羰基)-2-氧代吡咯烷-3-基]-3-({n-[(4-甲氧基-1h
‑ꢀ
吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代丁酯;哌啶-1-甲酸 (3s)-4-[(3s)-1-乙酰基-2-氧代吡咯烷-3-基]-3-({n-[(4-甲氧基-1h-吲哚
ꢀ‑
2-基)羰基]-l-亮氨酰基}氨基)-2-氧代丁酯;和哌啶-1-甲酸 (3s)-4-[(3s)-1-{(1s)-1-[(甲氧基羰基)氧基]乙基}-2-氧代吡咯烷-3
‑ꢀ
基]-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代丁 酯;或其药学上可接受的盐。
[0029]
本发明的第一方面的第十五个实施方案是式ie的根据权利要求1 所述的化合物
[0030][0031]
或其药学上可接受的盐。
[0032]
本发明的第一方面的第十六个实施方案是第一方面的第十五个实 施方案的化合物,其中r2选自氢、-c(o)ch3、-co2ch3、
ꢀ‑
ch2oc(o)och3和-ch(ch3)oc(o)och3;和r5在每次出现时独立 地选自氢、甲基、乙基、异丙基和叔丁基;或两个r5基团一起为-ch(苯 基)ch2ch
2-;或其药学上可接受的盐。
[0033]
本发明的第一方面的第十七个实施方案是第一方面的第十五个实 施方案的化合物,其选自:碳酸(1s)-1-{(3s)-3-[(2s)-4-[(二甲氧基磷酰 基)氧基]-2-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-3
‑ꢀ
氧代丁基]-2-氧代吡咯烷-1-基}乙基甲酯;磷酸二氢(3s)-3-({n-[(4-甲氧 基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯 烷-3-基]丁酯;磷酸(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨 酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基二甲酯;磷酸 (3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁基二丙烷-2-基酯;磷酸(3s)-4-[(3s)-1
‑ꢀ
乙酰基-2-氧代吡咯烷-3-基]-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l
‑ꢀ
亮氨酰基}氨基)-2-氧代丁基二甲酯;4-甲氧基-n-[(2s)-4-甲基
ꢀ‑
1-({(2s)-4-[(2-氧(oxido)-4-苯基-1,3,2-二氧杂磷杂环己烷 (dioxaphosphinan)-2-基)氧基]-3-氧代-1-[(3s)-2-氧代吡咯烷-3-基] 丁-2-基}氨基)-1-氧代戊烷-2-基]-1h-吲哚-2-甲酰胺;磷酸二乙基 (3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁酯;和(3s)-3-[(2s)-4-[(二甲氧基磷酰基) 氧基]-2-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-3-氧代 丁基]-2-氧代吡咯烷-1-甲酸甲酯;或其药学上可接受的盐。
[0034]
本发明的第一方面的第十八个实施方案是第一方面的第一个实施 方案的化合物,其中r1是-p(o)(c
1-c6烷基)(or5);或其药学上可接受 的盐。本发明的第一方面的第十九个实施方案是第一方面的第十八个 实施方案的化合物,其为甲基膦酸(3s)-3-({n-[(4-甲氧基-1h-吲哚-2
‑ꢀ
基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基甲 酯;或其药学上可接受的盐。
[0035]
本发明的第一方面的第二十个实施方案是式if的第一方面的第一 个实施方案的化合物
[0036][0037]
或其药学上可接受的盐。
[0038]
本发明的第一方面的第二十一个实施方案是第一方面的第二十个 实施方案的化合物,其中r2选自氢、-c(o)ch3、-co2ch3、
ꢀ‑
ch2oc(o)och3和-ch(ch3)oc(o)och3;或其药学上可接受的盐。
[0039]
本发明的第一方面的第二十二个实施方案是第一方面的第二十个 实施方案的化合物,其选自:碳酸(1s)-1-{(3s)-3-[(2s)-2-({n-[(4-甲氧 基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-(2-氧代-1,3-二氧杂环戊 烯-4-基)乙基]-2-氧代吡咯烷-1-基}乙基甲酯;4-甲氧基-n-[(2s)-4-甲基
ꢀ‑
1-氧代-1-({(1s)-1-(2-氧代-1,3-二氧杂环戊烯-4-基)-2-[(3s)-2-氧代吡咯 烷-3-基]乙基}氨基)戊烷-2-基]-1h-吲哚-2-甲酰胺; n-[(2s)-1-{[(1s)-2-[(3s)-1-乙酰基-2-氧代吡咯烷-3-基]-1-(2-氧代-1,3
‑ꢀ
二氧杂环戊烯-4-基)乙基]氨基}-4-甲基-1-氧代戊烷-2-基]-4-甲氧基
ꢀ‑
1h-吲哚-2-甲酰胺;和(3s)-3-[(2s)-2-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-(2-氧代-1,3-二氧杂环戊烯-4-基)乙基]-2-氧代 吡咯烷-1-甲酸甲酯;或其药学上可接受的盐。
[0040]
本发明的第一方面的第二十三个实施方案是式ig的化合物
[0041][0042]
其中r2选自-c(o)r7、-co2r7和-c
1-c6烷基-oc(o)or7;或其药 学上可接受的盐。
[0043]
本发明的第一方面的第二十四个实施方案是第一方面的第二十三 个实施方案的化合物,其选自:碳酸(1s)-1-{(3s)-3-[(2s)-4-羟基
ꢀ‑
2-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-3-氧代丁 基]-2-氧代吡咯烷-1-基}乙基甲酯;n-[(2s)-1-({(2s)-1-[(3s)-1-乙酰基-2
‑ꢀ
氧代吡咯烷-3-基]-4-羟基-3-氧代丁-2-基}氨基)-4-甲基-1-氧代戊烷-2
‑ꢀ
基]-4-甲氧基-1h-吲哚-2-甲酰胺;(3s)-3-[(2s)-4-羟基-2-({n-[(4-甲氧基
ꢀ‑
1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-3-氧代丁基]-2-氧代吡咯烷-1
‑ꢀ
甲酸甲酯;和碳酸{(3s)-3-[(2s)-4-羟基-2-({n-[(4-甲氧基-1h-吲哚-2-基) 羰基]-l-亮氨酰基}氨基)-3-氧代丁基]-2-氧代吡咯烷-1-基}甲基甲基 酯;或其药学上可接受的盐。
[0044]
本发明的第二方面的第一个实施方案是药物组合物,其包含治疗 有效量的第一方面的第一至第二十四个实施方案中的任一个的化合物 或其药学上可接受的盐以及药学上可接受的载体。本发明的第二方面 的第二个实施方案是第二方面的第一个实施方案的药物组合物,其中 所述组合物呈口服剂型的形式。本发明的第二方面的第三个实施方案 是第二方面的第一个实施方案的药物组合物,其中所述组合物是在鼻 内剂型或吸入剂型中。本发明的第二方面的第四个实施方案是第二方 面的第一个实施方案的药物组合物,进一步包含另外的治疗剂。本发 明的第二方面的第五个实施方案是第二方面的第四个实施方案的药物 组合物,其中所述药物组合物进一步包含氯喹、羟氯喹、阿奇霉素和 瑞德西韦中的一种或多种。
[0045]
本发明的另一个实施方案是治疗患者中的covid-19的方法,所 述方法包括给有此需要的患者施用治疗有效量的第一方面的第一至第 二十四个实施方案中的任一个的化合物或其药学上可接受的盐。
[0046]
本发明的另一个实施方案是治疗患者中的covid-19的方法,所 述方法包括给有此需要的患者施用本发明的第二方面的第一至第五个 实施方案中的任一个的药物组合物。
[0047]
本发明的另一个实施方案是抑制或预防sars-cov-2病毒复制的 方法,包括使sars-cov-2冠状病毒3cl蛋白酶与治疗有效量的第一 方面的第一至第二十四个实施方案中的任一个的化合物或其药学上可 接受的盐或其药学上可接受的盐或所述化合物或药学
上可接受的盐的 代谢物接触。
[0048]
本发明的另一个实施方案是抑制或预防患者中的sars-cov-2病 毒复制的方法,所述方法包括给需要抑制或预防sars-cov-2病毒复 制的患者施用治疗有效量的第一方面的第一至第二十四个实施方案中 的任一个的化合物或其药学上可接受的盐。
[0049]
本发明的另一个实施方案是第一方面的第一至第二十四个实施方 案中的任一个的化合物或其药学上可接受的盐用于治疗covid-19的 用途。本发明的另一个实施方案是第一方面的第一至第二十四个实施 方案中的任一个的化合物或其药学上可接受的盐用于制备药物的用 途,所述药物可用于治疗covid-19。
[0050]
本发明的另一个实施方案是治疗患者中的covid-19的方法,所 述方法包括给有此需要的患者施用治疗有效量的磷酸二氢 (3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁酯或其药学上可接受的盐。
[0051]
本发明的另一个实施方案是前面紧挨着的实施方案,进一步包括 给有此需要的患者施用一种或多种另外的治疗剂。
[0052]
本发明的另一个实施方案是前面紧挨着的实施方案,其中所述另 外的治疗剂选自瑞德西韦和阿奇霉素。
[0053]
本发明的下述实施方案e1-e49是本发明的特别优选的实施方案。
[0054]
e1是式i的化合物
[0055][0056]
其中
[0057]
‑‑‑‑‑‑
不存在或是键;
[0058]
r1选自-ch(r
4a
)-oc(o)r4、-c(o)or4、-ch(r
4a
)-oc(o)or4、
ꢀ‑
p(o)(or5)2、-p(o)(c
1-c6烷基)(or5)和-c(o)n(r6)2;
[0059]
r2选自氢、-c(o)r7、-co2r7和-c
1-c6烷基-oc(o)or7;
[0060]
且当r2是-c(o)r7、-co2r7或-c
1-c6烷基-oc(o)or7时;则r1选自氢、-ch(r
4a
)-oc(o)r4、-c(o)or4、-ch(r
4a
)-oc(o)or4、
ꢀ‑
p(o)(or5)2、-p(o)(c
1-c6烷基)(or5)和-c(o)n(r6)2;
[0061]
当
‑‑‑‑‑
不存在时r3是氧代,或当
‑‑‑‑‑
是键时,r3与r1以及r1所连接的氧一起是-oc(o)o-;
[(3s)-2-氧代吡咯烷-3-基]丁基二丙烷-2-基酯;磷酸 (3s)-4-[(3s)-1-乙酰基-2-氧代吡咯烷-3-基]-3-({n-[(4-甲氧基-1h-吲哚
ꢀ‑
2-基)羰基]-l-亮氨酰基}氨基)-2-氧代丁基二甲酯;4-甲氧基
ꢀ‑
n-[(2s)-4-甲基-1-({(2s)-4-[(2-氧-4-苯基-1,3,2-二氧杂磷杂环己烷-2-基) 氧基]-3-氧代-1-[(3s)-2-氧代吡咯烷-3-基]丁-2-基}氨基)-1-氧代戊烷-2
‑ꢀ
基]-1h-吲哚-2-甲酰胺;磷酸二乙基(3s)-3-({n-[(4-甲氧基-1h-吲哚-2
‑ꢀ
基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯; 和(3s)-3-[(2s)-4-[(二甲氧基磷酰基)氧基]-2-({n-[(4-甲氧基-1h-吲哚
ꢀ‑
2-基)羰基]-l-亮氨酰基}氨基)-3-氧代丁基]-2-氧代吡咯烷-1-甲酸甲 酯;或其药学上可接受的盐、溶剂化物或水合物。
[0074]
e5是e4的化合物,其为磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚
ꢀ‑
2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁 酯;或其药学上可接受的盐、溶剂化物或水合物。
[0075]
e6是e5的化合物,其为水合物的形式。
[0076]
e7是e6的化合物,其为结晶磷酸二氢(3s)-3-({n-[(4-甲氧基-1h
‑ꢀ
吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基] 丁酯水合物。
[0077]
e8是根据权利要求e7所述的化合物,其为结晶磷酸二氢 (3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁酯形式1水合物,其具有一个或多个选 自粉末x-射线衍射图谱、
13
c固态nmr波谱和拉曼光谱的特征;其 中所述粉末x-射线衍射图谱特征选自
[0078]
a)包含在4.1
±
0.2和7.2
±
0.2度2-θ处的峰的粉末x-射线衍射图 谱;
[0079]
b)包含在4.1
±
0.2、7.2
±
0.2和10.4
±
0.2度2-θ处的峰的粉末x
‑ꢀ
射线衍射图谱;和
[0080]
c)包含在4.1
±
0.2、7.2
±
0.2、10.4
±
0.2和14.5
±
0.2度2-θ处的峰 的粉末x-射线衍射图谱;
[0081]
其中所述
13
c固态nmr波谱特征选自
[0082]
a)包含在21.7、153.8和172.2ppm处的峰的
13
c固态nmr波 谱;每个峰
±
0.2ppm;
[0083]
b)包含在21.7、153.8、172.2和118.6ppm处的峰的
13
c固态 nmr波谱;每个峰
±
0.2ppm;
[0084]
c)包含在21.7、153.8、172.2、118.6和57.8ppm处的峰的
13
c 固态nmr波谱;每个峰
±
0.2ppm;和
[0085]
其中所述拉曼光谱特征选自
[0086]
a)包含在1271、1421和1217cm-1
处的拉曼峰的拉曼光谱;每 个峰
±
2cm-1
;
[0087]
b)包含在1271、1421、1217和1640cm-1
处的拉曼峰的拉曼光 谱;每个峰
±
2cm-1
;和
[0088]
c)包含在1271、1421、1217、1640和3074cm-1
处的拉曼峰的 拉曼光谱;每个峰
±
2cm-1
。
[0089]
e9是e5的化合物,其为甲基乙基酮溶剂化物的形式。
[0090]
e10是e9的化合物,其为结晶磷酸二氢(3s)-3-({n-[(4-甲氧基-1h
‑ꢀ
吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基] 丁酯甲基乙基酮溶剂化物。
[0091]
e11是e10的化合物,其为结晶磷酸二氢(3s)-3-({n-[(4-甲氧基
ꢀ‑
1h-吲哚-2-基)
羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷
ꢀ‑
3-基]丁酯甲基乙基酮溶剂化物,其具有一个或多个选自粉末x-射线 衍射图谱、
13
c nmr波谱和拉曼光谱的特征;
[0092]
其中所述粉末x-射线衍射图谱特征选自
[0093]
a)包含在7.7
±
0.2、8.1
±
0.2和23.1
±
0.2度2-θ处的峰的粉末x
‑ꢀ
射线衍射图谱;
[0094]
b)包含在7.7
±
0.2、8.1
±
0.2、23.1
±
0.2和17.0
±
0.2度2-θ处的峰 的粉末x-射线衍射图谱;和
[0095]
c)包含在7.7
±
0.2、8.1
±
0.2、23.1
±
0.2、17.0
±
0.2和25.8
±
0.2度 2-θ处的峰的粉末x-射线衍射图谱;
[0096]
其中所述
13
c固态nmr波谱特征选自
[0097]
a)包含在7.2、206.4和215.8ppm处的峰的
13
c固态nmr波 谱;每个
±
0.2ppm;
[0098]
b)包含在7.2、206.4、215.8和42.2ppm处的峰的
13
c固态nmr 波谱;每个
±
0.2ppm;和
[0099]
c)包含在7.2、206.4、215.8、42.2和101.2ppm处的峰的
13
c 固态nmr波谱;每个
±
0.2ppm;和
[0100]
其中所述拉曼光谱特征选自
[0101]
a)包含在1511、1644和3081cm-1
处的峰的拉曼光谱;每个
±
2 cm-1
;
[0102]
b)包含在1511、1644、3081和1265cm-1
处的峰的拉曼光谱; 每个
±
2cm-1
;和
[0103]
c)包含在1511、1644、3081、1265和446cm-1
处的峰的拉曼 光谱;每个
±
2cm-1
。
[0104]
e12是e5的化合物,其为二甲基亚砜溶剂化物的形式。
[0105]
e13是e12的化合物,其为结晶磷酸二氢(3s)-3-({n-[(4-甲氧基
ꢀ‑
1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷
ꢀ‑
3-基]丁酯二甲基亚砜溶剂化物。
[0106]
e14是e13的化合物,其为结晶磷酸二氢(3s)-3-({n-[(4-甲氧基
ꢀ‑
1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷
ꢀ‑
3-基]丁酯二甲基亚砜溶剂化物,其具有一个或多个选自粉末x-射线 衍射图谱、
13
c固态nmr波谱和拉曼光谱的特征;
[0107]
其中所述粉末x-射线衍射图谱特征选自
[0108]
a)包含在7.4
±
0.2、14.8
±
0.2和26.2
±
0.2度2-θ处的峰的粉末x
‑ꢀ
射线衍射图谱;
[0109]
b)包含在7.4
±
0.2、14.8
±
0.2、26.2
±
0.2和10.8
±
0.2度2-θ处的 峰的粉末x-射线衍射图谱;和
[0110]
c)包含在7.4
±
0.2、14.8
±
0.2、26.2
±
0.2、10.8
±
0.2和22.3
±
0.2度 2-θ处的峰的粉末x-射线衍射图谱;
[0111]
其中所述
13
c固态nmr波谱特征选自
[0112]
a)包含在173.4
±
0.2、210.7
±
0.2和26.2
±
0.2ppm处的峰的
13
c 固态nmr波谱;
[0113]
b)包含在173.4
±
0.2、210.7
±
0.2、26.2
±
0.2和22.8
±
0.2ppm处 的峰的
13
c固态nmr波谱;和
[0114]
c)包含在173.4
±
0.2、210.7
±
0.2、26.2
±
0.2、22.8
±
0.2和25.5
±
0.2 ppm处的峰的
13
c固态nmr波谱;和
[0115]
其中所述拉曼光谱特征是
[0116]
a)包含在1717
±
2和675
±
2cm-1
处的峰的拉曼光谱。
[0117]
e15是根据权利要求e5所述的化合物,其为二甲基亚砜溶剂化 物水合物的形式。
[0118]
e16是e15的化合物,其为结晶磷酸二氢(3s)-3-({n-[(4-甲氧基
ꢀ‑
1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷
ꢀ‑
3-基]丁酯二甲基亚砜溶剂化物水合物。
[0119]
e17是e13的化合物,其为结晶磷酸二氢(3s)-3-({n-[(4-甲氧基
ꢀ‑
1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷
ꢀ‑
3-基]丁酯二甲基亚砜溶剂化物水合物,其具有粉末x-射线衍射图谱 特征;
[0120]
其中所述x-射线粉末衍射图谱特征选自
[0121]
a)包含在14.5
±
0.2、25.6
±
0.2和26.6
±
0.2度2-θ处的峰的粉末 x-射线衍射图谱;
[0122]
b)包含在14.5
±
0.2、25.6
±
0.2、26.6
±
0.2和21.9
±
0.2度2-θ处的 峰的粉末x-射线衍射图谱;和
[0123]
c)包含在14.5
±
0.2、25.6
±
0.2、26.6
±
0.2、21.9
±
0.2、17.8
±
0.2度2-θ处的峰的粉末x-射线衍射图谱。
[0124]
e18是e5的化合物,其为磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲 哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁 酯。
[0125]
e19是e18的化合物,其为无定形磷酸二氢(3s)-3-({n-[(4-甲氧基
ꢀ‑
1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷
ꢀ‑
3-基]丁酯。
[0126]
e20是e19的化合物,其为无定形磷酸二氢(3s)-3-({n-[(4-甲氧基
ꢀ‑
1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷
ꢀ‑
3-基]丁酯,其具有一个或多个选自
13
c固态nmr波谱以及
13
c固态 nmr波谱和
31
p固态nmr波谱的组合的特征;
[0127]
其中所述
13
c固态nmr波谱特征选自
[0128]
a)包含在175.0
±
0.4、204
±
1.5和181.8
±
0.4ppm处的峰的
13
c固 态nmr波谱;
[0129]
b)包含在175.0
±
0.4、204
±
1.5、181.8
±
0.4和54.8
±
0.2ppm处的 峰的
13
c固态nmr波谱;和
[0130]
c)包含在175.0
±
0.4、204
±
1.5、181.8
±
0.4、54.8
±
0.2和162.9
±
0.2 ppm处的峰的
13
c固态nmr波谱;和
[0131]
13
c固态nmr波谱和
31
p固态nmr波谱的组合是包含在 175.0
±
0.4和204
±
1.5处的峰的
13
c固态nmr波谱和具有在-0.8
±
0.2 ppm处的峰的
31
p固态nmr波谱。
[0132]
e21是e5的化合物,其为磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲 哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁 酯钠盐。
[0133]
e22是e21的化合物,其为无定形磷酸二氢(3s)-3-({n-[(4-甲氧基
ꢀ‑
1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷
ꢀ‑
3-基]丁酯钠盐。
[0134]
e23是根据权利要求e22所述的化合物,其为无定形磷酸二氢 (3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁酯钠盐,其具有一个或多个选自
13
c固 态nmr波谱以及
13
c固态nmr波谱和
31
p固态nmr波谱的组合的 特征;
[0135]
其中所述
13
c固态nmr波谱特征选自
[0136]
a)包含在126.0
±
0.4ppm、181.0
±
0.4ppm和208.0
±
1.5ppm处 的峰的
13
c固态nmr波谱;
[0137]
b)包含在126.0
±
0.4ppm、181.0
±
0.4ppm、208.0
±
1.5ppm和 174.1
±
0.4ppm 175.0
±
0.4ppm处的峰的
13
c固态nmr波谱;和
[0138]
c)包含在126.0
±
0.4ppm、181.0
±
0.4ppm、208.0
±
1.5ppm、 174.1
±
0.4ppm和163.1
±
0.2ppm处的峰的
13
c固态nmr波谱;和
[0139]
13
c固态nmr波谱和
31
p固态nmr波谱的组合是包含在 126.0
±
0.4ppm、181.0
±
0.4ppm处的峰的
13
c固态nmr波谱和具有在 1.9
±
0.2ppm处的峰的
31
p固态nmr波谱。
[0140]
e24是药物组合物,其包含治疗有效量的根据权利要求e5至e23 中的任一项的磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l
‑ꢀ
亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯、或其药学上 可接受的盐、溶剂化物或水合物以及药学上可接受的载体。
[0141]
e25是e24的药物组合物,其中所述药物组合物进一步包含缓冲 剂。
[0142]
e26是e25的药物组合物,其中:
[0143]
a)所述药学上可接受的盐选自苄星(benzathine)、钙、胆碱、 二乙胺、二乙醇胺、镁、葡甲胺、赖氨酸、哌嗪、钾、三(羟基甲基) 氨基甲烷和钠;
[0144]
b)药学上可接受的盐中盐抗衡离子与磷酸二氢(3s)-3-({n-[(4
‑ꢀ
甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代 吡咯烷-3-基]丁酯的摩尔比是大约0.5:1至大约3:1;和
[0145]
c)所述缓冲剂选自磷酸、柠檬酸、马来酸、酒石酸、乳酸和乙 酸。
[0146]
e27是e26的药物组合物,其中:
[0147]
a)所述药学上可接受的盐是钠;
[0148]
b)所述药学上可接受的盐中钠抗衡离子与磷酸二氢 (3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁酯的摩尔比是大约0.5:1至大约2:1;
[0149]
c)所述缓冲剂是柠檬酸;和
[0150]
d)磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨 酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯与柠檬酸的摩尔比 是大约2:1至大约10:1。
[0151]
e28是e25至e27中的任一项的药物组合物,其中所述组合物呈 粉末或亲液物(lyophile)的形式,其中重构制剂的溶液ph是在2-6 的范围内。
[0152]
e29是e28的药物组合物,其中重构制剂的溶液ph是在3-5的 范围内。
[0153]
e30是e24至e29中的任一项的药物组合物,其中所述药物组合 物进一步包含一种或多种稳定剂。
[0154]
e31是e30的药物组合物,其中所述一种或多种稳定剂选自葡聚 糖、蔗糖、乳糖、海藻糖、甘露醇、山梨醇、葡萄糖、棉子糖、甘氨 酸、组氨酸、聚乙烯吡咯烷酮和聚乙二醇。
[0155]
e32是e30的药物组合物,其中所述一种或多种稳定剂选自聚乙 二醇300、聚乙二醇400和聚乙二醇3350。
[0156]
e33是e32的药物组合物,其中一种或多种稳定剂的总量至多为 制剂的大约15%w/w。
[0157]
e34是权利要求e24至e33中的任一项的药物组合物,其中所述 药物组合物进一步包含一种或多种增溶剂。
[0158]
e35是e34的药物组合物,其中所述增溶剂选自聚山梨酯20、聚 乙氧基化的蓖麻油、聚乙二醇(15)-羟基硬脂酸酯、羟丙基-β-环糊精、 磺丁基醚-β环糊精、γ环糊精和聚山梨酯80。
[0159]
e36是e35的药物组合物,其中所述增溶剂是聚山梨酯80和所 述缓冲剂是柠檬酸。
[0160]
e37是e32的药物组合物,其中所述组合物是粉末或亲液物,其 当用注射用水、0.9%盐水或5%w/v重构时提供一种水溶液,其中磷 酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨 基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯或其药学上可接受的盐的 浓度是约1mg/ml至约200mg/ml。
[0161]
e38是e37的药物组合物,其中在重构后制剂的溶液ph是在约 3至约5的范围内。
[0162]
e39是e38的药物组合物,其在重构后具有至多为大约5%w/w 的聚山梨酯80浓度。
[0163]
e40是e37的药物组合物,其中所述药物粉末或亲液物具有小于 约1%的含水量。
[0164]
e41是e24至e40中的任一项的药物组合物,其为适合用于胃肠 外施用的水溶液,或用注射用水、0.9%盐水或5%w/v右旋糖重构以 形成适合用于胃肠外施用的水溶液。
[0165]
e42是治疗患者中的冠状病毒感染的方法,所述方法包括给有此 需要的患者施用治疗有效量的根据e5至e23中的任一项的磷酸二氢 (3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁酯;或其药学上可接受的盐、溶剂化物 或水合物。
[0166]
e43是e42的方法,其中所述冠状病毒感染是covid-19。
[0167]
e44是e43的方法,其中每天施用约0.1g至约5g磷酸二氢 (3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁酯;或其药学上可接受的盐、溶剂化物 或水合物。
[0168]
e45是e44的方法,其中每天静脉内施用约0.1至约1g磷酸二 氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧 代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯;或其药学上可接受的盐、溶剂化 物或水合物。
[0169]
e46是治疗患者中的covid-19的方法,所述方法包括给需要此 治疗的患者施用根据e41的药物组合物。
[0170]
e47是e42至e46中的任一项的方法,其中将一种或多种另外的 治疗剂施用给患者。
[0171]
e48是e47的方法,其中所述一种或多种另外的治疗剂选自瑞德 西韦、加利西韦(galidesivir)、法维拉韦(favilavir)/阿韦法韦、 mulnupiravir(mk-4482/eidd 2801)、at-527、at-301、 bld-2660、法匹拉韦、卡莫司他、slv213恩曲他滨/替诺福韦、克 拉夫定(clevudine)、达塞曲匹、波普瑞韦、abx464、地塞米松、 氢化可的松、恢复期血浆、凝溶胶蛋白(rhu-p65n)、单克隆抗体、 瑞丹维单抗(regdanvimab)(regkirova)、雷夫利珠单抗(ultomiris)、 vir-7831/vir-7832、brii-196/brii-198、covi-amg/covidrops(sti-2020)、班兰尼维单抗(bamlanivimab)(ly-cov555)、 mavrilimab、乐昂立单抗(leronlimab)(pro140)、azd7442、仑兹 鲁单抗(lenzilumab)、英夫利昔单抗、阿达木单抗、js 016、 sti-1499(coviguard)、拉那利尤单抗(lanadelumab) (takhzyro)、卡那奴单抗
(ilaris)、瑾司鲁单抗(gimsilumab)、 otilimab、卡西瑞单抗(casirivimab)/伊德单抗(imdevimab) (regn-cov2)、mk-7110(cd24fc/saccovid)、肝素、阿派沙班、 托珠单抗(actemra)、sarilumab(kevzara)、阿匹莫德二甲磺酸盐 (apilimod dimesylate)、dnl758、pb1046、达格列净 (dapaglifozin)、abivertinib、atr-002、bemcentinib、阿卡替尼 (acalabrutinib)、巴瑞替尼、托法替尼、洛吡莫德、法莫替丁、氯 硝柳胺和二脒那秦。
[0172]
e49是e48的方法,其中所述一种或多种另外的药剂选自瑞德西 韦、地塞米松、malnupiravir、班兰尼维单抗、托法替尼和巴瑞替尼。
[0173]
应当理解,治疗方法权利要求还可以解释为适当的用途型权利要 求。
[0174]
本发明也提供了一种通过给患者施用药学有效量的如本文中所述 的sars-cov-2蛋白酶抑制剂治疗所述患者中由sars-cov-2冠状病 毒3c样蛋白酶活性介导的病症的方法。
[0175]
本发明也提供了一种靶向sars-cov-2抑制的方法,作为治疗由 sars-cov-2相关的病毒感染造成的适应症的方式。
[0176]
本发明也提供了一种识别细胞或病毒途径的方法,干扰所述途径 的功能可用于通过施用本文所述的sars-cov-2蛋白酶抑制剂来治疗 由sars-cov-2感染引起的适应症。
[0177]
本发明也提供了一种使用本文所述的sars-cov-2蛋白酶抑制剂 作为了解其它sars-cov-2抑制剂作用机制的工具的方法。
[0178]
本发明也提供了一种使用sars-cov-2 3c样蛋白酶抑制剂进行 基因谱分析实验的方法,用于监测基因的上调或下调,目的是确定用 于治疗由sars-cov-2感染引起的适应症(诸如covid-19)的抑制 剂。
[0179]
本发明进一步提供了一种用于治疗哺乳动物中covid-19的药物 组合物,其含有有效治疗covid-19的量的sars-cov-2 3c样蛋白 酶抑制剂和药学上可接受的载体。
[0180]
本发明的另一个实施方案是一种治疗患者中的covid-19的方 法,其中给患者施用大约500mg/天的磷酸二氢(3s)-3-({n-[(4-甲氧基
ꢀ‑
1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷
ꢀ‑
3-基]丁酯。施用可以是静脉内的例如通过连续静脉内输注。施用可以 是在每天250ml或更少的溶液体积中。
[0181]
本发明的另一个实施方案是治疗患者中的covid-19的方法,其 中通过连续静脉内输注将0.25g至5g磷酸二氢(3s)-3-({n-[(4-甲氧基
ꢀ‑
1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷
ꢀ‑
3-基]丁酯施用给患者。施用可以是以每天250ml或更少的静脉内的 溶液体积。所述方法可以包括给患者共同施用一种或多种另外的治疗 剂。所述方法可以包括共同施用一种或多种选自以下的另外的治疗剂 给患者:瑞德西韦、(2r,3r,4s,5r)-2-(4-氨基吡咯并[2,1-f][1,2,4]三嗪
ꢀ‑
7-基)-3,4-二羟基-5-(羟基甲基)氧杂戊环(oxolane)-2-甲腈 (gs-441524)、(2s)-2-((s)-2-(((苄氧基)羰基)氨基)-4-甲基戊酰氨基)-1
‑ꢀ
羟基-3-(2-氧代吡咯烷-3-基)丙烷-1-磺酸钠(gc376)、地塞米松、阿奇 霉素、umifenovir和法匹拉韦。
[0182]
本发明的另一个实施方案f1是药物组合物,其包含磷酸二氢 (3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁酯或其药学上可接受的盐以及药学上可 接受的载体。
[0183]
本发明的另一个实施方案f2是前面紧挨着的实施方案f1的药物 组合物,其中所
述药物组合物进一步包含缓冲剂。
[0184]
本发明的另一个实施方案f3是前面紧挨着的实施方案的根据权 利要求所述的药物组合物,其中所述缓冲剂选自磷酸、柠檬酸、马来 酸、酒石酸、乳酸和乙酸。
[0185]
本发明的另一个实施方案f4是前面紧挨着的实施方案f3的药物 组合物,其中所述缓冲剂是柠檬酸。
[0186]
本发明的另一个实施方案f5是三个前面紧挨着的实施方案f2-f4 中的任一个的药物组合物,其中所述组合物呈水溶液的形式且制剂的 溶液ph是在2-6的范围内。
[0187]
本发明的另一个实施方案f6是前面紧挨着的实施方案f5的药物 组合物,其中制剂的溶液ph是在3-5的范围内。
[0188]
本发明的另一个实施方案f7是前面紧挨着的实施方案f6的药物 组合物,其中制剂的溶液ph是在3.5-4.5的范围内。
[0189]
本发明的另一个实施方案f8是f1-f4的药物组合物,其中所述 药物组合物进一步包含填充剂。
[0190]
本发明的另一个实施方案f9是f8的药物组合物,其中所述填充 剂选自:蔗糖、乳糖、海藻糖、甘露醇、山梨醇、葡萄糖、棉子糖、 甘氨酸、组氨酸、聚乙烯吡咯烷酮。
[0191]
本发明的另一个实施方案f10是f9的药物组合物,其中所述填 充剂选自:海藻糖、蔗糖、乳糖、甘露醇和聚乙二醇400。
[0192]
本发明的另一个实施方案是f1至f10中的任一个的药物组合物, 其呈亲液物或粉末的形式。
[0193]
本发明的另一个实施方案是f1至f10中的任一个的药物组合物, 其呈水溶液的形式。
[0194]
本发明的另一个实施方案是f1至f10中的任一个的药物组合物, 其中所述药物组合物进一步包含增溶剂。
[0195]
本发明的另一个实施方案是前面紧挨着的实施方案的药物组合 物,其中所述增溶剂选自聚山梨酯20、克米莫佛el、kolliphor hs-15、 羟丙基-β-环糊精、磺丁基醚-β环糊精、γ环糊精和聚山梨酯80。
[0196]
本发明的另一个实施方案是前面紧挨着的实施方案的药物组合 物,其中所述增溶剂是聚山梨酯80和所述缓冲剂是柠檬酸盐。
[0197]
本发明的另一个实施方案是前面紧挨着的实施方案的药物组合 物,其中所述组合物是水溶液。
[0198]
本发明的另一个实施方案是前面紧挨着的实施方案的药物组合 物,其中制剂的溶液ph是在约3.5至约4.5的范围内。
[0199]
本发明的另一个实施方案是前面紧挨着的实施方案的药物组合 物,其中所述聚山梨酯80浓度是约5mg/ml和柠檬酸盐缓冲液浓度 是约40mm。
[0200]
本发明的另一个实施方案是包含增溶剂聚山梨酯80和缓冲剂柠 檬酸盐的药物组合物,且其中所述药物组合物是粉末或亲液物。
[0201]
本发明的另一个实施方案m1是治疗患者中的冠状病毒感染的方 法,所述方法包括给有此需要的患者施用治疗有效量的磷酸二氢 (3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁酯或其药学上可
接受的盐。
[0202]
本发明的另一个实施方案m2是m1的方法,其中所述冠状病毒 感染是sars-cov-2、mers-cov、229e-cov-2、nl63-cov、 oc43-cov或hku1-cov感染。
[0203]
本发明的另一个实施方案m3是m2的方法,是sars-cov-2感 染。
[0204]
本发明的另一种方法m4是根据权利要求m1所述的方法,其中 所述磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯或其药学上可接受的盐 作为药物组合物施用,所述药物组合物包含磷酸二氢(3s)-3-({n-[(4-甲 氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡 咯烷-3-基]丁酯或其药学上可接受的盐以及药学上可接受的载体。
[0205]
本发明的另一种方法m5是治疗患者中的冠状病毒感染的方法, 所述方法包括施用根据权利要求f1至f10中的任一项的药物组合物。
[0206]
本发明的另一种方法m6是根据权利要求m5所述的方法,其中 所述药物组合物是胃肠外的溶液,其静脉内施用给患者。
附图说明
[0207]
图1:sars-cov和sars-cov-2之间的残基差异,在活性位点 显示了抑制剂化合物。
[0208]
图2:sars-cov-2 3cl与核心对接配体(化合物b, n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙 基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺)的同源性模 型的结合位点。
[0209]
图3:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨 酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯水合物的形式1的 pxrd图谱。
[0210]
图4:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨 酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯水合物的形式1的 13
c固态nmr波谱。
[0211]
图5:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨 酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯水合物的形式1的 15
n固态nmr波谱。
[0212]
图6:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨 酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯水合物的形式1的 拉曼光谱。
[0213]
图7:母体化合物n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代 吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2
‑ꢀ
甲酰胺与sars-cov-2 3clpro的代表性热迁移结合数据。
[0214]
图8:与瑞德西韦组合的母体化合物n-((1s)-1-{[((1s)-3-羟基-2
‑ꢀ
氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁 基)-4-甲氧基-1h-吲哚-2-甲酰胺对sars-cov-2的抗病毒活性的等效 线图解法。
[0215]
图9:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨 酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯甲基乙基酮溶剂化 物的pxrd。
[0216]
图10:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯甲基乙基酮溶剂 化物在重新处理以后的pxrd。
[0217]
图11:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨
基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯无定形钠盐的 pxrd图谱。
[0218]
图12:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯无定形钠盐的 13
c固态nmr波谱。
[0219]
图13:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯无定形钠盐的 15
n固态nmr波谱。
[0220]
图14:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯无定形钠盐的 31
p固态nmr波谱。
[0221]
图15:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯无定形钠盐的拉 曼光谱。
[0222]
图16:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯无定形钠盐的用 于玻璃化转变测定的调制dsc。
[0223]
图17:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯dmso溶剂化物 的pxrd图谱。
[0224]
图18:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯dmso溶剂化物 的
13
c ssnmr波谱。
[0225]
图19:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯dmso溶剂化物 的拉曼光谱。
[0226]
图20:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯dmso溶剂化物 水合物的pxrd图谱。
[0227]
图21:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯dmso溶剂化物 水合物和磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨 酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯dmso溶剂化物的 混合物的pxrd图谱。
[0228]
图22:pf-07304814的冷冻干燥的药物产品的代表性pxrd衍射 图谱。
[0229]
图23:用5mg/ml聚山梨酯80制备的pf-07304814亲液物的 pxrd表征。
[0230]
图24:具有钾抗衡离子的pf-07304814的冷冻干燥的药物产品的 pxrd衍射图谱。
[0231]
图25:具有哌嗪抗衡离子的pf-07304814的冷冻干燥的药物产品 的pxrd衍射图谱。
[0232]
图26:具有a)10mg/ml peg400,b)10mg/ml peg3350,c)10 mg/ml peg 400/10mg/ml peg3350的pf-07304814的冷冻干燥的药 物产品的pxrd衍射图谱。
[0233]
图27:显示了从来自dmso纯化过程的pf-07304814的3个不 同批制备的冷冻干燥的药物产品的pxrd衍射图谱。a)pf-07304814 批1(0.12%dmso),b)pf-07304814批2(6%dmso),c)pf-07304814 批3(12%dmso)。
[0234]
图28:pf-07304814无定形游离酸的
13
c固态nmr波谱。
[0235]
发明详述
[0236]
出于本文所述和要求保护的本发明的目的,以下术语定义如下:
[0237]
本文中使用的术语“包含”和“包括”以其开放的、非限制性的含义 使用。除非另有说明,否则本文所用的术语“治疗”是指逆转、减轻、 抑制该术语适用的病症的进展或预防
该病症的障碍或该障碍或病症的 一种或多种症状。除非另有说明,否则本文使用的术语“治疗”表示治 疗的行为,如在上面刚刚定义的“治疗”。
[0238]
本文中使用的术语“烷基”表示直链或支链饱和烃基取代基(即,通 过除去氢从烃得到的取代基);在一个实施方案中,含有1-6个碳原子。 这样的取代基的非限制性例子包括甲基、乙基、丙基(包括正丙基和异 丙基)、丁基(包括正丁基、异丁基、仲丁基和叔丁基)、戊基、异戊基、 己基等。
[0239]
术语“烷氧基”表示连接至氧残基的直链或支链饱和烃基取代基 (即,通过从oh除去氢从烃醇得到的取代基);在一个实施方案中, 含有1-6个碳原子。这样的取代基的非限制性例子包括甲氧基、乙氧 基、丙氧基(包括正丙氧基和异丙氧基)、丁氧基(包括正丁氧基、异丁 氧基、仲丁氧基和叔-丁氧基)、戊氧基、己氧基等。连接至烷基的烷 氧基被称作烷氧基烷基。烷氧基烷基的一个例子是甲氧基甲基。
[0240]
术语“亚烷基”表示烷二基(即通过除去2个氢从烃得到的取代基); 在一个实施方案中,含有3-5个碳。这样的基团的非限制性例子包括 亚丙基、亚丁基和亚戊基。
[0241]
在某些情况下,烃基取代基(即,烷基、环烷基等)中的碳原子的 数目通过前缀“c
x-cy‑”
或“c
x-y”指示,其中x为该取代基中的碳原子的 最小数量,且y为最大数量。因而,例如,“c
1-c
6-烷基”或“c
1-6
烷基
”ꢀ
表示含有1-6个碳原子的烷基取代基。进一步举例说明,c
3-c6环烷基 或c
3-6-环烷基表示含有3-6个碳环原子的饱和环烷基。
[0242]
术语“环烷基”表示通过从饱和碳环分子除去氢而得到的碳环取代 基,例如具有3-7个碳原子的碳环取代基。术语“环烷基”包括单环饱 和碳环。术语“c
3-c7环烷基”是指3-7元环系的残基,其包括基团环丙 基、环丁基、环戊基、环己基和环庚基。术语“c
3-6
环烷基”是指3-6 元环系的残基,其包括基团环丙基、环丁基、环戊基和环己基。术语
ꢀ“c3-6
环烷氧基”表示连接至氧自由基的3-6元环烷基。例子包括环丙 氧基、环丁氧基、环戊氧基和环己氧基。术语“c
5-c
12
二环烷基”是指 二环环烷基部分,诸如二环戊基、二环己基、二环庚基、二环辛基、 二环壬基、螺戊基、螺己基、螺庚基、螺辛基和螺壬基。
[0243]
在某些情况下,含有一个或多个杂原子的环状取代基(即,杂芳基 或杂环烷基)中的原子的数目通过前缀“x-y元”指示,其中x是形成取 代基的环状部分的原子的最小数目且y是最大数目。因而,例如,“4-6 元杂环烷基”表示在杂环烷基的环状部分中含有4-6个原子(包括1-3 个杂原子)的杂环烷基。类似地,短语“5-6元杂芳基”表示含有5-6个 原子的杂芳基,且“5-10元杂芳基”表示在杂芳基的环状部分中含有 5-10个原子的杂芳基,每个包括一个或多个杂原子。此外,短语“5元 杂芳基”和“6元杂芳基”分别表示5元杂芳族环系和6元杂芳族环系。 存在于这些环系中的杂原子选自n、o和s。
[0244]
术语“羟基(hydroxy)”或“羟基(hydroxyl)”表示-oh。当与另一个 术语组合使用时,前缀“羟基”指示,前缀所连接的取代基被一个或多 个羟基取代基取代。携带一个或多个羟基取代基所连接的碳的化合物 包括例如醇、烯醇和苯酚。术语氰基表示-cn基团。术语“氧代”是指 通过双键连接至碳的氧(即当r3是氧代时,那么r3与它所连接的碳一 起为c=o部分)。
[0245]
术语“卤代(halo)”或“卤素(halogen)”表示氟(其可以描绘为-f)、 氯(其可以描绘为-cl)、溴(其可以描绘为-br)或碘(其可以描绘为-i)。
[0246]
术语“杂环烷基”表示通过从饱和的或部分饱和的环结构除去氢而 得到的取代
基,所述环结构共含有指定数目的原子,诸如4-6个环原 子,其中环原子中的至少一个是杂原子(即,氧、氮或硫),剩余的环 原子独立地选自碳、氧、氮和硫。在具有杂环烷基取代基的基团中, 结合至基团的杂环烷基取代基的环原子可以是氮杂原子,或它可以是 环碳原子。类似地,如果杂环烷基取代基又被基团或取代基取代,所 述基团或取代基可以结合至氮杂原子,或它可以结合至环碳原子。
[0247]
术语“杂芳基”表示含有指定数目的环原子的芳族环结构,其中环 原子中的至少一个是杂原子(即,氧、氮或硫),剩余的环原子独立地 选自碳、氧、氮和硫。杂芳基取代基的例子包括6元杂芳基取代基诸 如吡啶基、吡嗪基、嘧啶基和哒嗪基;和5元杂芳基取代基诸如三唑 基、咪唑基、呋喃基、噻吩基、吡唑基、吡咯基、唑基、异唑基、 噻唑基、1,2,3-、1,2,4-、1,2,5-、或1,3,4-二唑基和异噻唑基。杂芳 基还可以是二环杂芳族基团诸如吲哚基、苯并呋喃基、苯并噻吩基、 苯并咪唑基、苯并噻唑基、苯并唑基、苯并异唑基、唑并吡啶 基、咪唑并吡啶基、咪唑并嘧啶基等。在具有杂芳基取代基的基团中, 结合至基团的杂芳基取代基的环原子可以是杂原子之一,或它可以是 环碳原子。类似地,如果杂芳基取代基又被基团或取代基取代,所述 基团或取代基可以结合至杂原子之一,或它可以结合至环碳原子。术 语“杂芳基”也包括吡啶基n-氧化物和含有吡啶n-氧化物环的基团。 另外,杂芳基可以含有氧代基团诸如存在于吡啶酮基团中的氧代基团。 其它例子包括呋喃基、噻吩基、唑基、噻唑基、咪唑基、吡唑基、 三唑基、四唑基、异唑基、异噻唑基、二唑基、噻二唑基、吡啶 基、哒嗪基、嘧啶基、吡嗪基、吡啶-2(1h)-酮基、哒嗪-2(1h)-酮基、 嘧啶-2(1h)-酮基、吡嗪-2(1h)-酮基、咪唑并[1,2-a]吡啶基和吡唑并 [1,5-a]吡啶基。杂芳基可以如本文中定义进一步被取代。
[0248]
单环杂芳基和杂环烷基的例子包括呋喃基、二氢呋喃基、四氢呋 喃基、噻吩基、二氢噻吩基、四氢噻吩基、吡咯基、异吡咯基、吡咯 啉基、吡咯烷基、咪唑基、异咪唑基、咪唑啉基、咪唑烷基、吡唑基、 吡唑啉基、吡唑烷基、三唑基、四唑基、二硫杂环戊烯基、氧杂硫杂 环戊烯基、唑基、异唑基、噻唑基、异噻唑基、噻唑啉基、异噻 唑啉基、噻唑烷基、异噻唑烷基、硫杂二唑基(thiaoxadiazolyl)、噻唑基、二唑基(包括二唑基、1,2,4-二唑基、1,2,5-二唑基, 或1,3,4-二唑基)、吡喃基(包括1,2-吡喃基或1,4-吡喃基)、二氢吡喃 基、吡啶基、哌啶基、二嗪基(包括哒嗪基、嘧啶基、哌嗪基、三嗪基 (包括s-三嗪基、as-三嗪基和v-三嗪基)、嗪基(包括2h-1,2-嗪基、6h-1,3-嗪基,或2h-1,4-嗪基)、异嗪基(包括邻-异嗪基或对
‑ꢀ
异嗪基)、唑烷基、异唑烷基、噻嗪基(包括1,2,5-噻嗪基 或1,2,6-噻嗪基)、二嗪基(包括2h-1,2,4-二嗪基或2h-1,2,5-二嗪基)、吗啉基。
[0249]
术语“杂芳基”还可以包括,当这样指定时,具有2个环的环系, 其中这样的环可以稠合,且其中一个环是芳族且另一个环不是共轭的 芳族系统的完整部分(即,杂芳族环可以稠合至环烷基或杂环烷基环)。 这样的环系的非限制性例子包括5,6,7,8-四氢异喹啉基、5,6,7,8-四氢
‑ꢀ
喹啉基、6,7-二氢-5h-环戊二烯并[b]吡啶基、6,7-二氢-5h-环戊二烯并 [c]吡啶基、1,4,5,6-四氢环戊二烯并[c]吡唑基、2,4,5,6-四氢环戊二烯并 [c]吡唑
基、5,6-二氢-4h-吡咯并[1,2-b]吡唑基、6,7-二氢-5h-吡咯并 [1,2-b][1,2,4]三唑基,5,6,7,8-四氢-[1,2,4]三唑并[1,5-a]吡啶基、 4,5,6,7-四氢吡唑并[1,5-a]吡啶基、4,5,6,7-四氢-1h-吲唑基和4,5,6,7
‑ꢀ
四氢-2h-吲唑基。应当理解,如果碳环的或杂环的部分可以通过不同 的环原子键合或以其它方式连接至指定的基质(没有指定特定连接 点),那么意指所有可能的点,无论是通过碳原子还是例如三价氮原 子。例如,术语“吡啶基”是指2-、3-或4-吡啶基,术语“噻吩基”是指 2-或3-噻吩基,诸如此类。
[0250]
如果多个取代基被描述为“独立地”具有超过一个变量,那么每个 取代基独立于其它取代基从可用变量列表中进行选择。因此,每个取 代基可以与其它取代基相同或不同。
[0251]
如果多个取代基被描述为“独立地选自”组,那么每个取代基彼此 独立地选择。因此,每个取代基可以与其它取代基相同或不同。
[0252]
本文中使用的术语“式i”在下文中可以被称作“本发明的化合物”、
ꢀ“
本发明”和“式i的化合物”。这样的术语也定义为包括式i的化合物 的所有形式,包括其水合物、溶剂化物、异构体、结晶性和非结晶形 式、同晶型物、多晶型物和代谢物。例如,本发明的化合物或其药学 上可接受的盐可以以未溶剂化和溶剂化形式存在。当溶剂或水紧密结 合时,复合物将具有明确的化学计量学,与湿度无关。但是,当溶剂 或水弱结合时(如在通道溶剂化物和吸湿性化合物中),水/溶剂含量 将取决于湿度和干燥条件。在这样的情况下,非化学计量学将是常态。
[0253]
本发明的化合物可以作为笼形包合物或其它复合物存在。在本发 明范围内包括复合物诸如笼形包合物、药物-主体包合络合物,其中药 物和主体以化学计量的或非化学计量的量存在。还包括本发明化合物 的含有两种或更多种有机和/或无机组分的复合物,其可以呈化学计量 的或非化学计量的量。所得复合物可以是离子化的、部分离子化的或 非离子化的。关于这样的复合物的综述,参见j.pharm.sci.,64(8), 1269-1288by haleblian(august 1975)。
[0254]
本发明的化合物具有不对称的碳原子。在本文中可以使用实线 (——)、实心楔形或点状楔形描绘本发明的化合物的碳
‑ꢀ
碳键。用实线描绘不对称碳原子的键意指包括在该碳原子处的所有可 能的立体异构体(例如,特定的对映异构体、外消旋混合物等)。用实 心或点状楔形描绘不对称碳原子的键意味着表示仅意图包括所示的立 体异构体。式i的化合物可能含有超过一个不对称的碳原子。在那些 化合物中,用实线描绘不对称碳原子的键意味着表示意图包括所有可 能的立体异构体。例如,除非另外说明,式i的化合物意图可以作为 对映异构体和非对映异构体存在,或作为外消旋体及其混合物存在。 用实线描绘式i的化合物中的一个或多个不对称碳原子的键以及用实 心或点状楔形描绘同一化合物中其它不对称碳原子的键意味着表示存 在非对映异构体的混合物。
[0255]
式i的立体异构体包括本发明的化合物的顺式和反式异构体、光 学异构体诸如r和s对映异构体、非对映异构体、几何异构体、旋转 异构体、构象异构体和互变异构体,包括表现出超过一种类型的异构 现象的化合物;及其混合物(诸如外消旋体和非对映体对)。还包括酸 加成或碱加成盐,其中抗衡离子是光学活性的,例如d-乳酸根或l
‑ꢀ
赖氨酸,或外消旋的,例如,dl-酒石酸根或dl-精氨酸。
[0256]
当任何外消旋体结晶时,两种不同类型的晶体是可能的。第一类 是上文所述的外
消旋化合物(真外消旋体),其中产生一种均质形式的 晶体,其含有等摩尔量的两种对映异构体。第二类是外消旋混合物或 聚集物(conglomerate),其中两种形式的晶体以等摩尔量产生,各自包 含单一对映异构体。
[0257]
式i的化合物可以存在互变异构现象;这样的互变异构体也被视 作本发明的化合物。所有这样的互变异构形式及其混合物被包括在式 i的化合物的范围内。互变异构体作为在溶液中的互变异构集合的混 合物存在。在固体形式中,通常一种互变异构体占优势。尽管可能描 述一种互变异构体,本发明包括式i的化合物的所有互变异构体及其 盐。
[0258]
除非另有说明,否则本文所用的短语“药学上可接受的盐”包括可 存在于本文所述化合物中的酸性或碱性基团的盐。本发明方法中使用 的性质上为碱性的化合物能够与各种无机和有机酸形成多种盐。可用 于制备此类碱性化合物的药学上可接受的酸加成盐的酸是那些形成无 毒酸加成盐的酸,所述盐即含有药理学上可接受的阴离子的盐,诸如 乙酸盐、苯磺酸盐、苯甲酸盐、碳酸氢盐、硫酸氢盐、酒石酸氢盐、 硼酸盐、溴化物、依地酸钙、樟脑磺酸盐、碳酸盐、氯化物、克拉维 酸盐、柠檬酸盐、二盐酸盐、依地酸盐、edislyate、依托酸盐(estolate)、 乙磺酸盐、乙基琥珀酸盐、富马酸盐、葡庚糖酸盐、葡萄糖酸盐、谷 氨酸盐、乙醇酰基对氨基苯胂酸盐(glycollylarsanilate)、己基间苯二酚 盐(hexylresorcinate)、哈胺(hydrabamine)、氢溴酸盐、盐酸盐、碘化 物、羟乙基磺酸盐、乳酸盐、乳糖酸盐、月桂酸盐、苹果酸盐、马来 酸盐、扁桃酸盐、甲磺酸盐、甲基硫酸盐、粘酸盐、萘磺酸盐、硝酸 盐、油酸盐、草酸盐、扑酸盐(双羟萘酸盐)、棕榈酸盐、泛酸盐、磷 酸盐/二磷酸盐、聚半乳糖醛酸盐(polygalacturonate)、水杨酸盐、硬 脂酸盐、次醋酸盐、琥珀酸盐、鞣酸盐、酒石酸盐、8-氯茶碱盐、甲 苯磺酸盐、triethiodode和戊酸盐。
[0259]
关于本发明方法中使用的本发明的化合物,如果化合物也以互变 异构形式存在,则本发明涉及那些互变异构体以及所有此类互变异构 体及其混合物的用途。
[0260]
本发明也包括化合物和用同位素标记的化合物治疗covid-19的 方法和抑制sars-cov-2的方法,所述同位素标记的化合物与本文所 述的那些相同,但一个或多个原子被其原子质量或质量数与通常在自 然界中发现的原子质量或质量数不同的原子替换。可结合到本发明化 合物的同位素的例子包括氢、碳、氮、氧、磷、氟和氯的同位素,分 别例如2h、3h、
13
c、
14
c、
15
n、
18
o、
17
o、
31
p、
32
p、
35
s、
18
f和
36
cl。 含有上述同位素和/或其它原子的同位素的本发明的化合物、其前药, 以及所述化合物或所述前药的药学上可接受的盐,均在本发明的范围 内。本发明的某些同位素标记的化合物,例如结合有放射性同位素如 3
h和
14
c的那些,可用于药物和/或底物组织分布测定。氚化(即3h) 和碳-14(即
14
c)同位素是特别优选的,因为它们易于制备和检测。 此外,用更重的同位素(例如氘,即2h)取代可以提供某些治疗优势, 因为代谢稳定性更高,例如增加体内半衰期或减少剂量需求,因此在 某些情况下可能是优选的。在本发明的方法中使用的同位素标记的化 合物及其前药通常可以通过用容易获得的同位素标记的试剂代替非同 位素标记的试剂来进行本领域公开的化合物的制备程序来制备。
[0261]
本发明还包括使用药物组合物的方法和通过施用本发明的前药化 合物来治疗covid-19感染的方法。具有游离酰氨基或羟基的化合物 可以转化为前药。前药包括这样的化合物,其中氨基酸残基或两个或 更多个(例如,两个、三个或四个)氨基酸残基的多肽链通过酯键共 价连接至用于本发明方法的化合物的羟基。氨基酸残基包括但不限于 通常由
三个字母符号指定的20个天然存在的氨基酸,并且还包括4
‑ꢀ
羟脯氨酸、羟赖氨酸、锁链素(demosine)、异锁链素(isodemosine)、3
‑ꢀ
甲基组氨酸、正缬氨酸(norvalin)、β-丙氨酸、γ-氨基丁酸、瓜氨酸高 半胱氨酸、高丝氨酸、鸟氨酸和甲硫氨酸砜。还包括其它类型的前药。 例如,如advanced drug delivery reviews,1996,19,115中所述,可 以使用包括但不限于半琥珀酸酯、磷酸酯、二甲基氨基乙酸酯和磷酰 氧基甲氧基羰基的基团来衍生游离羟基。还包括羟基和氨基的氨基甲 酸酯前药,以及羟基的碳酸酯前药、磺酸酯和硫酸酯。也包括羟基衍 生为(酰氧基)甲基和(酰氧基)乙基醚,其中酰基可以是烷基酯, 任选被包括但不限于醚、胺和羧酸官能团的基团取代,或者其中酰基 是如上所述的氨基酸酯。这种类型的前药描述于j.med.chem.,1996, 29,10。游离胺也可以衍生为酰胺、磺酰胺或膦酰胺。所有这些前药部 分都可以引入包括但不限于醚、胺和羧酸官能团的基团。
[0262]
本发明的化合物也可以与其它药物组合用在本发明的方法中。例 如,将本发明的sars-cov-2冠状病毒3cl蛋白酶抑制剂和干扰素(如 干扰素α)或聚乙二醇化干扰素(如peg-intron或pegasus)施用于 sars-cov-2冠状病毒感染患者(即covid-19患者),可能比单独 施用干扰素、聚乙二醇化干扰素或sars-cov-2冠状病毒抑制剂提供 更大的临床益处。可以用在本发明的方法中的其它额外药剂包括氯喹、 羟氯喹、阿奇霉素和瑞德西韦。更大的临床益处的例子可能包括 covid-19症状的大幅减少、症状缓解的时间更快、肺部病理减少、 患者体内sars-cov-2冠状病毒的数量(病毒载量)大幅减少,以及 死亡率降低。
[0263]
sars-cov-2冠状病毒感染表达p-糖蛋白的细胞。本发明的一些 sars-cov-2冠状病毒3cl蛋白酶抑制剂是p-糖蛋白底物。抑制 sars-cov-2冠状病毒的化合物(也是p-糖蛋白底物)可以与p-糖蛋 白抑制剂一起施用。p-糖蛋白抑制剂的例子是维拉帕米、长春碱、酮 康唑、奈非那韦、利托那韦或环孢菌素。p-糖蛋白抑制剂通过抑制本 发明的sars-cov-2冠状病毒抑制剂外流出细胞而起作用。抑制基于 p-糖蛋白的外流将防止sars-cov-2冠状病毒抑制剂由于p-糖蛋白外 流而降低细胞内浓度。抑制p-糖蛋白外流将导致sars-cov-2冠状病 毒抑制剂的细胞内浓度较高。给感染sars-cov-2冠状病毒的患者施 用本发明的sars-cov-2冠状病毒3cl蛋白酶抑制剂和p-糖蛋白抑 制剂可通过增加sars-cov-2冠状病毒3cl蛋白酶抑制剂的细胞内浓 度来降低达到有效剂量所需的sars-cov-2冠状病毒3cl蛋白酶抑制 剂的量。
[0264]
可用于增加哺乳动物对本发明化合物的暴露的药剂包括可作为细 胞色素p450(cyp450)酶的至少一种同种型的抑制剂的药剂。可能被有 益抑制的cyp450同种型包括、但不限于cyp1a2、cyp2d6、 cyp2c9、cyp2c19和cyp3a4。本发明方法中使用的化合物包括可 以是cyp3a4底物并被cyp3a4代谢的化合物。向感染sars-cov-2 冠状病毒的患者施用为cyp3a4底物的sars-cov-2冠状病毒抑制剂 (例如sars-cov-2冠状病毒3cl蛋白酶抑制剂)和cyp3a4抑制剂(例 如利托那韦、奈非那韦或地拉韦定),将减少sars-cov-2冠状病毒抑 制剂通过cyp3a4的代谢。这将导致sars-cov-2冠状病毒抑制剂的 清除率降低并增加sars-cov-2冠状病毒抑制剂的血浆浓度。清除率 降低和血浆浓度升高可能导致sars-cov-2冠状病毒抑制剂的有效剂 量较低。
[0265]
可在本发明的方法中与sars-cov-2抑制剂组合使用的其它治疗 剂包括以下:
[0266]
plpro抑制剂:利巴韦林、伐昔洛韦、β-胸苷、阿司帕坦、氧烯 洛尔、多西环素、醋奋乃静、碘普胺、核黄素、瑞普特罗、2,2
′‑
环胞 苷、氯霉素、氯苯甘油氨酯、左羟丙哌嗪、头孢
(β-d-吡喃葡萄糖基氧基)-1,3,5-三羟基-9h-呫吨-9-酮,
[0269]
可以用在本发明的方法中的另外的治疗剂包括地奥司明、橙皮苷、mk-3207、维奈托克、双氢麦角汀、勃拉嗪、r428、ditercalinium、依托泊苷、替尼泊苷、uk-432097、伊立替康、lumacaftor、维帕他韦、eluxadoline、ledipasvir、洛匹那韦/利托那韦+利巴韦林、alferon和泼尼松。可用在本发明的方法中的其它额外药剂包括氯喹、羟氯喹、阿奇霉素和瑞德西韦。
[0270]
可以用在本发明的方法中的其它额外药剂包括命名为11r、13a和13b的α-酮酰胺化合物,如下所示,如在zhang,l.;lin,d.;sun,x.;rox,k.;hilgenfeld,r.;x-raystructureofmainproteaseofthenovelcoronavirussars-cov-2enablesdesignofα-ketoamideinhibitors;biorxivpreprintdoi:https://doi.org/10.1101/2020.02.17.952879中描述的
[0271][0272]
可以用在本发明的方法中的其它药剂包括rig1途径活化剂诸如在美国专利号9,884,876中描述的那些。
[0273]
可以用在本发明的方法和组合物中的另外的治疗剂包括一种或多种选自以下的药剂:瑞德西韦、加利西韦、法维拉韦/阿韦法韦、mulnupiravir(mk-4482/eidd2801)、at-527、at-301、bld-2660、法匹拉韦、卡莫司他、slv213恩曲他滨/替诺福韦、克拉夫定、达塞曲匹、波普瑞韦、abx464、地塞米松、氢化可的松、恢复期血浆(convalescentplasma)、溶凝胶蛋白(rhu-p65n)、单克隆抗体、瑞丹维单抗(regkirova)、雷夫利珠单抗(ultomiris)、vir-7831/vir-7832、brii-196/brii-198、covi-amg/covidrops(sti-2020)、班兰尼维单抗(ly-cov555)、mavrilimab、乐昂立单抗(pro140)、azd7442、仑兹鲁单抗、英夫利昔单抗、阿达木单抗、js016、sti-1499(coviguard)、lanadelumab(takhzyro)、卡那奴单抗(ilaris)、瑾司鲁单抗、otilimab、卡西瑞单抗/伊德单抗(regn-cov2)、mk-7110(cd24fc/saccovid)、肝素、阿派沙班、托珠单抗(actemra)、sarilumab(kevzara)、apilimod二甲磺酸酯、dnl758、pb1046、dapaglifozin、abivertinib、atr-002、bemcentinib、acalabrutinib、巴瑞替尼、托法替尼、洛吡莫德、法莫替丁、氯硝柳胺和二脒那秦。
[0274]
术语“sars-cov-2抑制剂”是指本文描述的任何sars-cov-2相关的冠状病毒3c样蛋白酶抑制剂化合物或其药学上可接受的盐、水合物、前药、活性代谢物或溶剂化物,或以任何方式抑制sars-cov-2的复制的化合物。
[0275]
术语“干扰或预防”细胞中sars-cov-2相关的冠状病毒(“sars-cov-2”)病毒复制是指与未被核酶或编码核酶的载体瞬时或稳定转导的细胞相比,减少细胞中sars-cov-2复制或子代病毒所需的sars-cov-2组分产生。确定sars-cov-2病毒复制是否减少的简单而方便的检测包括elisa检测受试者血液中抗sars-cov-2抗体的存在、缺失或减少(nasoff等人,pnas88:5462-5466,1991),rt-pcr(yu,等人,inviralhepatitisand
liver disease 574-577,nishioka, suzuki and mishiro(eds.);springer-verlag,tokyo,1994年)。这 样的方法是本领域普通技术人员众所周知的。可替换地,可以分离转 导和感染的“对照”细胞的总rna,并通过斑点印迹或rna印迹进行 分析,并用sars-cov-2特异性dna进行探测,以确定sars-cov-2 复制是否减少。可替换地,sars-cov-2蛋白表达的减少也可以作为 抑制sars-cov-2复制的指标。与对照细胞相比sars-cov-2复制减 少超过50%通常可以用数量说明对sars-cov-2复制的预防。
[0276]
如果本发明方法中使用的sars-cov-2抑制剂化合物是碱,则可 以通过本领域已知的任何合适的方法制备所需的盐,包括用以下酸处 理游离碱:无机酸(诸如盐酸、氢溴酸、硫酸、硝酸、磷酸等),或有 机酸(诸如乙酸、马来酸、琥珀酸、扁桃酸、富马酸、丙二酸、丙酮酸、 草酸、羟乙酸、水杨酸、吡喃糖苷酸(诸如葡糖醛酸或半乳糖醛酸)、α
‑ꢀ
羟酸(诸如柠檬酸或酒石酸)、氨基酸(诸如天冬氨酸或谷氨酸)、芳香酸 (诸如苯甲酸或肉桂酸)、磺酸(诸如对甲苯磺酸或乙磺酸)等。
[0277]
如果本发明方法中使用的sars-cov-2抑制剂化合物是酸,则可 以通过本领域已知的任何合适的方法制备所需的盐,包括用无机或有 机碱(例如胺(伯胺、仲胺或叔胺))、碱金属氢氧化物或碱土金属 氢氧化物处理游离酸。合适的盐的说明性实例包括衍生自氨基酸(例 如甘氨酸和精氨酸)、氨、伯胺、仲胺、叔胺和环胺(例如哌啶、吗 啉和哌嗪)的有机盐,以及衍生自钠、钙、钾、镁、锰、铁、铜、锌、 铝和锂的无机盐。
[0278]
在sars-cov-2抑制剂化合物、前药、盐或溶剂化物为固体的情 况下,本领域技术人员理解,本发明方法中使用的化合物、前药、盐 和溶剂化物可以以不同的多晶型物或晶体形式存在,所有这些都意图 在本发明和特定分子式的范围内。此外,本发明方法中使用的化合物、 盐、前药和溶剂化物可以作为互变异构体存在,所有这些都在本发明 的广泛范围内。
[0279]
增溶剂也可以与本发明的化合物一起使用以增加化合物在生理上 可接受的溶液的水中的溶解度。这些增溶剂包括环葡聚糖 (cyclodextran)、丙二醇、二乙基乙酰胺、聚乙二醇、吐温、乙醇和 胶束形成剂。提供的增溶剂是环葡聚糖,特别是β环葡聚糖,且尤其 是羟丙基β环葡聚糖和磺丁基醚β环葡聚糖。
[0280]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯的制剂
[0281]
本发明的一种特别优选的化合物,磷酸二氢(3s)-3-({n-[(4-甲氧基
ꢀ‑
1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷
ꢀ‑
3-基]丁酯(在某些情况下被称作pf-07304814),可以作为基于溶液或 粉末的制剂来提供,其含有或不含赋形剂以产生适合用于胃肠外施用 的药物组合物。pf-07304814溶液制剂的浓度,或在重构后冷冻干燥 或粉末填充制剂的浓度,优选地是在25-200mg/ml的范围内。所述 制剂可以在无菌注射用水、0.9%w/v氯化钠或5%w/v右旋糖溶液中重 构或稀释用于静脉内(iv)施用。例如,为了静脉内施用的目的,在 大约250ml或大约500ml的输注体积中大约3g pf-0730814的每日 剂量将分别产生12mg/ml或6mg/ml的输注浓度。作为另一个例子, 为了静脉内施用的目的,在大约250ml或大约500ml的输注体积中 大约1g pf-0730814的每日剂量将分别产生4mg/ml或2mg/ml的 输注浓度。作为另一个例子,为了静脉内施用的目的,在大约250ml 或大约500ml的输注体积中大约500mg pf-0730814的每日剂量将 分别产生2mg/ml或1mg/ml
的输注浓度。
[0282]
对于pf-07304814,存在多种具有ph依赖性机制的降解物,且 产生最小降解的ph对于每种降解物是不同的。pf-07304814制剂(包 括任何溶液制剂、在冷冻干燥前溶液、在冷冻干燥后的重构溶液和用 于静脉内施用的稀释溶液)的优选ph值是在大约ph 2.0至大约ph 6.0 的范围内,且最优选的ph范围是大约ph 3.0至大约ph 5.0。为了维 持所需的ph,任选地加入缓冲剂,优选的缓冲剂是乳酸、磷酸、乙 酸和酒石酸,最优选的缓冲剂是柠檬酸。pf-07304814与柠檬酸盐缓 冲剂的优选摩尔比是大约1:1至大约20:1,更优选摩尔比是大约2:1 至大约10:1,和最优选摩尔比是大约4.5:1。通过加入合适的碱性赋形 剂可以调节和控制制剂的ph,优选的碱包括苄星、氢氧化钙、胆碱、 二乙胺、二乙醇胺、氢氧化镁和葡甲胺;更优选的碱是赖氨酸、哌嗪、 氢氧化钾和三(羟基甲基)氨基甲烷;和最优选的碱是氢氧化钠 (naoh)。
[0283]
在制剂中使用的pf-07304814形式可以是游离酸或合适的盐。在 溶液中,pf-07304814的磷酸根基团预期在目标ph范围发生离子化 和带负电荷,且因而溶液中的阳离子物质预期充当与磷酸根基团相互 作用的抗衡离子。令人惊奇地,我们发现,抗衡离子不会显著地影响 冷冻干燥的粉末的固态结构,如通过粉末x-射线衍射(pxrd)或调制 型示差扫描量热法(mdsc)所测得的,但是可以显著影响初级降解物的 降解速率。形成pf-07304814的盐的优选抗衡离子包括苄星、钙、胆 碱、二乙胺、二乙醇胺、镁、葡甲胺,更优选的抗衡离子包括赖氨酸、 哌嗪、钾和三(羟基甲基)氨基甲烷,且最优选的抗衡离子是钠。在药 物组合物制剂(包括任何溶液制剂、在冷冻干燥前溶液、在冷冻干燥后 的重构溶液和用于静脉内施用的稀释溶液)中抗衡离子与pf-07304814 的优选摩尔比是大约0.5:1至大约3:1,且最优选摩尔比是大约0.5:1 至大约2:1。
[0284]
意外地,我们发现,一种或多种稳定赋形剂的添加可以产生具有 可比较的含水量、结晶度和外观的冷冻干燥制剂,但是可以显著降低 降解物1(磷酸酯切割的化合物)的形成速率。优选的稳定赋形剂包括 糖、多元醇、聚合物和氨基酸;更优选的赋形剂包括葡聚糖、甘氨酸、 乳糖、甘露醇、聚乙烯吡咯烷酮、蔗糖和海藻糖;和最优选的赋形剂 包括聚乙二醇(pegs;例如peg300、peg400、peg3350)。冷冻干燥 的粉末中稳定赋形剂的优选量至多为大约30%w/w,且最优选量至多 为大约15%w/w。冷冻干燥后的重构溶液中总稳定赋形剂的优选量至 多为大约50mg/ml,且最优选量至多为大约20mg/ml。用于静脉内 施用的稀释溶液中总稳定赋形剂的优选量至多为10mg/ml,且最优 选量至多为4mg/ml。
[0285]
对于pf-07304814,我们发现,少量增溶赋形剂的添加可以防止 难溶性杂质的沉淀。优选的增溶赋形剂包括表面活性剂和络合赋形剂 (例如环糊精);更优选的增溶赋形剂包括聚乙氧基化的蓖麻油、聚乙 二醇(15)-羟基硬脂酸酯、羟丙基-β-环糊精(hp-β-cd)、磺丁基醚-β-环 糊精(sbe-β-cd)、γ-环糊精;和最优选的增溶赋形剂包括聚山梨酯20 (ps20)或聚山梨酯80(ps80)。冷冻干燥的粉末中增溶赋形剂的优选量 至多为大约15%w/w,且最优选量至多为大约5%w/w。冷冻干燥后的 重构溶液中总增溶赋形剂的优选量至多为大约20mg/ml,且最优选 量至多为大约5mg/ml。用于静脉内施用的稀释溶液中增溶赋形剂的 优选量至多为4mg/ml,且最优选量至多为1mg/ml。
[0286]
对于pf-07304814,制备冷冻干燥产品以降低药物产品中的含水 量。我们发现,冷冻干燥循环的优化可以导致低含水量,这会显著改 善化学稳定性。优选的含水量小于2%
w/w,更优选地小于1%w/w,和最优选地小于0.5%w/w。pf-07304814可以制备为溶液制剂,其可以填充进适当的容器封闭系统中。可以将溶液制剂作为溶液储存和供给,或随后冷冻干燥以制备冷冻干燥制剂。可替换地,pf-07304814可以在适当的容器封闭系统中制备为粉末,用标准或专业稀释剂来制备溶液。
[0287]
在某些情况下,在本发明的方法中使用的sars-cov-2抑制剂化合物、盐、前药和溶剂化物可以具有手性中心。当手性中心存在时,所述异羟肟酸酯化合物、盐、前药和溶剂化物可以作为单一立体异构体、外消旋体和/或对映异构体和/或非对映异构体的混合物存在。所有这些单一的立体异构体、外消旋体和它们的混合物都在本发明的广泛范围内。
[0288]
如本领域技术人员通常理解的,光学纯的化合物是对映体纯的化合物。本文中使用的术语“光学纯”旨在表示包含至少足够活性的化合物。优选地,产生具有所需药理学纯的本发明化合物的化合物的光学纯量单一对映异构体包含至少90%的单一异构体(80%对映异构体过量),更优选地至少95%(90%e.e.),甚至更优选地至少97.5%(95%)e.e.),和最优选地至少99%(98%e.e.)。
[0289]
除非另有说明,否则本文所用的术语“治疗”意指逆转、减轻、抑制这样的术语所适用的障碍或病症的进展、或这样的障碍或病症的一种或多种症状或预防这样的术语所适用的障碍或病症、或这样的障碍或病症的一种或多种症状。除非另有说明,否则本文所用的术语“治疗”是指治疗(如上定义的“治疗”)的行为。在本发明的一个优选实施方案中,“治疗”是指至少减轻人的疾病病症,所述病症通过抑制sars-cov-23c样蛋白酶的活性而减轻,该蛋白酶是sars-cov-2的主要蛋白酶,是covid-19的病原体。sars-cov-2病毒应当理解为涵盖该病毒的最初发现的株以及出现的突变株,诸如但不限于株诸如b.1.1.7(uk变体)、b.1.351(南非变体)和p.1(巴西变体)。对于患有covid-19的患者,发烧、疲劳和干咳是该病的主要表现,而鼻塞、流鼻涕和其它上呼吸道症状很少见。北京市疾病预防控制中心表示,典型的covid-19病例有一个逐步加重的过程。根据疾病的严重程度可将covid-19分为轻型、正常、重型和危重型nationalhealthcommissionofthepeople’srepublicofchina.diagnosisandtreatmentofpneumoniacausedby2019-ncov(trialversion4).availableonline:http://www.nhc.gov.cn/jkj/s3577/202002/573340613ab243b3a7f61df260551dd4/files/c791e5a7ea5149f680fdcb34dac0f54e.pdf(accessedon6february2020):(1)轻度病例-轻度临床症状,胸部计算机断层扫描(ct)未发现肺炎;(2)正常病例
‑‑
有发热、呼吸道症状、发现有肺炎影像学表现的患者;(3)重型病例
‑‑
以下三种情况之一:呼吸窘迫、呼吸频率≥30次/分钟(静息状态,指氧饱和度≤93%)、动脉血氧分压
[0290]
(pao2)/吸氧浓度(fio2))≤300mmhg(1mmhg=0.133kpa);(4)危重病例
‑‑
以下三种情况之一:呼吸衰竭和需要机械通气、休克或其它需要重症监护病房的器官相关衰竭。目前的临床数据显示,大多数死亡发生在老年患者中。但是,在具有独特因素的年轻人中记录了严重病例,特别是那些患有慢性疾病的人,例如糖尿病或乙型肝炎。那些长期使用激素或免疫抑制剂和免疫功能下降的人很可能会受到严重感染。
[0291]
减轻疾病病症例如covid-19的治疗方法包括以任何常规可接受的方式使用本发明中的一种或多种化合物。根据本发明的某些优选实施方案,将本发明方法中使用的一种或多种化合物施用于有需要的哺乳动物,例如人。优选地,有此需要的哺乳动物感染了冠
状病毒,例 如covid-19的病原体,即sars-cov-2。
[0292]
本发明还包括预防方法,包括将有效量的本发明的sars-cov-2 抑制剂或其药学上可接受的盐、前药、药学活性代谢物或溶剂化物施 用于哺乳动物,例如处于sars-cov-2感染危险中的人。根据某些优 选实施方案,将有效量的一种或多种本发明化合物或其药学上可接受 的盐、前药、药学活性代谢物或溶剂化物施用于有感染sars-cov-2 (covid-19的病原体)风险的人。本发明的预防方法包括以任何常规可 接受的方式使用本发明中的一种或多种化合物。
[0293]
以下是本发明具体实施方案的例子:
[0294]
本发明方法中使用的某些化合物是已知的并且可以通过本领域已 知的方法制备。
[0295]
最近的证据表明,新型冠状病毒sars-cov-2是covid-19的病 原体。sars-cov-2冠状病毒的核苷酸序列以及最近确定的l-和s
‑ꢀ
亚型最近已确定并公开可用。
[0296]
抑制剂化合物作为sars-cov-2病毒活性抑制剂的活性可以通过 本领域可用的任何合适方法测量,包括体内和体外测定。本发明化合 物作为冠状病毒3c样蛋白酶活性(例如sars-cov-2冠状病毒的3c 样蛋白酶)抑制剂的活性可以通过本领域技术人员已知的任何合适方 法测量,包括体内和体外测定。用于活性测量的合适测定的例子包括 本文所述的抗病毒细胞培养测定以及本文描述的抗蛋白酶测定,例如 实施例部分中描述的测定。
[0297]
sars-cov-2抑制剂化合物及其药学上可接受的前药、盐、活性 代谢物和溶剂化物的施用可根据本领域技术人员可获得的任何可接受 的施用模式进行。合适的施用方式的说明性实例包括口服、鼻、肺、 胃肠外、局部、静脉内、注射、透皮和直肠。优选口服、静脉内和经 鼻递送。
[0298]
sars-cov-2抑制剂可以任何合适的药物形式作为药物组合物施 用。合适的药物形式包括固体、半固体、液体或冻干制剂,例如片剂、 粉末、胶囊剂、栓剂、混悬剂、脂质体和气雾剂。sars-cov-2抑制 剂可以使用多种方法中的任一种制备成溶液。例如,sars-cov-2抑 制剂可以用酸(例如1m hcl)溶解,并用足量的5%右旋糖水溶液 (d5w)稀释,以产生所需的sars-cov-2-抑制剂终浓度(例如,约 15mm)。可替换地,可以使用含有约15mm hcl的d5w溶液来提 供适当浓度的sars-cov-2抑制剂溶液。此外,sars-cov-2抑制剂 可以使用例如羧甲基纤维素(cmc)的1%溶液制备为混悬剂。
[0299]
制备药物组合物的合适药物形式的可接受方法是已知的或可由本 领域技术人员常规确定。例如,药物制剂可以按照药物化学家的常规 技术制备,包括以下步骤,例如混合、制粒和在需要片剂形式时压制, 或适当地混合、填充和溶解成分,以得到用于静脉内、口服、胃肠外、 局部、阴道内、鼻内、支气管内、眼内、耳内和/或直肠施用的所需产 品。
[0300]
根据预期用途,本发明的药物组合物还可以包括合适的赋形剂、 稀释剂、媒介物和载体,以及其它药物活性剂。药物组合物中可以使 用固体或液体药学上可接受的载体、稀释剂、媒介物或赋形剂。示例 性固体载体包括淀粉、乳糖、硫酸钙二水合物、石膏粉、蔗糖、滑石 粉、明胶、果胶、阿拉伯胶、硬脂酸镁和硬脂酸。示例性的液体载体 包括糖浆、花生油、橄榄油、盐水溶液和水。载体或稀释剂可以包括 合适的缓释材料,例如单硬脂酸甘油酯或二硬脂酸甘油酯,单独使用 或与蜡一起使用。当使用液体载体时,制剂可以是糖浆剂、酏剂、乳 剂、软明胶胶囊、无菌可注射液体(例如溶液)或非水性或水性液体 悬浮液的形式。
[0301]
化学和物理稳定性影响药物组合物的贮存条件的选择和贮存期 限,并决定药物产品的生存力。化学稳定性通常涉及在药物组合物内 的组分的化学性质的变化,这可能包括活性药物成分(api)的降解、赋 形剂的降解、api(或它的有关的降解物)与赋形剂(或它们的有关降解 物)的反应、或在药物组合物内的组分与容器封闭系统的反应。药物组 合物中降解物的可接受性要求研究,这可以包括降解物结构的鉴定、 降解物溶解度的评价(对于胃肠外产品)和降解物安全性的评估(计算 机平台实验、体外和体内)。具体地,前药部分的化学稳定性依赖于活 性代谢物上的前药部分的身份、位置和局部的环境,以及药物产品的 制剂和贮存条件。具体地,磷酸酯前药的水解对前药部分周围的空间 和电子环境、制剂ph和制剂中的水的量是敏感的。物理稳定性通常 涉及药物组合物的相变化,这可能包括粉末固态结构的变化、难溶性 物质从溶液中沉淀或分散体系结构的变化。为了控制药物组合物的化 学和物理稳定性,研究人员可以研究api的制备方法、药物组合物 的制剂设计或药物组合物的制备方法。
[0302]
在制剂设计中,控制稳定性相关挑战的一种可能方法是添加ph 调节剂或缓冲剂以调节和保持ph。ph值调节可能会改变在溶液中的 物质(例如api、赋形剂或降解物)的溶解度,或者可能会改变特定 降解物的形成速率。然而,ph值偏离中性的肠胃外药物组合物可能 在注射部位引起局部刺激。此外,由于存在多种ph依赖性降解机制, 因此ph优化可能并非易事。因此,药物组合物的ph选择需要仔细 研究和考虑。
[0303]
另一种控制可电离api稳定性的制剂设计方法是使用抗衡离 子。抗衡离子可以与带相反电荷的可电离基团发生静电相互作用,并 且可以在电子或空间上稳定键的降解。抗衡离子还可以改变api在 冻干或粉末制剂中形成晶体结构的能力,这可能会影响化学和物理稳 定性。然而,抗衡离子的影响难以预测,需要对化学和物理稳定性、 化学相容性和安全性评估进行实验研究。
[0304]
另一种控制稳定性的制剂设计方法是在冻干制剂中添加稳定赋形 剂。稳定赋形剂可以提高制剂在冷冻干燥过程的整个冷冻和干燥步骤 或者在药物产品的整个保质期内的储存中的化学和物理稳定性。稳定 赋形剂可能会改变冻干制剂的结晶度和/或玻璃化转变温度(tg),这 可能会影响固态物质的取向和迁移率,从而影响降解的动力学和热力 学。对于对水敏感的降解机制(例如水解),稳定赋形剂也可以取代 与api相互作用的水,从而保护api免于降解,或者可以有效地隔离 水,从而防止其与api反应。稳定赋形剂的选择和优化需要仔细考虑 多种因素来生产具有更高化学稳定性的制剂,包括评估固态结构的结 晶度和物理稳定性、亲液物的吸水-解吸特性、api和稳定赋形剂的相 容性,以及赋形剂的安全性。
[0305]
如果化学和物理稳定性无法提高,那么可替换的由制剂驱动的方 法是添加增溶赋形剂,其防止在肠胃外组合物中溶解性差的降解物沉 淀。增溶赋形剂也可能有助于防止难溶性api相关杂质的沉淀,这些 杂质很难经由api分离和纯化方法去除。增溶赋形剂可包括溶剂、络 合赋形剂、表面活性剂或其他赋形剂。然而,许多增溶赋形剂的肠胃 外施用会导致不利的安全影响,其限制了可用于特定患者群体的赋形 剂的量。例如,在欧洲药品管理局截至2018年11月19日的“关于聚 山梨酯用作人用医药产品赋形剂的包装说明书信息”中,静脉内聚山 梨酯的剂量水平超过每剂10mg/kg可能对心血管产生不利影响和剂 量水平超过35mg/kg/天可能有不利的肝毒性作用。高水平的表面活性 剂也会对药品的生产或
性能产生负面影响,这可能包括药品生产过程 中或用于胃肠外施用的药品制备过程中的发泡,或冻干过程中形成的 固态结构的改变。因此,必须研究和优化增溶赋形剂的量,以防止溶 解性差的物质沉淀,而不会给药物产品带来额外的风险。
[0306]
在药物组合物的制备中,制造单元操作(例如混合、冷冻干燥) 可使制剂暴露于导致降解的应激源。此外,该制备可以产生具有不同 组成或影响稳定性的结构的药物产品。对于对水解降解敏感的api, 例如磷酸酯前药,粉末中残留的水含量会显著影响制剂的化学稳定性。
[0307]
一剂药物组合物可以包含至少治疗有效量的sars-cov-2抑制 剂,并且优选由一个或多个药物剂量单位组成。可以通过任何已知的 或合适的施用药剂方法将选择的剂量施用于需要通过抑制 sars-cov-2相关冠状病毒活性介导的治疗的哺乳动物,例如人类患 者,所述施用药剂方法包括局部施用,例如作为软膏或霜剂;口服施 用;直肠施用,例如作为栓剂;通过注射胃肠外施用;静脉内施用; 或连续通过阴道内、鼻内、支气管内、耳内或眼内输注。
[0308]
短语“治疗有效量”和“有效量”旨在表示当施用于需要治疗的哺乳 动物时,足以对通过抑制sars-cov-2病毒复制而减轻的损伤或疾病 病症进行治疗的本发明药剂的量。本发明的方法中使用的给定 sars-cov-2抑制剂的将是治疗有效的量将根据诸如特定的 sars-cov-2抑制剂、疾病病症及其严重程度、有此需要的哺乳动物 的身份和特征的因素而变化,其量可由本领域技术人员常规确定。
[0309]
应当理解,用于本发明药物组合物中的sars-cov-2抑制剂的实 际剂量将根据所用特定药剂的性质、配制的特定组合物、施用方式和 特定部位,以及所治疗的宿主和病症来选择。本领域技术人员可以使 用常规剂量测定试验确定一组给定条件的最佳剂量。对于口服施用, 例如,可以采用的剂量为从约0.01至约1000mg/kg体重,优选地从 约0.1至约500mg/kg体重,且甚至更优选地从约1至约500mg/kg 体重,以适当的时间间隔重复治疗疗程。对于静脉内施用,可以使用 至多为每天5克的剂量。静脉内施用可以在一天中间歇性进行,也可 以在24小时内连续进行。
[0310]
本文中使用的术语“细胞色素p450-抑制量”和“细胞色素p450酶 活性-抑制量”是指在存在降低细胞色素p450酶或特定细胞色素p450 酶同种型的活性所需的化合物的情况下这种化合物降低细胞色素 p450酶或特定细胞色素p450酶同种型的活性所需的量。可以通过本 领域普通技术人员已知的方法和本文所述的方法确定特定化合物是否 降低细胞色素p450酶活性,以及这样做所需的此类化合物的量。
[0311]
冠状病毒复制和转录所需的蛋白功能由所谓的“复制酶”基因编 码。两个重叠的多聚蛋白从该基因翻译而来,并被病毒蛋白酶广泛加 工。冠状病毒主要或“3c样蛋白酶”在11个保守的域间连接处加工c
‑ꢀ
近端区域。“3c样”蛋白酶的名称源于冠状病毒酶与众所周知的小核糖 核酸病毒3c蛋白酶之间的某些相似之处。这些包括底物偏好、使用 半胱氨酸作为催化中的活性位点亲核试剂,以及它们推定的整体多肽 折叠的相似性。将sars-cov-2相关冠状病毒3c样蛋白酶的氨基酸 序列与其它已知冠状病毒(如sars-cov)的氨基酸序列进行比较, 表明氨基酸序列具有大约96%的共享同源性。
[0312]
蛋白酶切割位点中底物的氨基酸从n到c末端编号如下:
ꢀ‑
p3-p2-p1-p1
’‑
p2
’‑
p3’,在p1和p1'残基之间发生切割(schechter& berger,1967)。底物特异性主要由p2、p1和
p1'位置决定。冠状病 毒主要蛋白酶切割位点特异性高度保守,需要p1处的谷氨酰胺和p1' 处的小氨基酸(journal of general virology,83,pp.595-599(2002))。
[0313]
根据在下面反应方案1-17中所述的方法,可以制备在本发明的方 法中使用的化合物。
[0314]
方案1
[0315][0316]
方案1说明了用于制备如显示的式ia的化合物的合成顺序,其中 将式a的化合物用式b的化合物处理,其中x是卤素原子,最常见 氯(参见pct国际申请公开wo 2005/113580)。在该情况下,式b的 化合物被称作氯甲酸酯,且此类方法是本领域技术人员众所周知的。 在有合适的碱存在下进行反应,以消耗作为反应副产物产生的卤化氢 hx。合适的碱的例子包括但不限于叔胺诸如n-甲基吗啉(nmm),2,6
‑ꢀ
二甲基吡啶或二异丙基乙胺(diea)或无机碱诸如氧化镁(mgo)、碳酸 钠(na2co3)或碳酸氢钾(khco3)。合适的溶剂包括、但不限于非质子 溶剂诸如二氯甲烷(ch2cl2)、四氢呋喃(thf)或乙腈(ch3cn)。本领域 技术人员会明白,在式a的化合物具有为h的r2的情况下,以上转 化可以得到式ia的产物化合物,其中r2可以是h或可以是roc(o), 取决于反应参数诸如时间、温度、溶剂和采用的式e的化合物的当量 的选择。
[0317]
方案2
[0318][0319]
方案2说明了用于制备如显示的式1a的化合物的合成顺序,其中 将式a的化合物用式c的化合物(经常被本领域技术人员称作焦碳酸 酯)处理。经常在有亲核催化剂存在下进行反应以加速反应。这样的亲 核的催化剂的例子包括但不限于4-(二甲基氨基)吡啶,咪唑或1,8-二 氮杂双环[5.4.0]十一碳-7-烯(dbu)。合适的溶剂包括但不限于ch2cl2、 thf、吡啶或ch3cn。本领域技术人员会明白,在式a的化合物具 有为h的r2的情况下,以上转化可以得到式ia的产物化合物,其中 r2可以是或可以是r4oc(o),取决于反应参数诸如时间、温度、溶剂 和采用的式c的化合物的当量的选择。
[0320]
方案3
[0321][0322]
方案3说明了用于制备如显示的式id的化合物的合成顺序,其中 将式a的化合物用式d的化合物处理,其中x是卤素原子,最常见 氯。在该情况下,式d的化合物被称作氨甲酰氯,且此类方法是本领 域技术人员众所周知的。在有碱存在下进行反应以消耗作为反应副产 物产生的卤化氢hx。合适的碱的例子包括但不限于叔胺诸如n-甲基 吗啉,2,6-二甲基吡啶或二异丙基乙胺,或无机碱诸如mgo、na2co3或khco3。合适的溶剂包括但不限于ch2cl2、thf或ch3cn。在 另一个实施方案中,x可以是通过杂环n原子之一连接的咪唑、吡唑 或三唑环。这样的试剂是本领域技术人员已知的,且通常从对应的胺 (r6)2nh和1,1
’‑
羰基二咪唑、1,1'-羰基二-1h-吡唑,或1,1'-羰基二-1h-1,2,3-三唑制备,最常见作为合成顺序中的初步步骤。本领域技术 人员会明白,在式a的化合物含有r2=h的情况下,以上转化可以 得到式1d的产物化合物,其中r2可以是h或可以是(r6)2nc(o),取 决于反应参数诸如时间、温度、溶剂和采用的式d的化合物的当量的 选择。
[0323]
方案4
[0324][0325]
方案4说明了用于制备如显示的式1d的化合物的合成顺序,其中 将式a的化合物用式e的化合物处理,随后在一段时间以后用(r6)2nh 处理。在该实施方案中,x可以是通过杂环n原子之一连接的咪唑、 吡唑或三唑环,或x可以是通过o-n氧原子连接的n-氧基-酰亚胺。 本领域技术人员常用的这样的试剂的例子包括1,1
’‑
羰基二咪唑,1,1'
‑ꢀ
羰基二-1h-吡唑,1,1'-羰基二-1h-1,2,3-三唑和1,1'-[羰基双(氧基)]二
ꢀ‑
2,5-吡咯烷二酮。可以在有亲核催化剂存在下进行反应以加速反应。 这样的亲核催化剂的例子包括但不限于4-(二甲基氨基)吡啶、咪唑或 dbu。合适的溶剂包括但不限于ch2cl2、thf、dmf、dmso或 ch3cn。本领域技术人员会明白,在式a的化合物含有r2=h的情 况下,以上转化可以得到式1d的产物化合物,其中r2可以是h或可 以是(r6)2nc(o),取决于反应参数诸如时间、温度、溶剂和采用的式 e的化合物的当量的选择。
[0326]
方案5
[0327][0328]
方案5说明了用于制备如所示的式ib的化合物的合成顺序,其中 r4是h、甲基或乙基,其中将式a的化合物用式f的化合物处理,其 中r
4a
是h、甲基或乙基且x是卤素原子,经常是氯。这样的化合物 f描述在化学文献中且可以是商购可得的。通过在合适的溶剂中用碱 例如碳酸铯(cs2co3)处理进行反应,所述溶剂可以包括但不限于 thf、dmf、dmso或ch3cn。本领域技术人员会明白,在式a的 化合物含有r2=h的情况下,以上转化可以得到式1b的产物化合物, 其中r2可以是h和/或可以是ch(r
4a
)oc(o)or4,取决于反应参数诸 如时间、温度、溶剂和采用的式f的化合物的当量的选择。
[0329]
方案6
[0330][0331]
方案6说明了用于制备如所示的式1b的化合物的合成顺序,其中 r
4a
不等于h,其中将式a的化合物用式g的烯属化合物处理。这样 的化合物g描述在化学文献中且可以是商购可得的。通过用本领域技 术人员已知的催化剂处理进行反应,所述催化剂可以包括但不限于酸、 钯的化合物或汞的化合物。合适的溶剂可以包括但不限于乙酸、thf 或ch3cn。本领域技术人员会明白,在式a的化合物含有r2=h的 情况下,以上转化可以得到式1b的产物化合物,其中r2可以是h或 可以是ch(r
4a
)oc(o)or4,取决于反应参数诸如时间、温度、溶剂和 采用的式g的化合物的当量的选择。
[0332]
方案7
[0333][0334]
方案7说明了用于制备如所示的式1c的化合物的合成顺序,其中 r
4a
是h、甲基或乙基,其中将式a的化合物用式h的化合物处理, 其中r
4a
是h、甲基或乙基且x是卤素原子。在有碱存在下进行反应 以消耗作为反应副产物产生的卤化氢hx。合适的碱的例子包括但不 限于叔胺诸如n-甲基吗啉、2,6-二甲基吡啶或二异丙基乙胺,或无机 碱诸如mgo、cs2co3或khco3。合适的溶剂可以包括但不限于thf、 dmf、dmso或ch3cn。本领域技术人员会明白,在式a的化合物 含有r2=h的情况下,以上转化可以得到式1的产物化合物,其中 r2可以是h或可以是ch(r
4a
)oc(o)r4,取决于反应参数诸如时间、 温度、溶剂和采用的式h的化合物的当量
的选择。
[0335]
方案8
[0336][0337]
方案8说明了用于制备如所示的式ic的化合物的合成顺序,其中 r
4a
不等于h,其中将式a的化合物用式i的烯属化合物处理。这样 的化合物i描述在化学文献中且可以是商购可得的。通过用本领域技 术人员已知的催化剂处理进行反应,所述催化剂可以包括但不限于酸、 钯的化合物或汞的化合物。合适的溶剂可以包括但不限于乙酸、thf 或ch3cn。本领域技术人员会明白,在式a的化合物含有r2=h的 情况下,以上转化可以得到式1c的产物化合物,其中r2可以是h或 可以是ch(r
4a
)oc(o)r4,取决于反应参数诸如时间、温度、溶剂和 采用的式i的化合物的当量的选择。
[0338]
方案9
[0339][0340]
方案9说明了用于制备如显示的式ie的化合物的合成顺序,其中 将式a的化合物用式j的化合物处理,其中x通常是卤素原子且z 可以是直接连接至磷的c
1-c6烷基或通过o原子连接至磷的r5o基 团。上述的产物ie是这样的,其中z是r5o-,但是应当理解,当z 替代性地为烷基时,则显示的-or5基团之一替代性地为该烷基。此类 方法是本领域技术人员众所周知的。化合物j描述在化学文献中且可 以是商购可得的。在有碱存在下进行反应以消耗作为反应副产物产生 的卤化氢hx。合适的碱的例子包括但不限于叔胺诸如n-甲基吗啉、 吡啶、三乙胺或二异丙基乙胺。合适的溶剂包括但不限于ch2cl2、 dmf、thf或ch3cn。本领域技术人员会明白,在式a的化合物含 有r2=h的情况下,以上转化可以得到式1e的产物化合物,其中r2可以是h或可以是p(o)z(or5),取决于反应参数诸如时间、温度、 溶剂和采用的式j的化合物的当量的选择。
[0341]
方案10
[0342][0343]
方案10说明了用于制备如显示的式1e的化合物的合成顺序,其 中将式a的化合物
用式k的化合物处理,其中alk通常是烷基,诸如 甲基、乙基、异丙基、叔丁基或苄基。化合物k被本领域技术人员称 作亚磷酰胺(phosphoramidite)且可以是商购可得的。通常在有亲核催 化剂存在下进行反应,1h-四唑是特别常见的。在反应过程中,通常 在分离式1的化合物之前加入氧化剂。典型氧化剂包括但不限于间氯 过氧苯甲酸(mcpba)、过氧化氢(h2o2)和叔丁基氢过氧化物。合适的 溶剂包括但不限于ch2cl2、thf或ch3cn。本领域技术人员会明白, 在式a的化合物含有r2=h的情况下,以上转化可以得到式1的产物 化合物,其中r2可以是h或可以是p(o)(or5)2,取决于反应参数诸 如时间、温度、溶剂和采用的式k的化合物当量的选择。
[0344]
方案11
[0345][0346]
方案11说明了用于制备如显示的式1f的化合物的合成顺序,其 中将式a的化合物用式l的化合物处理,其中x是卤素原子(通常 为氯)或occl3基团。化合物o在化学文献中被称作光气衍生物且是 商购可得的。在有碱存在下进行反应以消耗作为反应副产物产生的卤 化氢hx。合适的碱的例子包括但不限于叔胺诸如n,n-二甲基苯胺、 吡啶或n-甲基吗啉。合适的溶剂包括但不限于ch2cl2、thf或 ch3cn。
[0347]
方案12
[0348][0349]
方案12说明了用于制备如显示的式ie’的化合物的合成顺序,其 中将式1e”的化合物(例如如在方案10中所示制备)用一种或多种试 剂处理,造成磷上的r5’
o基团的切割以释放所示的oh基团。这样的 方法是本领域技术人员众所周知的,且条件的选择取决于附着于磷上 的r6o基团的性质。例如,当r5’
o基团是phch2o时,反应可以通 过在钯催化剂上的氢化反应来进行。可替换地,当r5’
o基团是 phch2o、叔丁基或ch2ch2cn时,反应可以通过使式1e的化合物 暴露于酸来进行,其中三氟乙酸是特别常用的。合适的溶剂包括但不 限于ch2cl2、dmf、thf或ch3cn。
[0350]
本领域技术人员会明白,可能制备本发明的化合物,其中r2可以 是h以外的某种其它基团。下述方案以非限制性的方式说明了如何将 这样的其它r2基团引入以提供式a的化合物并最终提供其中r2不等 于h的本发明的化合物。
[0351]
方案13
[0352][0353]
方案13说明了用于制备如显示的式r的化合物的合成顺序,其 中将式r1的化合物(pct国际申请公开wo 2005/113580)用试剂处 理,所述试剂将显示的oh基团甲硅烷基化。这样的方法是本领域技 术人员众所周知的,且解释的反应可以通过式r1的化合物暴露于叔 丁基二甲基氯硅烷来完成,通常在有咪唑存在下。合适的溶剂包括但 不限于ch2cl2、dmf、thf或ch3cn。本领域技术人员会明白,其 它试剂可以用于引入叔丁基二甲基甲硅烷基,且通过选择其它适当的 甲硅烷化剂,可以制备与式r的化合物非常类似的其它甲硅烷基醚, 例如三异丙基甲硅烷基或叔丁基二苯基甲硅烷基醚。
[0354]
方案14
[0355][0356]
方案14说明了用于制备如显示的式a的化合物的合成顺序,其 中将式r的化合物转化(通常在2个合成操作中)成式a的化合物, 其中如图示的r2等于c(o)or7。在第一个操作中,可以将式r的化 合物用式b的化合物(r7oc(o)x,方案1)处理,其中x是卤素原子, 最常见氯。在该情况下,式b的化合物被称作氯甲酸酯,且此类方法 是本领域技术人员众所周知的。在有碱存在下进行反应以消耗作为反 应副产物产生的卤化氢hx。合适的碱的例子包括但不限于叔胺诸如 n-甲基吗啉、2,6-二甲基吡啶或二异丙基乙胺或无机碱诸如mgo、 na2co3或khco3。合适的溶剂包括但不限于ch2cl2、thf或 ch3cn。可替换地,在第一个操作中,可以将式r的化合物用式c 的化合物(r7oc(o)oc(o)or7,方案2)(经常被本领域技术人员称作 焦碳酸酯)处理。经常在有亲核催化剂存在下进行反应以加速反应。这 样的亲核催化剂的例子包括但不限于4-(二甲基氨基)吡啶、咪唑或 dbu。合适的溶剂包括但不限于ch2cl2、thf、吡啶或ch3cn。
[0357]
在第二个操作中,可以除去甲硅烷基醚以得到所示的式a的化合 物。本领域技术人员会理解,实现该转化的试剂和条件的选择将取决 于在第一个操作中引入的特定c(o)or7基团的性质,使得第二个操作 的条件与在第一个操作中引入的c(o)or7基团的完整性不会不相容。 为了除去甲硅烷基醚常用的条件包括暴露于例如酸,诸如三氟乙酸、 乙酸、氢氟酸或盐酸,或者可替代地,暴露于氟离子源,其中四丁基 氟化铵是特别常用的。本领域技术人员会明白,第二个操作的合适溶 剂的选择将取决于实现该转化所选择的试剂且可以包括但不限于 ch2cl2、thf或ch3cn。
[0358]
方案15
[0359][0360]
方案15说明了用于制备如显示的式a的化合物的合成顺序,其 中将式r的化合物转化(通常在2个合成操作中)成式a的化合物, 其中如图示的r2等于c(o)r。在第一个操作中,将式r的化合物用 式o的化合物处理,其中x通常是卤素原子、oh或oc(o)r7。此类 方法是本领域技术人员众所周知的。例如,当x=卤素原子时,在有 碱存在下进行反应以消耗作为反应副产物产生的卤化氢hx。合适的 碱的例子包括但不限于叔胺诸如n-甲基吗啉、2,6-二甲基吡啶或二异 丙基乙胺,或无机碱诸如mgo、na2co3或khco3。合适的溶剂包括 但不限于ch2cl2、dmf、thf或ch3cn。当x=oh时,式o的化 合物是羧酸且常规地使用试剂或试剂的组合来加速羧酸o的反应。本 领域技术人员可以选择使用例如碳二亚胺试剂诸如edc或dcc,任 选地在有辅助亲核体诸如hobt或hopo存在下。进一步,当x=oh 时,本领域技术人员可以选择使用适合用于形成混合的羧基/碳酸酐的 试剂,诸如cdi、氯甲酸异丁酯或氯甲酸乙酯,经常在有碱诸如上述 的碱存在下。合适的溶剂包括但不限于ch2cl2、thf或ch3cn。当 x=oh时,本领域技术人员常用的另一个方案是在有碱诸如上述的 碱存在下用例如羧酸氯化物(诸如me3ccocl)处理式o的化合物以 产生式r7c(o)o(o)ccme3的混合羧酸酸酐。合适的溶剂包括但不限 于ch2cl2、thf或ch3cn。在许多情况下可能使用式o的期望羧酸 的对称酸酐以实现方案15的反应,任选地在有诸如上述碱存在下,在 该情况下x=o(o)cr且式o的化合物因此是r7c(o)o(o)r7。合适 的溶剂包括但不限于ch2cl2、thf或ch3cn。
[0361]
在第二个操作中,可以除去甲硅烷基醚以得到所示的式a的化合 物。本领域技术人员会理解,实现该转化的试剂和条件的选择将取决 于在第一个操作中引入的特定c(o)r基团的性质,使得第二个操作的 条件与在第一个操作中引入的c(o)r基团的完整性不会不相容。为了 除去甲硅烷基醚常用的条件包括暴露于例如酸,诸如三氟乙酸、乙酸、 氢氟酸或盐酸,或者可替代地,暴露于氟离子源,其中四丁基氟化铵 是特别常用的。本领域技术人员会明白,第二个操作的合适溶剂的选 择将取决于进行该转化所选择的试剂且可以包括但不限于ch2cl2、 thf或ch3cn。
[0362]
方案16
[0363][0364]
方案16说明了用于制备如显示的式a的化合物的合成顺序,其 中将式r的化合物转化(通常在2个合成操作中)成式a的化合物, 其中如图示的r2等于ch2oc(o)or或chmeoc
(o)or7。在第一个 操作中,可以将式r的化合物用式f的化合物(xch2oc(o)or7或 xchmeoc(o)or7,方案5)处理,其中x是卤素原子。这样的式f的 化合物描述在化学文献中且可以是商购可得的。通过在合适的溶剂中 用碱例如kotbu或cs2co3处理来实现反应,所述溶剂可以包括但不 限于thf、dmf、dmso或ch3cn。
[0365]
在第二个操作中,可以除去甲硅烷基醚以得到所示的式a的化合 物。本领域技术人员会理解,实现该转化的试剂和条件的选择可以取 决于在第一个操作中引入的特定ch2oc(o)or7或chmeoc(o)or7基团的性质,使得第二个操作的条件与在第一个操作中引入的 ch2oc(o)or7或chmeoc(o)or7基团的完整性不会不相容。通过 暴露于氟离子源可以除去甲硅烷基醚,其中四丁基氟化铵是特别合适 的。第二个操作的合适的溶剂可以包括但不限于dmf、ch2cl2、thf 或ch3cn。
[0366]
方案17
[0367][0368]
方案17说明了用于制备如显示的式a的化合物的合成顺序,其 中将式r的化合物转化(通常在2个合成操作中)成式a的化合物, 其中如图示的r2等于chmeoc(o)or7。在第一个操作中,可以将式 r的化合物用式g的烯属化合物(ch2=choc(o)or7,方案6)处理。 这样的式g的化合物描述在化学文献中且可以是商购可得的。通过用 本领域技术人员已知的催化剂处理来实现反应,所述催化剂可以包括 但不限于酸、钯的化合物或汞的化合物。合适的溶剂可以包括但不限 于ch2cl2、thf或ch3cn。
[0369]
在第二个操作中,可以除去甲硅烷基醚以得到所示的式a的化合 物。本领域技术人员会理解,实现该转化的试剂和条件的选择可以取 决于在第一个操作中引入的特定chmeoc(o)or7基团的性质,使得 第二个操作的条件与在第一个操作中引入的chmeoc(o)or7基团的 完整性不会不相容。通过暴露于氟离子源可以除去甲硅烷基醚,其中 四丁基氟化铵是特别合适的。第二个操作的合适的溶剂可以包括但不 限于dmf、ch2cl2、thf或ch3cn。
实施例
[0370]
根据在上文方案1-17中和关于实施例1、2、5、7、8、43、44、 49、57、64和65描述的方法可以制备下述实施例,可以如在下文中 具体所述制备。
[0371]
实验规程
[0372]
下文举例说明本发明的各种化合物的合成。单独地或与本领域通 常已知的技术组合地使用在这些实施例中举例说明的方法,可以制备 在本发明范围内的额外化合物。在这些制备和实施例部分中的全部起 始原料是商购可得的,或可以通过本领域已知的或如本文中所述的方 法制备。
[0373]
除非另外指出,在氮或氩气气氛下使用连续搅拌进行所有反应。 当适当时,使用
热枪在动态真空下干燥反应设备,并采用无水溶剂(来 自aldrich chemical company,milwaukee,威斯康辛州sure-seal
tm
产品或来自emd chemical,gibbstown,nj的drisolv
tm
产品)。在某 些情况下,使市售溶剂穿过填充了分子筛的柱,直到达到下述水 qc标准:a)对于二氯甲烷、甲苯、n,n-二甲基甲酰胺和四氢呋喃《100 ppm;b)对于甲醇、乙醇、1,4-二氧杂环己烷和二异丙胺《180ppm。 对于非常敏感的反应,用金属钠、氢化钙或分子筛进一步处理溶剂, 并且在即将使用前蒸馏。其它市售溶剂和试剂不经进一步纯化地使用。 对于参照在其它实施例或方法中的规程的合成,反应条件(反应时间 和温度)可以变化。通常将产品在真空下干燥,然后转移至进一步反 应或进行生物学试验。
[0374]
当指出时,使用biotage initiator或personal chemistry emrysoptimizer微波通过微波辐射加热反应物。使用薄层色谱法(tlc)、液 相色谱法-质谱法(lcms)、高效液相色谱法(hplc)和/或气相色谱法
‑ꢀ
质谱法(gcms)分析监测反应进程。在具有荧光指示剂(254nm激发波 长)的预包被的硅胶平板上进行tlc,并在紫外线下和/或用i2、 kmno4、cocl2、磷钼酸和/或钼酸铵铈染料显影。在agilent 1100series 仪器上获取lcms数据,其具有leap technologies自动采样器、 gemini c18柱、乙腈/水梯度和三氟乙酸、甲酸或氢氧化铵调节剂。 在100-1200da以阳性和阴性离子模式使用waters zq质谱仪扫描分 析柱洗脱液。也使用其它类似的仪器。通常在agilent 1100series仪 器上获取hplc数据,使用指示的柱、乙腈/水梯度、和三氟乙酸或氢 氧化铵调节剂。使用具有hp 6890注射器、hp-1柱(12m x 0.2mm x 0.33μm)和氦载气的hewlett packard 6890烘箱获取gcms数据。使 用电子电离,在hp 5973质量选择性检测器(扫描50-550da)上分 析样品。使用isco combiflash companion,analogix intelliflash 280, biotage sp1,或biotage isolera one仪器和预填充的isco redisep或 biotage snap硅胶(silica)柱,通过中效液相色谱法(mplc)进行纯化。 通过手性超临界流体色谱法(sfc)进行手性纯化,通常使用berger或 thar仪器;柱诸如chiralpak-ad、-as、-ic、chiralcel-od或-oj 柱;和具有甲醇、乙醇、2-丙醇或乙腈的co2混合物,其为单独的或 使用三氟乙酸或丙烷-2-胺调节。使用紫外检测触发级分收集。对于参 考在其它实施例或方法中的规程的合成,纯化可以变化:一般而言, 选择用于洗脱液/梯度的溶剂和溶剂比以提供适当的rf或保留时间。
[0375]
从lcms分析报告质谱法数据。通过大气压化学电离(apci)、电 喷射电离(esi)、电子碰撞电离(ei)或电子散射电离(es)源进行质谱法 (ms)。将质子核磁光谱法(1h nmr)化学位移以相对于四甲基硅烷往底 场的百万分之份数给出,并在300、400、500或600mhz varian、 bruker或jeol波谱仪上记录。以参照氘化溶剂残余峰(氯仿,7.26ppm; cd2hod,3.31ppm;乙腈-d2,1.94ppm;二甲亚砜-d5,2.50ppm;dho, 4.79ppm)的百万分之份数(ppm,δ)表达化学位移。峰形状被描述如下: s,单峰;d,双峰;t,三重峰;q,四重峰;quin,五重峰;m,多 重峰;br s,宽单峰;app,明显。通常如上所述在berger分析仪器 上获取分析sfc数据。使用1dm池子在perkinelmer 343型旋光计 上获取旋光度数据。由quantitative technologies inc.进行微分析,并 且是在计算值的0.4%内。
[0376]
除非另外指出,在室温(约23摄氏度)进行化学反应。
[0377]
除非另外指出,所有反应物商业得到且不经进一步纯化地使用, 或使用文献中已知的方法制备。
[0378]
术语“浓缩”、“蒸发”和“在真空中浓缩”表示用小于60℃的浴温度 在旋转蒸发器
上在减压下除去溶剂。缩写“min”和“h”分别代表“分钟
”ꢀ
和“小时”。术语“tlc”表示薄层色谱法,“室温或环境温度”是指 18-25℃之间的温度,“gcms”表示气相色谱法-质谱法,“lcms”表示 液相色谱法-质谱法,“uplc”表示超效液相色谱法,“hplc”表示高 效液相色谱法,和“sfc”表示超临界流体色谱法。
[0379]
可以在parr振荡器中在增压氢气下进行氢化,或在thales-nanoh-cube流式氢化设备中在全氢和1-2ml/min之间的流速在指定的温 度进行氢化。
[0380]
使用在规程中指出的方法测量hplc、uplc、lcms、gcms 和sfc保留时间。
[0381]
在某些实施例中,进行手性分离以分离本发明的某些化合物的对 映异构体或非对映异构体(在一些实施例中,分离的对映异构体根据其 洗脱顺序命名为ent-1和ent-2;类似地,分离的非对映异构体指定 为diast-1和diast-2,根据它们的洗脱顺序)。在一些实施例中, 使用旋光计测量对映异构体的旋光度。根据其观察到的旋光数据(或其 比旋光数据),具有顺时针旋光的对映异构体被命名为(+)-对映异构体, 且具有逆时针旋光的对映异构体被命名为(-)-对映异构体。外消旋的化 合物通过绘制的或描述的立体化学的缺失来指示,或通过邻近结构的 (
±
)的存在来指示;在该后一种情况下,指示的立体化学代表构成外消 旋混合物的两种对映异构体中的仅一种。
[0382]
使用由acd/chemsketch 2019.1.1,file version c05h41,build110712(advanced chemistry development,inc.,toronto,ontario, 加拿大)提供的命名约定,命名下面描述的化合物和中间体。由 acd/chemsketch 2019.1.1提供的命名约定是本领域技术人员众所周 知的,且据信由acd/chemsketch 2019.1.1提供的命名约定通常符合 iupac(international union for pure and applied chemistry)有机化 学命名推荐和cas index规则。
[0383]
实施例1:碳酸(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基甲酯(1)
[0384][0385]
将n-[(2s)-1-({(2s)-4-羟基-3-氧代-1-[(3s)-2-氧代吡咯烷-3-基]丁
ꢀ‑
2-基}氨基)-4-甲基-1-氧代戊烷-2-基]-4-甲氧基-1h-吲哚-2-甲酰胺(c1) (参见hoffman,r.l.等人,pct国际申请2005113580,2005年12月 1日;30mg,63μmol)在四氢呋喃(0.64ml)中的0℃溶液用n,n-二异丙 基乙胺(11μl,63μmol)处理,随后用氯甲酸甲酯(4.91μl,63.5μmol)处 理。使反应混合物温热至室温过夜,此后加入额外当量的氯甲酸甲酯。 3天后,因为反应仍然不完全,加入n,n-二甲基甲酰胺(0.2ml);4 小时后,将反应混合物用二氯甲烷稀释,并用1m盐酸洗涤。将有机 层经硫酸钠干燥,过滤,并在真空中浓缩。通过硅胶色谱法(梯度:0% 至10%的甲醇在二氯甲烷中的溶液)纯化,得到作为固体的碳酸 (3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁基甲酯(1)。收率:24mg,45μmol,71%. lcms m/z 531.4[m+h]
+
.1h nmr(400mhz,甲醇-d4)δ
7.28(br s, 1h),7.14(dd,abx系统的组分,j=8,8hz,1h),7.02(d,ab四重峰的 一半,j=8.3hz,1h),6.50(d,j=7.7hz,1h),4.91(ab四重峰,j
ab
= 17.4hz,δν
ab
=10.1hz,2h),4.66-4.57(m,2h),3.92(s,3h),3.76(s, 3h),3.29-3.20(m,2h),2.61-2.50(m,1h),2.33-2.22(m,1h),2.09 (ddd,j=14.2,11.2,4.7hz,1h),1.88-1.66(m,5h),1.03(d,j=6.1hz, 3h),0.99(d,j=6.2hz,3h)。
[0386]
实施例2:碳酸(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基丙烷-2-基酯(2)
[0387][0388]
将c1(15mg,32μmol)在四氢呋喃(0.32ml)中的0℃溶液用4-甲 基吗啉(4.2μl,38μmol)处理,随后用氯甲酸2-丙酯在甲苯中的溶液(1.0 m;34.8μl,34.8μmol)处理。将反应混合物温热至室温;5小时以后, 加热,并继续在40℃搅拌过夜,此后将反应混合物用二氯甲烷稀释和 用10%硫酸氢钾水溶液处理。在有机层已经经硫酸钠干燥以后,将它 过滤,并将滤液在真空中浓缩。通过反相hplc(柱:waters sunfirec18,19x 100mm,5μm;流动相a:含有0.05%三氟乙酸的水;流动 相b:含有0.05%三氟乙酸的乙腈;梯度:5%至95%b历时8.54分钟, 然后95%b保持1.46分钟;流速:25ml/分钟)纯化,得到碳酸 (3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁基丙烷-2-基酯(2)。收率:14.5mg, 26.0μmol,81%.lcms m/z 559.5[m+h]
+
.保留时间:2.73分钟(分析 条件。柱:waters atlantis dc18,4.6x 50mm,5μm;流动相a:含有 0.05%三氟乙酸的水(v/v);流动相b:含有0.05%三氟乙酸的乙腈 (v/v);梯度:5.0%至95%b,线性历时4.0分钟,然后95%b保持1.0 分钟;流速:2ml/分钟)。
[0389]
实施例3:碳酸(3s)-4-[(3s)-1-乙酰基-2-氧代吡咯烷-3
‑ꢀ
基]-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代丁 基甲酯
[0390]
实施例4:碳酸(3s)-4-[(3s)-1-{(1s)-1-[(甲氧基羰基)氧基]乙基}-2
‑ꢀ
氧代吡咯烷-3-基]-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代丁基甲酯
[0391]
实施例5:碳酸乙基(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯(5)
[0392][0393]
将氯甲酸乙酯(29.9mg,0.276mmol)和三乙胺(42.8mg,0.423 mmol)加入c1
(100mg,0.212mmol)在二氯甲烷(4.0ml)中的0℃溶液 中。将反应混合物在20℃搅拌2小时,此后将它用水(3ml)稀释,并 用二氯甲烷(3x 3ml)萃取。将合并的有机层用饱和氯化钠水溶液洗 涤,经硫酸钠干燥,在真空中浓缩,并与使用c1进行的类似反应的 产物(50.0mg,0.106mmol)合并。使用反相hplc(柱:agela durashellc18,40x 150mm,5μm;流动相a:0.225%甲酸在水中;流动相b:乙 腈;梯度:26%至66%b;流速:50ml/分钟)纯化,得到碳酸乙基(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁酯(5)作为白色固体。合并的收率:48.0 mg,88.1μmol,28%.lcms m/z 545.3[m+h]
+
.1h nmr(400mhz, dmso-d6)δ11.59(d,j=2.4hz,1h),8.59(d,j=7.9hz,1h),8.46(d, j=7.7hz,1h),7.66(s,1h),7.37(d,j=2.4hz,1h),7.09(dd,abx系 统的组分,j=8,8hz,1h),7.00(d,ab四重峰的一半,j=8.2hz,1h), 6.50(d,j=7.6hz,1h),4.89(ab四重峰,j
ab
=17.3hz,δν
ab
=21.6hz, 2h),4.52-4.37(m,2h),4.13(q,j=7.1hz,2h),3.88(s,3h),3.19
‑ꢀ
3.03(m,2h),2.37-2.25(m,1h),2.13-2.03(m,1h),1.98(ddd,j=14, 11,4hz,1h),1.79-1.50(m,5h),1.21(t,j=7.1hz,3h),0.94(d,j= 6.2hz,3h),0.89(d,j=6.2hz,3h)。
[0394]
实施例6:(3s)-3-[(2s)-4-[(甲氧基羰基)氧基]-2-({n-[(4-甲氧基
ꢀ‑
1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-3-氧代丁基]-2-氧代吡咯烷-1
‑ꢀ
甲酸甲酯
[0395]
lcms m/z 589.5[m+h]
+
.保留时间:2.77分钟(分析条件。柱: waters atlantis dc18,4.6x 50mm,5μm;流动相a:含有0.05%三氟 乙酸的水(v/v);流动相b:含有0.05%三氟乙酸的乙腈(v/v);梯度: 5.0%至95%b,线性历时4.0分钟,然后95%b保持1.0分钟;流速: 2ml/分钟)。
[0396]
实施例7和8:碳酸叔丁基(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基) 羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯(7)和 (3s)-3-[(2s)-4-[(叔丁氧基羰基)氧基]-2-({n-[(4-甲氧基-1h-吲哚-2-基) 羰基]-l-亮氨酰基}氨基)-3-氧代丁基]-2-氧代吡咯烷-1-甲酸叔丁酯(8)
[0397][0398][0399]
将4-(二甲基氨基)吡啶(0.13mg,1.10μmol)加入c1(26.8mg, 56.7μmol)和二碳酸
二叔丁酯(12mg,55μmol)在四氢呋喃(0.55ml)中 的溶液中。已经将反应混合物搅拌1小时40分钟以后,将它在真空中 浓缩和通过硅胶色谱法(梯度:0%至100%的乙酸乙酯在庚烷中)纯化 以得到碳酸叔丁基(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨 酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯(7)作为固体。收率: 7.4mg,13μmol,24%.lcms m/z 573.4[m+h]
+
.1h nmr(400mhz, 氯仿-d),特征峰:δ9.53(br s,1h),8.64(d,j=5.9hz,1h),7.17 (dd,abx系统的组分,j=8,8hz,1h),7.10(br d,j=2hz,1h),6.99 (d,ab四重峰的一半,j=8.3hz,1h),6.83(br d,j=8.2hz,1h),6.48 (d,j=7.8hz,1h),6.10(br s,1h),4.84(ab四重峰,j
ab
=17.2hz, δν
ab
=41.0hz,2h),4.83-4.74(m,1h),4.55-4.46(m,1h),3.93(s, 3h),3.34-3.16(m,2h),2.47-2.25(m,2h),1.48(s,9h),1.01-0.94(m, 6h)。
[0400]
也分离了8,作为固体。1h nmr(400mhz,氯仿-d),特征峰:δ 8.04(br s,1h),7.76(br d,j=6.5hz,1h),7.09(d,ab四重峰的一半,j =8.3hz,1h),6.70(br s,1h),6.49(d,j=7.8hz,1h),4.78(ab四重峰, j
ab
=17.8hz,δν
ab
=33.3hz,2h),4.38-4.28(m,1h),3.94(s,3h), 3.82-3.69(m,1h),3.38-3.28(m,1h),3.27-3.15(m,1h),2.30-2.17 (m,1h),2.04-1.88(m,2h),1.63(s,9h),1.61(s,9h),1.03(d,j=6.6 hz,3h),0.96(d,j=6.5hz,3h)。
[0401]
通过反相hplc(柱:waters sunfire c18,19x 100mm,5μm;流 动相a:含有0.05%三氟乙酸的水;流动相b:含有0.05%三氟乙酸的 乙腈;梯度:45%至85%b历时8.5分钟,然后85%至95%b历时0.5 分钟,然后95%b保持1.0分钟;流速:25ml/分钟)进一步纯化该批 次的8,得到(3s)-3-[(2s)-4-[(叔丁氧基羰基)氧基]-2-({n-[(4-甲氧基-1h
‑ꢀ
吲哚-2-基)羰基]-l-亮氨酰基}氨基)-3-氧代丁基]-2-氧代吡咯烷-1-甲酸 叔丁酯(8)。收率:13.5mg,20.1μmol,36%.lcms m/z 673.7[m+h]
+
. 保留时间:3.43分钟(分析条件。柱:waters atlantis dc18,4.6x 50mm, 5μm;流动相a:含有0.05%三氟乙酸的水(v/v);流动相b:含有0.05% 三氟乙酸的乙腈(v/v);梯度:5.0%至95%b,线性历时4.0分钟,然后 95%b保持1.0分钟;流速:2ml/分钟)。
[0402]
该反相hplc纯化也提供了三-叔丁基氧基羰基衍生物 2-{[(2s)-1-({(2s)-1-[(3s)-1-(叔丁氧基羰基)-2-氧代吡咯烷-3-基]-4-[(叔 丁氧基羰基)氧基]-3-氧代丁-2-基}氨基)-4-甲基-1-氧代戊烷-2-基]氨甲 酰基}-4-甲氧基-1h-吲哚-1-甲酸叔丁酯.收率:6.2mg,8.0μmol,14%. 1
h nmr(400mhz,氯仿-d)δ7.70(br d,j=8.4hz,1h),7.55(d,j= 8.5hz,1h),7.33-7.25(m,1h,假定;被溶剂峰部分地遮蔽),6.95(s, 1h),6.77-6.66(m,1h),6.67(d,j=8.0hz,1h),4.90(ab四重峰,j
ab
=17.5hz,δν
ab
=41.3hz,2h),4.72-4.62(m,2h),3.93(s,3h),3.71
‑ꢀ
3.61(m,1h),3.54-3.43(m,1h),2.54-2.42(m,1h),1.63(s,9h),1.48 (s,9h),1.48(s,9h),1.00(d,j=6.4hz,6h)。预纯化的样品中第2个 (非-8)峰的lcms:m/z 773.8[m+h]
+
。
[0403]
实施例9:碳酸(1r)-1-({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基) 乙基甲酯
[0404]
实施例10:碳酸(1r)-1-({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基) 乙基丙烷-2-基酯
[0405]
实施例11:碳酸(1r)-1-({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮
氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基) 丙基甲酯
[0406]
实施例12:碳酸(1r)-1-({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基) 丙基丙烷-2-基酯
[0407]
实施例13:碳酸({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基)甲基甲基 酯
[0408]
实施例14:碳酸({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基)甲基丙烷
ꢀ‑
2-基酯
[0409]
实施例15:碳酸乙基(1r)-1-({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2
‑ꢀ
基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基} 氧基)乙酯
[0410]
实施例16:碳酸乙基(1r)-1-({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2
‑ꢀ
基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基} 氧基)丙酯
[0411]
实施例17:碳酸乙基({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基) 甲酯
[0412]
实施例18:(3s)-3-[(2s)-4-{[(甲氧基羰基)氧基]甲氧基}-2-({n-[(4
‑ꢀ
甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-3-氧代丁基]-2-氧代吡 咯烷-1-甲酸甲酯
[0413]
实施例19:碳酸叔丁基(1r)-1-({(3s)-3-({n-[(4-甲氧基-1h-吲哚
ꢀ‑
2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基} 氧基)乙酯
[0414]
实施例20:碳酸叔丁基(1r)-1-({(3s)-3-({n-[(4-甲氧基-1h-吲哚
ꢀ‑
2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基} 氧基)丙酯
[0415]
实施例21:碳酸叔丁基({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基) 甲酯
[0416]
实施例22:碳酸{(3s)-3-[(2s)-4-{[(甲氧基羰基)氧基]甲氧 基}-2-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-3-氧代丁 基]-2-氧代吡咯烷-1-基}甲基甲基酯
[0417]
实施例23:碳酸{[(3s)-4-[(3s)-1-乙酰基-2-氧代吡咯烷-3
‑ꢀ
基]-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代丁 基]氧基}甲基甲基酯
[0418]
实施例24:碳酸{[(3s)-4-[(3s)-1-{(1r)-1-[(甲氧基羰基)氧基]乙 基}-2-氧代吡咯烷-3-基]-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨 酰基}氨基)-2-氧代丁基]氧基}甲基甲基酯
[0419]
实施例25:2,2-二甲基丙酸(1r)-1-({(3s)-3-({n-[(4-甲氧基-1h-吲 哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁 基}氧基)乙酯
[0420]
实施例26:2-甲基丙酸(1s)-1-({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2
‑ꢀ
基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基} 氧基)乙酯
[0421]
实施例27:丙酸(1s)-1-({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基) 乙酯
[0422]
实施例28:2,2-二甲基丙酸(1s)-1-({(3s)-3-({n-[(4-甲氧基-1h-吲 哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁 基}氧基)丙酯
[0423]
实施例29:2,2-二甲基丙酸(1s)-1-({(3s)-3-({n-[(4-甲氧基-1h-吲 哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁 基}氧基)丙酯
[0424]
实施例30:2-甲基丙酸(1s)-1-({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2
‑ꢀ
基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基} 氧基)丙酯
[0425]
实施例31:丙酸(1s)-1-({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基) 丙酯
[0426]
实施例32:2,2-二甲基丙酸({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基) 羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基) 甲酯
[0427]
实施例33:2,6-二甲基苯甲酸({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2
‑ꢀ
基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基} 氧基)甲酯
[0428]
实施例34:2-甲基丙酸({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基) 甲酯
[0429]
实施例35:d-缬氨酸({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基) 甲酯
[0430]
实施例36:n,n-二甲基甘氨酸({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2
‑ꢀ
基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基} 氧基)甲酯
[0431]
实施例37:丙酸({(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基}氧基)甲酯
[0432]
实施例38:(3s)-3-{(2s)-2-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l
‑ꢀ
亮氨酰基}氨基)-3-氧代-4-[(丙酰氧基)甲氧基]丁基}-2-氧代吡咯烷-1
‑ꢀ
甲酸甲酯
[0433]
实施例39:丙酸{[(3s)-4-[(3s)-1-乙酰基-2-氧代吡咯烷-3
‑ꢀ
基]-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代丁 基]氧基}甲酯
[0434]
实施例40:丙酸{[(3s)-4-[(3s)-1-{(1s)-1-[(甲氧基羰基)氧基]乙 基}-2-氧代吡咯烷-3-基]-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨 酰基}氨基)-2-氧代丁基]氧基}甲酯
[0435]
实施例41:1,4
’‑
联哌啶-1
’‑
甲酸(3s)-3-({n-[(4-甲氧基-1h-吲哚-2
‑ꢀ
基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯
[0436]
lcms m/z 667.6[m+h]
+
.保留时间:2.16分钟(分析条件。柱: waters atlantis dc18,4.6x 50mm,5μm;流动相a:含有0.05%三氟 乙酸的水(v/v);流动相b:含有0.05%三氟乙酸的乙腈(v/v);梯度: 5.0%至95%b,线性历时4.0分钟,然后95%b保持1.0分钟;流速: 2ml/分钟)。
[0437]
实施例42:[2-(二甲基氨基)乙基]氨基甲酸(3s)-3-({n-[(4-甲氧基
ꢀ‑
1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷
ꢀ‑
3-基]丁酯
[0438]
lcms m/z 587.6[m+h]
+
.保留时间:1.96分钟(分析条件。柱: waters atlantis dc18,4.6x 50mm,5μm;流动相a:含有0.05%三氟 乙酸的水(v/v);流动相b:含有0.05%三氟乙酸的乙腈(v/v);梯度: 5.0%至95%b,线性历时4.0分钟,然后95%b保持1.0分钟;流速: 2ml/分钟)。
[0439]
实施例43:[2-(二甲基氨基)乙基]甲基氨基甲酸(3s)-3-({n-[(4-甲 氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡 咯烷-3-基]丁酯三氟乙酸盐(43)
[0440][0441]
将1,1
’‑
羰基二咪唑(6.86mg,42.3μmol)加入c1(20mg,42μmol) 在二氯甲烷(0.42ml)中的溶液中。将反应混合物在室温搅拌1小时, 此后加入n,n,n'-三甲基乙烷-1,2-二胺(5.50μl,42.3μmol),并继续搅 拌过夜。将反应混合物已经在真空中浓缩以后,将残余物通过反相色 谱法(柱:waters sunfire c18,19x 100mm,5μm;流动相a:含有 0.05%三氟乙酸的水;流动相b:含有0.05%三氟乙酸的乙腈;梯度: 5%至95%b历时8.54分钟,然后95%b保持1.46分钟;流速:25ml/ 分钟)纯化,以得到[2-(二甲基氨基)乙基]甲基氨基甲酸(3s)-3-({n-[(4
‑ꢀ
甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代 吡咯烷-3-基]丁酯三氟乙酸盐(43)。收率:16.5mg,23.1μmol,55%. lcms m/z 601.6[m+h]
+
.保留时间:2.05分钟(分析条件。柱:watersatlantis dc18,4.6x 50mm,5μm;流动相a:含有0.05%三氟乙酸的 水(v/v);流动相b:含有0.05%三氟乙酸的乙腈(v/v);梯度:5.0%至 95%b,线性历时4.0分钟,然后95%b保持1.0分钟;流速:2ml/ 分钟)。
[0442]
实施例44:哌啶-1-甲酸(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯(44)
[0443][0444]
向c1(20mg,42μmol)在二氯甲烷(0.42ml)中的溶液中加入1,1
’‑ꢀ
羰基二咪唑(6.86mg,42.3μmol),随后加入4-甲基吗啉(4.65μl, 42.3μmol)。已经将反应混合物搅拌1小时以后,将它用哌啶(4.60μl, 46.5μmol)处理并将其搅拌过夜,此后将它在乙酸乙酯和10%硫酸氢钾 水溶液之间分配。将有机层经硫酸钠干燥,过滤,在真空中浓缩,并 进行反相hplc(柱:waters sunfire c18,19x 100mm,5μm;流动相 a:含有0.05%三氟乙酸的水;流动相b:含有0.05%三氟乙酸的乙腈; 梯度:5%至95%b历时8.54分钟,然后95%b保持1.46分钟;流速: 25ml/分钟),得到哌啶-1-甲酸(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯(44)。收 率:18.7mg,32.0μmol,76%.lcms m/z 584.5[m+h]
+
.保留时间:2.75 分钟(分析条件。柱:waters atlantis dc18,4.6x 50mm,5μm;流动相 a:含有0.05%三氟乙酸的水(v/v);流动相b:含有0.05%三氟乙酸的 乙腈(v/v);梯度:5.0%至95%b,线性历时4.0分钟,然后95%b保 持
1.0分钟;流速:2ml/分钟)。
[0445]
实施例45:哌啶-1-甲酸(3s)-4-[(3s)-1-(甲氧基羰基)-2-氧代吡咯 烷-3-基]-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧 代丁酯
[0446]
实施例46:哌啶-1-甲酸(3s)-4-[(3s)-1-乙酰基-2-氧代吡咯烷-3
‑ꢀ
基]-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代丁 酯
[0447]
实施例47:哌啶-1-甲酸(3s)-4-[(3s)-1-{(1s)-1-[(甲氧基羰基)氧基] 乙基}-2-氧代吡咯烷-3-基]-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代丁酯
[0448]
实施例48:碳酸(1s)-1-{(3s)-3-[(2s)-4-[(二甲氧基磷酰基)氧 基]-2-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-3-氧代丁 基]-2-氧代吡咯烷-1-基}乙基甲酯
[0449]
实施例49:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯(49)
[0450][0451]
步骤1.磷酸二叔丁基(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯(c2)的 合成。
[0452]
向n-[(2s)-1-({(2s)-4-羟基-3-氧代-1-[(3s)-2-氧代吡咯烷-3-基]丁
ꢀ‑
2-基}氨基)-4-甲基-1-氧代戊烷-2-基]-4-甲氧基-1h-吲哚-2-甲酰胺(c1) (参见hoffman,r.l.等人,pct国际申请2005113580,2005年12月 1日;2.82g,5.97mmol)和1h-四唑(1.25g,17.9mmol)在四氢呋喃(60 ml)中的0℃溶液中加入n,n-二丙烷-2-基亚磷酰胺二叔丁酯 (di-tert-butyl n,n-dipropan-2-ylphosphoramidoite)(7.53ml,6.62g, 23.9mmol)在四氢呋喃(0.5ml)中的溶液。将反应混合物历时30分钟 温热至室温并然后重新冷却至0℃。加入过氧化氢水溶液(50%w/w, 0.80ml,11.9mmol)并继续搅拌1小时。将反应混合物用水(30ml)稀 释并萃取进二氯甲烷(3x 20ml)中。将合并的有机层用硫代硫酸钠水 溶液(1m,20ml)和水(20ml)洗涤,然后经硫酸镁干燥,过滤,并在 真空中浓缩。硅胶色谱法(梯度:0%
至15%的甲醇在二氯甲烷中)得到 c2作为固体。收率:3.60g,5.42mmol,91%.lcms m/z 663.5[m-h]-。 使用来自在相同条件下运行的较小规模中试反应(pilot reaction)的 批次,得到该化合物的1h nmr数据。
[0453]1h nmr(400mhz,甲醇-d4,
31
p-解耦(decoupled))δ7.27(s,1h), 7.15(t,j=8.1hz,1h),7.02(d,j=8.3hz,1h),6.51(d,j=7.6hz, 1h),4.75(ab四重峰,j
ab
=17.3hz,δν
ab
=26.3hz,2h),4.70(dd,j= 10.3hz,3.7hz,部分地重叠在4.75ppm处的ab四重峰,1h),4.64 (dd,j=9.3hz,5.1hz,1h),3.93(s,3h),3.33-3.20(m,2h,假定;被 甲醇峰部分地遮蔽),2.62-2.53(m,1h),2.34-2.25(m,1h),2.11-2.01(m, 1h),1.90-1.65(m,5h),1.51-1.43[多重峰(1h)重叠在1.49处的2个增 宽的单峰(18h),19h总计],1.03(d,j=6.1hz,3h),1.00(d,j=6.1hz, 3h)。
[0454]
步骤2.磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯(1)的合成
[0455]
将三氟乙酸(2.07ml,27.1mmol)加入c2(3.60g,5.42mmol)在二 氯甲烷(54ml)中的0℃溶液中。搅拌1小时以后,将反应混合物在真 空中浓缩。在此时的lcms分析指示转化成49:lcms m/z 553.3 [m+h]
+
。将残余物在乙醇(15ml)中在75℃制浆30分钟,并然后在室 温制浆2小时。将固体通过过滤进行收集以得到磷酸二氢 (3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁酯(49)作为固体。收率:1.60g,2.90mmol, 54%.1h nmr(400mhz,甲醇-d4,
31
p-解耦)δ7.27(s,1h),7.15(t,j= 8.0hz,1h),7.03(d,j=8.3hz,1h),6.51(d,j=7.7hz,1h),4.80-4.56 (m,3h),{4.25-4.19(m)和4.02-3.81[多重峰重叠在3.93处的单峰 (3h)],4h总计},3.31-3.18(m,2h,假定;被甲醇峰部分地遮蔽), [2.63-2.52(m)和2.51-2.38(m),1h总计],2.36-2.24(m,1h),2.10-1.98 (m,1h),1.94-1.65(m,5h),1.04(d,j=5.6hz,3h),1.00(d,j=5.9hz, 3h)。保留时间:6.48分钟(分析条件。柱:waters xbridge c18,4.6x 150mm,5μm;流动相a:含有0.1%三氟乙酸的水;流动相b:含有 0.1%三氟乙酸的乙腈;梯度:5%b保持1.5分钟,然后5%至100%b 历时8.5分钟;流速:1.5ml/分钟)。
[0456]
还可以如下所述制备实施例49的化合物作为水合物(命名形式1)。
[0457]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯一水合物的合成
[0458]
步骤1:(4-甲氧基-1h-吲哚-2-羰基)-l-亮氨酸甲酯(49-b)的合成
[0459][0460]
向在20℃的夹套反应器中装入4-甲氧基-1h-吲哚-2-甲酸(49-a) (1.0当量,100g),乙腈(7ml/g,700ml),n-甲基咪唑(3.5当量,145.8 ml)和l-亮氨酸甲酯盐酸盐(1.15当量,109g)。逐滴加入在乙腈中的 1-丙烷膦酸环状酸酐(t3p)(50质量%,1.25当量,457ml),维持温度 低于30℃。将得到的混合物搅拌2h或直到通过uplc分析确定剩余《1%的
4-甲氧基-1h-吲哚-2-甲酸。将反应混合物穿过硅藻土垫过滤, 用乙腈(2ml/g,200ml)漂洗并将得到的滤液在减压下浓缩至~10 ml/g,维持温度小于50℃。加入水(1.5ml/g,150ml)并搅拌混合物 直到固体开始沉淀。逐滴加入另外的水(8ml/g,800ml)并将得到的浆 料造粒4h然后过滤。将固体用水(5ml/g,500ml)漂洗并在50℃干 燥,以95%收率得到作为白色固体的(4-甲氧基-1h-吲哚-2-羰基)-l-亮 氨酸甲酯(49-b)(158g)。
[0461]1h nmr(400mhz,dmso-d6)δ11.57(s,1h),8.62(d,j=7.8hz, 1h),7.35(d,j=1.7hz,1h),7.10(t,j=7.9hz,1h),7.01(d,j=8.2 hz,1h),6.51(d,j=7.6hz,1h),4.58-4.44(m,1h),3.89(s,3h),3.65 (s,3h),1.85-1.62(m,2h),1.64-1.53(m,1h),0.93(d,j=6.4hz,3h), 0.89(d,j=6.4hz,3h)。
[0462]
13
c nmr(101mhz,dmso-d6)δ173.14,161.20,153.65,137.88, 129.54,124.57,118.05,105.41,101.16,99.26,55.09,51.91,50.51,39.38, 24.44,22.85,21.16。
[0463]
步骤2:(4-甲氧基-1h-吲哚-2-羰基)-l-亮氨酸(49-c)的合成
[0464][0465]
向夹套反应器装入(4-甲氧基-1h-吲哚-2-羰基)-l-亮氨酸甲酯 (49-b)(1.0当量,158g),乙酸(5ml/g,790ml)和水(1ml/g,158ml)。 历时30分钟加入硫酸(1.5当量,39.7ml)。将混合物温热至50℃并保 持18-24h或直到反应含有大约5%剩余的(4-甲氧基-1h-吲哚-2-羰 基)-l-亮氨酸甲酯。在减压下蒸馏反应物以除去副产物甲醇或乙酸甲 酯。当反应达到结束时(少于1%剩余(4-甲氧基-1h-吲哚-2-羰基)-l-亮 氨酸甲酯),历时2h加入水(7ml/g,1106ml)。搅拌1h以后,将混 合物历时30分钟冷却至20℃,然后在20℃保持3h。将固体通过过滤 分离,用水(2x 2ml/g,316ml)漂洗。将固体在过滤器上干燥,然后 在真空烘箱中,在50℃干燥以91%收率产生(4-甲氧基-1h-吲哚-2-羰 基)-l-亮氨酸,(49-c)(137g)作为白色固体。
[0466]1h nmr(400mhz,dmso-d6)δ11.55(s,1h),8.50(d,j=8.0hz, 1h),7.34(d,j=1.6hz,1h),7.10(t,j=7.9hz,1h),7.01(d,j=8.2 hz,1h),6.51(d,j=7.6hz,1h),4.54-4.36(m,1h),3.89(s,3h),1.81
ꢀ‑
1.66(m,2h),1.63-1.53(m,1h),0.93(d,j=6.3hz,3h),0.89(d,j= 6.3hz,3h)。
[0467]
13
c nmr(101mhz,dmso-d6)δ174.22,161.11,153.63,137.82, 129.80,124.43,118.06,105.40,100.96,99.22,55.07,50.41,24.51,22.96, 21.13.(约39.5的峰在dmso峰下)。
[0468]
步骤3:(s)-2-((s)-2-(4-甲氧基-1h-吲哚-2-甲酰氨基)-4-甲基戊酰 氨基)-3-((s)-2-氧代吡咯烷-3-基)丙酸甲酯(49-e)的合成
[0469][0470]
向夹套反应器装入(4-甲氧基-1h-吲哚-2-羰基)-l-亮氨酸(49-c) (1.0当量,10.0g),(s)-2-氨基-3-((s)-2-氧代吡咯烷-3-基)丙酸甲酯4
‑ꢀ
甲基苯磺酸盐(49-d)(1.05当量,12.4g),2-羟基吡啶n-氧化物(0.025 当量,0.91g)和甲基乙基酮(5ml/g,50ml)。将得到的浆料冷却至0℃ 并装入n,n-二异丙基乙胺(2.25当量,12.9)。一次性装入1-(3-二甲基 氨基丙基)-3-乙基-碳二亚胺盐酸盐(1.2当量,7.86g)并将混合物搅拌 20min,然后温热至20℃和搅拌至少8h,或直到通过uplc确定剩 余小于1%(4-甲氧基-1h-吲哚-2-羰基)-l-亮氨酸(49-c)。将饱和盐水的 水溶液(23.5质量%,3.5ml/g,35ml)加入反应物,随后加入磷酸(1.5 当量,5.68g)在水(3ml/g,30ml)中的溶液。将得到的两相混合物搅 拌15分钟,然后分离各层。将有机层用饱和盐水溶液(23.5质量%,3.5 ml/g,35ml)洗涤。将有机层在减压下浓缩(250毫巴,50℃)至5ml/g, 然后装入甲基叔丁基醚(5ml/g,50ml)并重复蒸馏。装入另外的甲基 叔丁基醚(5ml/g,50ml)并将混合物历时15分钟冷却至35℃。然后, 缓慢地装入另外部分的甲基叔丁基醚(2.5ml/g,25ml),产生沉淀。 将浆料造粒30分钟,然后将甲基叔丁基醚(2.5ml/g,25ml)的最终部 分装入以达到大约4:1甲基叔丁基醚:甲基乙基酮的最终溶剂比。将最 终的浆料造粒30分钟,然后以0.25℃/min的速率冷却至10℃并保持 4h。将最终的浆料过滤,用甲基叔丁基醚(2.5ml/g,25ml)漂洗和在 过滤器上干燥,然后在真空烘箱中在25℃干燥。以75-85%收率分离 产物(s)-2-((s)-2-(4-甲氧基-1h-吲哚-2-甲酰氨基)-4-甲基戊酰氨 基)-3-((s)-2-氧代吡咯烷-3-基)丙酸甲酯(49-e),为甲基叔丁基醚溶剂化 物。
[0471]
步骤4:n-((s)-1-(((s)-4-氯-3-氧代-1-((s)-2-氧代吡咯烷-3-基)丁-2
‑ꢀ
基)氨基)-4-甲基-1-氧代戊烷-2-基)-4-甲氧基-1h-吲哚-2-甲酰胺(49-f) 的合成
[0472][0473]
给在20℃的夹套反应器装入叔丁基氯化镁在四氢呋喃中的溶液 (1m,21当量,32g)和n-甲基哌啶(10.5当量,1.71g)。将(s)-2-((s)-2-(4
‑ꢀ
甲氧基-1h-吲哚-2-甲酰氨基)-4-甲基戊酰氨基)-3-((s)-2-氧代吡咯烷
ꢀ‑
3-基)丙酸甲酯(49-e)(1.0当量,1.0g),氯乙酸(2.5当量,0.40g)和thf (10ml/g,10ml)的混合物通过加料漏斗加入反应器,维持温度低于 25℃。加入结束以后,保持混合物直到反应结束((49-e)的98%消耗)。 然后将反应物在减压下浓缩(150毫巴,温度维持低于30℃)至~20 ml/g,然后冷却至20℃。给第二个反应
器装入柠檬酸水溶液(25重量%, 20ml/g,20ml)和2-甲基四氢呋喃(10ml/g,10ml)并冷却至10℃。 将反应混合物缓慢地加入柠檬酸和2-甲基四氢呋喃,维持反应温度低 于15℃。结束后,将混合物温热至20℃并分离各层。将有机层用碳酸 氢钠水溶液(1.14m,10ml/g,10ml)洗涤,然后用更稀的碳酸氢钠水 溶液(0.6m,10ml/g,10ml)洗涤,然后用盐水(12质量%,10ml/g,10 ml)洗涤。将有机溶液在常压下浓缩10ml/g并用2-甲基四氢呋喃置 换。将混合物历时4h冷却至20℃并在20℃造粒,然后过滤和用2
‑ꢀ
甲基四氢呋喃(3ml/g,3ml)洗涤。将固体在真空烘箱中在50℃干燥 以得到n-((s)-1-(((s)-4-氯-3-氧代-1-((s)-2-氧代吡咯烷-3-基)丁-2-基) 氨基)-4-甲基-1-氧代戊烷-2-基)-4-甲氧基-1h-吲哚-2-甲酰胺(49-f),为 2-甲基四氢呋喃溶剂化物(~10重量%),63%收率。
[0474]
步骤5:磷酸二叔丁基((s)-3-((s)-2-(4-甲氧基-1h-吲哚-2-甲酰氨 基)-4-甲基戊酰氨基)-2-氧代-4-((s)-2-氧代吡咯烷-3-基)丁基)酯(49-g) 的合成
[0475][0476]
向在25℃的夹套反应器中装入n-((s)-1-(((s)-4-氯-3-氧代
ꢀ‑
1-((s)-2-氧代吡咯烷-3-基)丁-2-基)氨基)-4-甲基-1-氧代戊烷-2-基)-4
‑ꢀ
甲氧基-1h-吲哚-2-甲酰胺(49-f)(1.0当量,7.30g)、甲基乙基酮(12.5 ml/g,91ml)和n,n-二甲基甲酰胺(2.5ml/g,18ml)。然后,装入二 叔丁基磷酸钾(2.0当量,7.5g)和碘化钠(0.20当量,0.45g)。将混合物 搅拌48-72h直到通过uplc确定n-((s)-1-(((s)-4-氯-3-氧代-1-((s)-2
‑ꢀ
氧代吡咯烷-3-基)丁-2-基)氨基)-4-甲基-1-氧代戊烷-2-基)-4-甲氧基
ꢀ‑
1h-吲哚-2-甲酰胺(49-f)和对应的碘化物化合物以小于2%存在。装入 水(10ml/g,73ml),随后装入甲基叔丁基醚(5ml/g,37ml)。将两相 混合物搅拌5分钟,然后抛弃水层。将有机层用水(2x 10ml/g,73ml) 洗涤2次,然后在减压下浓缩至5ml/g,维持温度低于35℃。将甲基 乙基酮(15ml/g,110ml)装入反应器并将混合物再次在减压下浓缩至 5ml/g,维持温度低于35℃。将其重复第三次,或直到含水量》0.5%。 将混合物用甲基乙基酮(5ml/g,36ml)稀释并用于下一步。
[0477]1hnmr(400mhz,dmso-d6):δ11.55(d,j=1.8hz,1h),8.59 (d,j=8.0hz,1h),8.43(d,j=8.0hz,1h),7.63(s,1h),7.36(d,j= 1.6hz,1h),7.09(t,j=7.9hz,1h),7.00(d,j=8.2hz,1h),6.50(d,j =7.6hz,1h),4.74(dd,j=17.5,7.9hz,1h),4.61(dd,j=17.5,7.2hz, 1h),4.53-4.43(m,2h),3.88(s,3h),3.15-3.03(m,2h),2.32(m,1h), 2.06(m,1h),1.97(m,1h),1.72(m,2h),1.62(m,2h),1.52(m,1h), 1.40(s,9h),1.39(s,9h),0.94(d,j=6.2hz,3h),0.90(d,j=6.2hz, 3h)。
[0478]
13
cnmr(101mhz,dmso-d6):δ203.1(d,j=7.4hz),178.8, 173.4,161.6,154.1,138.3,130.3,124.9,118.5,105.9,101.7,99.7,82.6 (dd,j=7.2,4.4hz),68.7(d,j=5.8hz),55.5,54.0,51.9,49.1,37.7, 31.8,29.9,29.8,27.6,27.3,24.9,23.5,21.9。
[0479]
步骤6:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯甲基乙基酮溶剂 化物的合成
[0480][0481]
向来自前一步的磷酸二叔丁基((s)-3-((s)-2-(4-甲氧基-1h-吲哚-2
‑ꢀ
甲酰氨基)-4-甲基戊酰氨基)-2-氧代-4-((s)-2-氧代吡咯烷-3-基)丁基)酯 (49-g)在甲基乙基酮中的溶液中装入三氟乙酸(20当量,23ml)。将混 合物温热至30℃,或直到》98%的磷酸二叔丁基((s)-3-((s)-2-(4-甲氧基
ꢀ‑
1h-吲哚-2-甲酰氨基)-4-甲基戊酰氨基)-2-氧代-4-((s)-2-氧代吡咯烷
ꢀ‑
3-基)丁基)酯(49-g)(或对应的膦酸单叔丁酯)消耗已经发生。将混合 物冷却至20℃并造粒1h,然后过滤,并用甲基乙基酮(3ml/g,22ml) 洗涤。将产物在40℃干燥5h以得到磷酸二氢(3s)-3-({n-[(4-甲氧基
ꢀ‑
1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷
ꢀ‑
3-基]丁酯甲基乙基酮溶剂化物作为白色固体(4.9g),53%收率。
[0482]1hnmr(400mhz,dmso-d6):δ11.57(d,j=1.8hz,1h),8.52 (d,j=8.0hz,1h),8.42(d,j=8.0hz,1h),7.63(s,1h),7.36(d,j= 1.6hz,1h),7.09(t,j=7.8hz,1h),7.00(d,j=8.2hz,1h),6.50(d,j =7.6hz,1h),4.68(dd,j=17.6,8.1hz,1h),4.57(dd,j=17.6,7.2hz, 1h),4.53-4.43(m,2h),3.88(s,3h),3.15-3.03(m,2h),2.42(q,j=7.3 hz,2h),2.32(m,1h),2.06(s和m,4h),1.95(m,1h),1.77-1.51(m, 5h),0.94(d,j=6.2hz,3h),0.90(t,j=7.1hz,3h),0.89(d,j=6.2 hz,3h)。
[0483]
13
cnmr(101mhz,dmso-d6):δ209.3,203.9(d,j=7.4hz), 178.8,173.3,161.6,154.1,138.3,130.4,124.9,118.5,105.9,101.6,99.7, 68.2(d,j=4.8hz),55.5,54.0,51.9,37.8,36.3,31.2,29.8,27.6,24.9, 23.5,21.9,8.1。
[0484]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯甲基乙基酮溶剂化物的 粉末x-射线衍射图谱提供在图9中。
[0485]
步骤7:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯水合物的合成
[0486][0487]
向夹套反应器中加入磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2
‑ꢀ
基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯甲 基乙基酮溶剂化物(1.0当量,35.7g)和无水乙醇(15ml/g,536ml)。 历时30分钟将浆料温热至40℃并保持至少1h。取浆料样品以证实期 望的多晶型物存在。如果转化不完全,将浆料保持另外的时间。历时2h将浆料冷却至10℃并造粒2h,然后过滤,用乙醇(4ml/g,140ml) 洗涤。将固体在50℃干燥
过夜,以98%收率得到磷酸二氢 (3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁酯一水合物。
[0488]
如下所述使用粉末x-射线衍射、固态nmr和拉曼光谱法表征上 面制备的磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨 酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯水合物,命名形式 1。
[0489]
粉末x-射线衍射
[0490]
使用配备cu辐射源的bruker axs d8 endeavor衍射仪产生粉末 x-射线衍射图谱。将管电压和安培数分别设定至40kv和40ma。将 电动发散狭缝设置为11毫米的恒定照明。使用lynxeye xe-t能量 色散x-射线检测器检测衍射辐射,将位置敏感检测器(psd)开口设定 在4.00
°
。使用0.019
°
2θ的步长和0.2秒的每步时间,在cu波长在θ-θ 测角仪上在2.0-55.0度2-θ(
°
2θ)的范围内收集数据。样品准备用于分 析,方法是将它们放置在硅低背景小平板支架中并在数据收集过程中 以15rpm旋转。在diffrac.eva v5.0软件中分析数据。使用相对 强度≥5%的每个相应衍射图谱中的最强谱带的反射制备峰列表(list)。
ꢀ±
0.2
°
2θ的峰位置典型误差(usp-941)适用于该数据。与此测量相关的 微小误差可能由于多种因素而发生,包括:(a)样品制备(例如样品高 度),(b)仪器特征,(c)仪器校准,(d)操作员输入(例如在确定峰位置时) 和(e)材料的性质(例如择优取向和透明效应)。
[0491]
为了得到绝对峰位置,应当将粉末图谱与参考进行比对。这可能 是来自在室温解析的相同形式的晶体结构的模拟粉末图谱,或内部标 准物例如二氧化硅或刚玉。将形式1磷酸二氢(3s)-3-({n-[(4-甲氧基
ꢀ‑
1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷
ꢀ‑
3-基]丁酯水合物的收集的粉末图谱与含有内部标准物si(srm 640e) 的相同材料的粉末图谱进行比对。
[0492]
形式1磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯水合物的pxrd 曲线提供在图3中,且对应的2-θ峰列表和相对强度提供在下面表 pxrd-1中(值为
±
0.2 2-θ)。
[0493]
表pxrd-1:形式1的pxrd峰列表.
[0494][0495]
形式1的特征pxrd峰是分别在以下位置的峰:4.1和7.2;在4.1、 7.2和10.4;和在4.1、7.2、10.4和14.5 2-θ位置(每个是
±
0.2 2-θ)。
[0496]
固态nmr
[0497]
在bruker-biospin avance neo 400mhz(1h频率)nmr波谱仪 上进行固态nmr(ssnmr)分析。在12.5khz的魔角旋转速率在4mmmas探针上收集
13
c ssnmr波谱。将温度调节到20℃。用3ms cp 接触时间和3.5秒的再循环延迟记录交叉极化(cp)波谱。在波谱采集 期间应用约100khz的相位调制质子解耦场(decoupling field)。碳 波谱参考是相对于纯的四甲基硅烷,通过将来自外部α-甘氨酸样品的 高频信号设置为176.5ppm来进行。使用与
13
c波谱相同的仪器和探 针收集
15
n ssnmr波谱,旋转速率为12.5khz并将温度调节至20℃。 用10ms cp接触时间和3.5秒的再循环延迟记录交叉极化(cp)波谱。 氮波谱参考是相对于纯的硝基甲烷,通过将来自外部甘氨酸样品的信 号设定至-346.8ppm来进行。
13
c和
15
n固态nmr波谱分别提供在图 4和5中。
[0498]
使用acd labs 2017spectrus processor软件进行自动峰拾取,3%相对强度的阈值用于初步峰值选择。目视检查自动峰拾取的输出以 确保有效性,并在必要时手动进行调整。尽管本文报告了特定
13
c和 15
n ssnmr峰值,但由于仪器、样品和样品制备的差异,这些峰值确 实存在一个范围。结晶固体的
13
c和
15
n化学位移x-轴值的典型变异 性是在加或减0.2ppm的量级。本文报告的ssnmr峰高是相对强度。 ssnmr强度可以随实验参数的实际设置和样品的热史而变化。
[0499]
表nmr-1:形式1的
13
c ssnmr峰列表.
[0500][0501]
形式1的特征
13
c峰是在21.7、153.8和172.2ppm;在21.7、153.8、 172.2和118.6ppm;和在21.7、153.8、172.2、118.6和57.8ppm(每 个
±
0.2ppm)。
[0502]
表nmr-2:形式1的
15
n ssnmr峰列表.
[0503][0504]
形式1的特征
15
n峰是在-260.8和-256.9ppm;在-260.8、-256.9 和-248.0ppm;和在-260.8、-256.9、-248.0和-252.1ppm(每个
±
0.2 ppm)。
[0505]
拉曼光谱法
[0506]
使用连接至vertex 70波谱仪(bruker optik gmbh)的ram iift-raman模块收集拉曼光谱。仪器配有1064nm固态(nd:yag)激 光和液氮冷却的锗检测器。在数据采集之前,使用白光源以及聚苯乙 烯和萘参照物进行仪器性能和校准验证。
[0507]
在截短的nmr管中制备和分析样品。在测量过程中使用样品旋 转器(ventacon,uk)以使在数据收集过程中暴露于激光的材料的体积 最大化。将来自样品的反向散射的拉曼光谱信号优化,并使用500mw 的激光功率在2cm-1
的波谱分辨率收集数据。应用blackmann-harris 4-term切趾函数以使波谱畸变最小化。产生在3500-50cm-1
之间的波 谱,相应地调节扫描数目以确保适当的信噪比。
[0508]
通过将最强峰的强度设定至2.00,使波谱标准化。然后使用opusv8.2软件(bruker optik gmbh)中的自动峰拾取功能鉴定峰,将灵敏 度设定至2%。将峰位置和相对峰强度提取并制表。采用该实验构型 峰位置的变异性是在
±
2cm-1
内。
[0509]
由于ft-拉曼光谱和色散拉曼光谱是类似的技术,预期在此文件 中关于ft-拉曼光谱报告的峰位置与使用色散拉曼光谱测量观察到的 那些是一致的,假定适当的仪器校准。
[0510]
表拉曼-1:从形式1收集的ft拉曼光谱所提取的峰列表.
[0511]
[0512][0513]
形式1的特征拉曼峰是在1271、1421和1217cm-1
;在1271、1421、 1217和1640cm-1
;在1271、1421、1217、1640和3074cm-1
(每个
±
2 cm-1
)。
[0514]
如上所述的粉末x-射线衍射、固态nmr和拉曼光谱法技术也用 于表征磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰 基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯甲基乙基酮溶剂化 物,其经过重新处理以提高其结晶度。在固态表征之前进行磷酸二氢 (3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁酯甲基乙基酮溶剂化物的重新处理以提 高样品的结晶度。这通过磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2
‑ꢀ
基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯甲 基乙基酮溶剂化物在甲基乙基酮中的40℃至10℃热-冷重新制浆循环 实现,使用0.5℃/min加热和冷却速率,在每个温度具有10分钟保持 阶段,历时24小时。然后表征得到的结晶磷酸二氢(3s)-3-({n-[(4-甲 氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡 咯烷-3-基]丁酯甲基乙基酮溶剂化物。
[0515]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯甲基乙基酮溶剂化物的 粉末x-射线衍射
[0516]
将磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰 基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯甲基乙基酮溶剂化物 的收集的粉末x-射线衍射图谱与含有内部标准物si(srm 640e)的相 同材料的粉末图谱比对。该材料的pxrd曲线提供在图10中且对应 的峰列表提供在表pxrd-2中。mek溶剂化物的特征峰是在7.7、8.1、17.0、23.1和25.8 2-θ位置的峰。
[0517]
表pxrd-2:mek溶剂化物的pxrd峰列表。
[0518][0519]
mek溶剂化物的特征pxrd峰包括但不限于7.7、8.1和23.1; 7.7、8.1、17.0和23.1;和7.7、8.1、17.0、23.1和25.8(每个度2-θ
±
0.2 度2-θ)。
[0520]
表nmr-3:mek溶剂化物的
13
c ssnmr峰列表.
[0521][0522]
mek溶剂化物的特征
13
c ssnmr峰包括7.2、206.4和215.8;7.2、 206.4、215.8和42.2;和7.2、206.4、215.8、42.2和101.2(每个ppm
±
0.2 ppm)。
[0523]
表nmr-4:mek溶剂化物的
15
n ssnmr峰列表.
[0524]
15
nδ(ppm)相对强度,%-272.996.5-266.4100.0-251.893.7-244.693.1
[0525]
表拉曼-2:从mek溶剂化物收集的ft拉曼光谱所提取的峰列表
[0526][0527]
mek溶剂化物的特征拉曼峰包括但不限于在以下位置的那些: 1511、1644和3081cm-1
;1511、1644、3081和1265cm-1
;和1511、 1644、3081、1265和446cm-1
;每个
±
2cm-1
。
[0528]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯无定形游离酸形式 (pf-07304814的无定形游离酸形式)的制备;
[0529]
无定形游离酸pf-07304814-00如下制备:在具有搅拌棒的duran 烧瓶中,将220ml水加入1g磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲 哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁 酯(pf-07304814)。将样品在500rpm在环境条件搅拌1小时,然后将 溶液使用注射器式滤器过滤。随后将来自过滤溶液的水使用genevacez-2elite蒸发器通过在真空下离心蒸发在18小时中除去。回收了约 0.9g固体材料。
[0530]
粉末x-射线衍射
[0531]
使用配备cu辐射源的bruker axs d8 endeavor衍射仪产生粉末 x-射线衍射图谱。将管电压和安培数分别设定至40kv和40ma。将 电动发散狭缝设置为11毫米的恒定照明。使用lynxeye xe-t能量 色散x-射线检测器检测衍射辐射,将位置敏感检测器(psd)开口设定 在4.00
°
。使用0.019
°
2θ的步长和0.2秒的每步时间,在cu波长在θ-θ 测角仪上在2.0-55.0度2-θ(
°
2θ)的范围内收集数据。样品准备用于分 析,方法是将它们放置在硅低背景小平板支架中并在数据收集过程中 以15rpm旋转。在diffrac.eva v5.0软件中分析数据。收集的api 的pxrd曲线提供在图1中,是无定形材料的典型。
[0532]
固态nmr
[0533]
使用4mm mas探针在8khz的魔角旋转速率,在bruker avanceiii hd 400mhz(1h
频率)nmr波谱仪上进行固态nmr(ssnmr)分 析,将温度调节到20℃。用1ms cp接触时间和2秒的再循环延迟记 录具有toss旋转旁带抑制的
13
c交叉极化(cp)波谱。在波谱采集期 间应用约100khz的相位调制质子解耦场。碳波谱参考是相对于纯的 四甲基硅烷,通过将来自外部金刚烷样品的高频信号设置为38.5ppm 来进行。用1ms cp接触时间和2秒的再循环延迟记录
15
n cp波谱。 氮波谱参考是相对于纯的硝基甲烷,通过将来自外部甘氨酸样品的信 号设定至-346.8ppm来进行。使用与
13
c和
15
n波谱相同的mas探针 收集
31
p波谱,旋转速率为10khz。用4ms cp接触时间和2秒的再 循环延迟记录
31
p cp波谱。磷波谱参考是相对于85%h3po4的外部样 品。
[0534]
使用acd labs 2019spectrus processor软件进行峰拾取。本文 报告的ssnmr峰高是相对强度。ssnmr强度可以随实验参数的实际 设置和样品的热史而变化。由于许多
13
c峰的相对高的线宽度和噪音, 一些线的引用峰位置存在估计的
±
0.4-0.5ppm范围。在204ppm的共 振是特别宽的且引用的峰位置可能是
±
1.5ppm。对于其余峰,估计误 差为
±
0.2ppm。对于
31
p峰,估计误差为
±
0.2ppm。
15
n化学位移信息 衍生自观察的波谱的解卷积(deconvolution),且引用的强度信息应 当仅用作引导。估计
15
n误差为
±
1.5ppm。
[0535]
pf-07304814无定形游离酸的
13
c ssnmr峰列表(估计误差为
ꢀ±
0.2ppm,除非另外说明)
[0536][0537]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯的无定形游离酸形式的 特征
13
c ssnmr峰是在175.0
±
0.4、204
±
1.5和181.8
±
0.4ppm的
13
cssnmr峰;在175.0
±
0.4、204
±
1.5、181.8
±
0.4和54.8
±
0.2ppm的峰; 和在175.0
±
0.4、204
±
1.5、181.8
±
0.4、54.8
±
0.2和162.9
±
0.2ppm的峰; 以及在175.0
±
0.4和204
±
1.5的
13
c ssnmr峰和在-0.8
±
0.2ppm的
31
p 峰的组合。
[0538]
无定形游离酸的
15
n ssnmr发现了在-264
±
1.5ppm(具有100% 的相对强度)和-249
±
1.5ppm的峰。
[0539]
无定形游离酸的
31
p ssnmr发现了在-0.8
±
0.2ppm处的峰。
[0540]
拉曼光谱法
[0541]
使用连接至vertex 70波谱仪(bruker optik gmbh)的ram iift-raman模块收集拉曼光谱。仪器配有1064nm固态(nd:yag)激 光和液氮冷却的锗检测器。在数据采集之前,使用白光源以及聚苯乙 烯和萘参照物进行仪器性能和校准验证。
[0542]
在截短的nmr管中制备和分析样品。在测量过程中使用样品旋 转器(ventacon,
(wfi)并混合。加入4.575ml 1m naoh并混合直到完全溶解。将溶 液用wfi补充至25ml体积并混合。使用0.2μm灭菌级过滤器过滤 溶液并填充进玻璃小瓶(目标体积为10.9ml)。将小瓶放在托盘上并将 托盘加载进冷冻干燥器(lyostar)。将冷冻干燥器密封并将存放温度 (shelf temperature)以0.5℃/分钟的速率冷却至-45℃并保持1小时。 将真空压设定至150mtorr并将冷冻干燥器保持1小时。然后将存放 温度以0.5℃/分钟的速率加热至25℃并保持20小时。初步干燥结束后, 将存放温度以0.5℃/分钟的速率加热至40℃并保持10小时。在第二次 干燥结束时,给腔室回填氮并将存放温度冷却至5℃。在冷冻干燥器 中给样品盖上塞子,释放真空,并将样品取出、盖帽和贴标签。
[0551]
在冷冻干燥以后,样品呈现为饼状至粉末状,具有白色至灰白色/ 黄色/棕色颜色。冷冻干燥的样品显示回熔、收缩和皱缩的微小迹象。
[0552]
粉末x-射线衍射
[0553]
使用配有铜(cu)辐射源的bruker axs d8 endeavor衍射仪产生 粉末x-射线衍射图谱。将管电压和安培数分别设定至40kv和40ma。 将电动发散狭缝设置为11毫米的恒定照明。使用lynxeye xe-t能 量色散x-射线检测器检测衍射辐射,将位置敏感检测器(psd)开口设 定在4.00
°
。使用0.019
°
2θ的步长和0.2秒的每步时间,在cu波长在 θ-θ测角仪上在2.0-55.0度2-θ(
°
2θ)的范围内收集数据。样品准备用于 分析,方法是将它们放置在硅低背景小平板支架中并在数据收集过程 中以15rpm旋转。在diffrac.eva v5.0软件中分析数据。收集的 api的pxrd曲线提供在图1中,并与具有在3.3
°
2θ在低角度观察到 的单个宽峰的无定形卤化物(halo)一致。
[0554]
固态nmr
[0555]
使用4mm mas探针在12.5khz的魔角旋转速率,在brukeravance neo 400mhz(1h频率)nmr波谱仪上进行固态nmr (ssnmr)分析,将温度调节到25℃。用3ms cp接触时间和3秒的再 循环延迟记录
13
c交叉极化(cp)波谱。在波谱采集期间应用约100khz 的相位调制质子解耦场。碳波谱参考是相对于纯的四甲基硅烷,通过 将来自l-丙氨酸外部样品的高频率信号设定至177.8ppm进行。用10 ms cp接触时间和3秒的再循环延迟记录
15
n cp波谱。氮波谱参考是 相对于纯的硝基甲烷,通过将来自外部甘氨酸样品的信号设定至
ꢀ‑
346.8ppm进行。用4ms cp接触时间和3秒的再循环延迟记录
31
p cp 波谱。磷波谱参考是相对于磷酸二氢铵的外部样品,将信号设定至0.8 ppm。
[0556]
使用acd labs 2019spectrus processor软件进行峰拾取。本文 报告的ssnmr峰高是相对强度。ssnmr强度可以随实验参数的实际 设置和样品的热史而变化。由于许多
13
c峰的相对高的线宽度,组合 共振重叠和噪音水平,一些峰的引用峰位置存在估计
±
0.4-0.5ppm范 围。在~208ppm的共振是特别宽的和有噪声的,所以引用的峰位置可 能是
±
1.5ppm。对于其余的
13
c峰,估计误差为
±
0.2ppm。对于
31
p 峰,估计误差为
±
0.2ppm。
15
n化学位移信息衍生自观察的波谱的解 卷积,且引用的强度信息应当仅用作引导。估计
15
n误差为
±
1.5ppm。
[0557]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯的无定形钠盐的特征峰 是126.0
±
0.4ppm、181.0
±
0.4ppm、208.0
±
1.5ppm、174.1
±
0.4ppm和 163.1
±
0.2ppm(对于
13
c)和1.9
±
0.2ppm(对于
31
p)。
[0558]
拉曼光谱法
[0559]
使用连接至vertex 70波谱仪(bruker optik,gmbh)的ram iift-raman模块收集拉曼光谱。仪器配有1064nm固态(nd:yag)激 光和液氮冷却的锗检测器。在数据采集之前,使用白光源以及聚苯乙 烯和萘参照物进行仪器性能和校准验证。
[0560]
从装它的玻璃小瓶直接分析样品。将来自样品的反向散射的拉曼 光谱信号优化,并使用750mw的激光功率在2cm-1
的波谱分辨率收 集数据。应用blackmann-harris 4-term切趾函数以使波谱畸变最小 化。产生在3500-50cm-1
之间的波谱,相应地调节扫描数目以确保适 当的信噪比。取三个单独测量以确保测量代表整批材料。
[0561]
使用opus v8.2软件中的平均功能将三个测量平均化,并通过将 最强峰的强度设定至2.00将该波谱标准化。然后使用opus v8.2软件 (bruker optik gmbh)中的自动峰拾取功能鉴定峰,将灵敏度设定至 2%。将峰位置和相对峰强度提取并制表。采用该实验构型的峰位置的 变异性是在
±
2cm-1
内。
[0562]
由于ft-拉曼光谱和色散拉曼光谱是类似的技术,预期在此文件 中关于ft-拉曼光谱报告的峰位置与使用色散拉曼光谱测量观察到的 那些是一致的,假定适当的仪器校准。
[0563]
调制dsc
[0564]
通过调制的示差扫描量热法(mdsc),测量磷酸二氢 (3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯的无定形钠盐的玻璃化转变温度。将 重1.8mg的样品放在ta instruments t zero aluminium pan,将它 轻轻下压以改善与盘底的接触并允许热通过样品更好的流动。使用tzero aluminium lid包封盘。使用ta instruments discovery dsc利 用下述规程进行分析。将温度在25℃等温地保持5分钟,并然后以 2℃/min增加至200℃,同时在60秒的阶段中应用0.636℃的温度调制。
[0565]
使用trios(4.5.0.42498版)进行分析。在反向热流中在152.8℃观 察到具有中点(半高度)的玻璃化转变。
[0566]
表nmr-5:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯无定形 钠盐的
13
c ssnmr峰列表.
[0567][0568]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯无定形钠盐的特征
13
cssnmr峰包括在126.0
±
0.4ppm、181.0
±
0.4ppm和208.0
±
1.5ppm的 峰;在126.0
±
0.4ppm、181.0
±
0.4ppm、208.0
±
1.5ppm和174.1
±
0.4ppm的峰;和在126.0
±
0.4ppm、181.0
±
0.4ppm、208.0
±
1.5ppm、174.1
±
0.4ppm和163.1
±
0.2ppm的峰。
[0569]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯无定形钠盐的
15
nssnmr产生在δ-263ppm(具有100%的相对强度)和-248ppm(具有37%的相对强度)的
15
n峰。
[0570]
由于差的信噪比(s/n)和宽的重叠信号,存在于所述误差内的不足的差异以相对于磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯的其它形式选择磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯无定形钠盐的诊断
15
nssnmr峰。
[0571]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯无定形钠盐的
31
pssnmr产生在δ1.9ppm
±
0.2ppm(具有100%的相对强度)的特征
31
p峰。
[0572]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯无定形钠盐的特征
13
cssnmr和
31
pssnmr峰包括在126.0
±
0.4ppm、181.0
±
0.4ppm的
13
c峰和在1.9ppm
±
0.2ppm的
31
p峰。
[0573]
表拉曼-3:从磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯无定形钠盐收集的ft拉曼光谱所提取的峰列表
[0574][0575]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯二甲基亚砜(dmso)溶剂化物的制备:给在20℃的夹套反应器装入磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯甲基乙基酮溶剂化物(1.0当量,50g),二甲基亚砜(100ml,2ml/g)和2-丙醇(100ml,2ml/g)。将混合物在20℃搅拌直到得到澄清溶液。将溶液加热至30℃并加入异丙醇(800ml,16ml/g)。将得到的浆料历时2h冷却至10℃并造粒最小1h,然后过滤并用2-丙醇(200ml,4ml/g)洗涤。将固体在真空烘箱中在60℃干燥过夜,以83%收率得到作为二甲基亚砜溶剂化物(~12重量%)的磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯。因为环境湿度《30%rh,分离出二甲基亚砜溶剂化物。更高的环境湿度导致二
甲基亚砜溶剂化物水合物 的分离。
[0576][0577]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯的二甲基亚砜溶剂化物 向50%相对湿度的暴露产生二甲基亚砜溶剂化物水合物。
[0578]
粉末x-射线衍射
[0579]
使用配有铜(cu)辐射源(波长)的bruker axs d8 endeavor衍射仪,产生磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基) 羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯的 dmso溶剂化物的粉末x-射线衍射图谱。将管电压和安培数分别设定 至40kv和40ma。将电动发散狭缝设置为11毫米的恒定照明。使用 lynxeye xe-t能量色散x-射线检测器检测衍射辐射,将位置敏感 检测器(psd)开口设定在4.00
°
。使用0.019
°
2θ的步长和0.2秒的每步 时间,在cu波长在θ-θ测角仪上在2.0-55.0度2-θ(
°
2θ)的范围内收集 数据。样品准备用于分析,方法是将它们放置在硅低背景小平板支架 中并在数据收集过程中以15rpm旋转。在该表征过程中的环境实验 室相对湿度是13.6%。
[0580]
使用配有anton-paar chc+样品腔室和波长cu辐射源 的bruker axs d8 discover衍射仪,产生dmso溶剂化物水合物的 粉末x-射线衍射图谱。将管电压和安培数分别设定至40kv和40ma。 将电动发散狭缝设置为10mm的恒定照明。使用lynxeye xe能量 色散x-射线检测器检测衍射辐射,将psd开口设定在2.95
°
。使用 0.01
°
2θ的步长和0.2秒的每步时间,在cu波长在θ-θ测角仪上在 5.0-40.0度
°
2θ的范围内收集数据。样品准备用于分析,方法是将它们 放置在具有硅低背景插入物的样品支架中并在数据收集之前在25℃ 和50%相对湿度(rh)平衡至少2小时。
[0581]
在diffrac.eva v5.0软件中分析数据。使用相对强度≥5%的每 个相应衍射图谱中的最强谱带的反射制备峰列表。
±
0.2
°
2θ的峰位置典 型误差(usp-941)适用于该数据。与此测量相关的微小误差可能由于多 种因素而发生,包括:(a)样品制备(例如样品高度),(b)仪器特征,(c) 仪器校准,(d)操作员输入(例如在确定峰位置时),和(e)材料的性质(例 如择优取向和透明效应)。
[0582]
为了得到绝对峰位置,应当将粉末图谱与参考进行比对。这可能 是来自在室温解析的相同形式的晶体结构的模拟粉末图谱,或内部标 准物例如二氧化硅或刚玉。将磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚
ꢀ‑
2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯 的dmso溶剂化物的收集的粉末图谱与含有内部标准物si(srm640e)的相同材料的粉末图谱比对。将磷酸二氢(3s)-3-({n-[(4-甲氧基
ꢀ‑
1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷
ꢀ‑
3-基]丁酯的dmso溶剂化物水合物的收集的粉末图谱与来自晶体结 构的模拟粉末图谱进行比对。dmso溶剂化物和dmso溶剂化物水 合物的pxrd波谱分别提供在图17和图20中,且对应的峰列表分别 提供在表
pxrd-3和表pxrd-4中。dmso溶剂化物的特征峰是在 7.4、10.8、14.8、22.3和26.2 2-θ位置(度2-θ
±
0.2度2-θ)的峰。dmso 溶剂化物水合物的特征峰是在14.5、17.8、21.9、25.6和26.6 2-θ位置 (度2-θ
±
0.2度2-θ)的峰。当存在两种固体形式的混合物时,可能用dmso溶剂化物和dmso溶剂化物水合物的特征峰的组合来表征物 质。dmso溶剂化物和dmso溶剂化物水合物的混合物的pxrd图 谱的一个例子显示在图21中。
[0583]
表pxrd-3:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯dmso 溶剂化物的pxrd峰列表.
[0584][0585]
表pxrd-4:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯dmso 溶剂化物水合物的pxrd峰列表.
[0586]
[0587][0588]
固态nmr
[0589]
在定位于bruker-biospin avance iii 500mhz(1h频率)nmr波 谱仪中的cpmas探针上进行固态nmr(ssnmr)分析。将材料填充 进zro2转子并用o-环帽盖上。使用15.0khz的魔角旋转速率。
[0590]
使用质子解耦交叉极化魔角旋转(cpmas)实验收集
13
c ssnmr 波谱。在波谱采集期间应用80-100khz的相位调制质子解耦场。将交 叉极化接触时间设定至2ms。用3.5秒的再循环延迟收集波谱。调制 扫描数目以得到适当的信噪比。使用在结晶性金刚烷的外标准物上的 13
c cpmas实验来将
13
c化学位移的大小定标,将其高场共振设定至 29.5ppm(如从纯tms所确定的)。
[0591]
使用bruker-biospin topspin version 3.6软件进行自动峰拾取。 通常,5%相对强度的阈值用于初步峰选择。目视检查自动峰拾取的输 出以确保有效性,并在必要时手动进行调整。尽管本文报告了特定固 态nmr峰值,但由于仪器、样品和样品制备的差异,这些峰值确实 存在一个范围。这是固态nmr领域中的常见实践,因为峰位置固有 的变异。
13
c化学位移x-轴值的典型变异性是在
±
0.2ppm(对于结晶 固体)和
±
0.5ppm(对于无定形固体)的量级。本文报告的固态nmr 峰高是相对强度。固态nmr强度可以随实验参数的实际设置和样品 的热史而变化。
[0592]
表nmr-6:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯dmso 溶剂化物的
13
c ssnmr峰列表.
[0593][0594]
拉曼光谱法
[0595]
使用连接至vertex 70波谱仪(bruker optik gmbh)的ram iift-raman模块收集拉曼光谱。仪器配有1064nm固态(nd:yag)激 光和液氮冷却的锗检测器。在数据采集之前,使用白光源以及聚苯乙 烯和萘参照物进行仪器性能和校准验证。
[0596]
从装它们的玻璃小瓶直接分析样品;为每个样品在不同的位置进 行两个单独测量以使在数据收集过程中暴露于激光的材料的体积最大 化。将来自样品的反向散射的拉曼光谱信号优化,并使用1000mw的 激光功率在2cm-1
的波谱分辨率收集数据。应用blackmann-harris4-term切趾函数以使波谱畸变最小化。产生在3500-50cm-1
之间的波 谱,相应地调制扫描数目以确保适当的信噪比。
[0597]
通过将最强峰的强度设定至2.00,使波谱标准化。然后使用opusv8.2软件(bruker optik gmbh)中的自动峰拾取功能鉴定峰,将灵敏 度设定至2%。将峰位置和相对峰强度提取并制表。采用该实验构型 峰位置的变异性是在
±
2cm-1
内。
[0598]
由于ft-拉曼光谱和色散拉曼光谱是类似的技术,预期在此文件 中关于ft-拉曼光谱报告的峰位置与使用色散拉曼光谱测量会观察到 的那些是一致的,假定适当的仪器校准。
[0599]
从dmso溶剂化物收集的拉曼光谱呈现在图19中。应当指出, dmso溶剂化物和dmso溶剂化物水合物的拉曼光谱数据是等同位 置,在
±
2cm-1
的所述误差内。
[0600]
表拉曼-4:从磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯dmso 溶剂化物收集的ft拉曼光谱所提取的峰列表
[0601][0602]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯dmso溶剂化物的特征 pxrd峰包括在7.4
±
0.2、14.8
±
0.2和26.2
±
0.2度2-θ的峰;在7.4
±
0.2、 10.8
±
0.2、14.8
±
0.2和26.2
±
0.2度2-θ的峰;和在7.4
±
0.2、10.8
±
0.2、 14.8
±
0.2、22.3
±
0.2和26.2
±
0.2度2-θ的峰。
[0603]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯dmso溶剂化物的特征 13
c ssnmr峰包括在173.4
±
0.2、210.7
±
0.2和26.2
±
0.2ppm的
13
c峰; 在173.4
±
0.2、210.7
±
0.2、26.2
±
0.2和22.8
±
0.2ppm的峰;和在 173.4
±
0.2、210.7
±
0.2、26.2
±
0.2、22.8
±
0.2和25.5
±
0.2ppm的峰。
[0604]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯dmso溶剂化物的特征 拉曼峰包括在1717
±
2和675
±
2cm-1
的拉曼峰。
[0605]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯dmso溶剂化物的 pxrd峰和
13
c ssnmr峰的特征组合包括在7.4
±
0.2、14.8
±
0.2和 26.2
±
0.2度2-θ的pxrd峰和在173.4
±
0.2、210.7
±
0.2和26.2
±
0.2ppm 的
13
c ssnmr峰。
[0606]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯dmso溶剂化物的 pxrd峰和拉曼峰的特征组合包括在7.4
±
0.2、14.8
±
0.2和26.2
±
0.2度 2-θ的pxrd峰和在1717
±
2和675
±
2cm-1
的拉曼峰。
[0607]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯dmso溶剂化物的
13
cssnmr峰和拉曼峰的特征组合包括在173.4
±
0.2、210.7
±
0.2和26.2
±
0.2 ppm的
13
c ssnmr峰和在1717
±
2和675
±
2cm-1
的拉曼峰。
[0608]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯dmso溶剂化物水合物 的特征pxrd峰包括在14.5
±
0.2、25.6
±
0.2和26.6
±
0.2度2-θ的峰; 在14.5
±
0.2、21.9
±
0.2、25.6
±
0.2和26.6
±
0.2度2-θ的峰;和在14.5
±
0.2、 17.8
±
0.2、21.9
±
0.2、25.6
±
0.2和26.6
±
0.2度2-θ的峰。
[0609]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯的制剂实施例
[0610]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯的亲液物(lyophile)制 剂:
[0611]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯优选地如下配制:形成溶 液,然后进行冷冻干燥过程以制备亲液物。磷酸二氢(3s)-3-({n-[(4-甲 氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡 咯烷-3-基]丁酯形式可以是作为游离酸或作为合适的盐。形成磷酸二氢 (3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁酯的盐(即磷酸根部分的盐)的优选抗衡 离子包括胆碱、葡甲胺、苄星、二乙胺、三(羟基甲基)氨基甲烷、二 乙醇胺(diolamine)、哌嗪,更优选抗衡离子包括钾、镁和钙,和最 优选抗衡离子是钠。冷冻干燥溶液优选地在ph 2至ph 6的范围内配 制,更优选ph 3至ph 5和最优选在ph 3.5至ph 4.5的范围内。为 了维持所需的ph,将制剂缓冲,优选的缓冲剂是乳酸、磷酸、乙酸 和酒石酸,最优选的缓冲剂是柠檬酸。通过加入合适的碱性赋形剂可 以调节和控制制剂的ph,优选的碱包括胆碱、葡甲胺、苄星、二乙 胺、三(羟基甲基)氨基甲烷、二乙醇胺、哌嗪,更优选的碱是氢氧化 钾、氢氧化镁和氢氧化钙,且最优选的碱是氢氧化钠。
[0612]
还可以包括填充剂、张度调节剂或水清除赋形剂,其中优选的赋 形剂包括糖、多元醇、聚合物和氨基酸,更优选的赋形剂包括葡聚糖、 聚乙烯基吡咯烷酮和甘氨酸,和最优选的赋形剂包括海藻糖、蔗糖、 乳糖、甘露醇、聚乙二醇400和聚乙二醇3350。此外,制剂可以包括 增溶剂,其中优选的赋形剂包括表面活性剂和络合剂(例如环糊精), 更优选的赋形剂是聚山梨酯20、cremophor el、kolliphor hs-15、 羟丙基-β-环糊精、磺丁基醚-β环糊精、γ环糊精,和最优选聚山梨酯 80。
[0613]
在制备后冷冻干燥制剂的含水量优选《2%w/w,更优选《1%w/w, 和最优选《0.5%w/w。在冷冻干燥之前和在重构之后磷酸二氢 (3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁酯的浓度优选在10-300mg/ml的范围 内,更优选25-150mg/ml,和最优选在50-125mg/ml的范围内。 可以将制剂在无菌注射用水、0.9%w/v氯化钠(生理盐水)或5%w/v右 旋糖溶液中重构和稀释。
[0614]
在瓶制剂中的粉末:
[0615]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯还可以配制为粉末。在该 情况下,磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰 基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯可以作为粉末填充进 瓶中并重构至合适的ph。磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2
‑ꢀ
基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]
丁酯可 以作为游离酸或作为合适的盐而加入。形成磷酸二氢(3s)-3-({n-[(4-甲 氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡 咯烷-3-基]丁酯的盐的优选抗衡离子包括胆碱、葡甲胺、苄星、二乙胺、 三(羟基甲基)氨基甲烷、二乙醇胺、哌嗪,更优选抗衡离子包括钾、 镁和钙,且最优选抗衡离子是钠。在重构溶液以后,优选范围为ph 2 至ph 6,更优选ph 3至ph 5,且最优选在ph 3.5至ph 4.5的范围 内。为了维持所需的ph,将制剂用缓冲剂缓冲,优选的缓冲剂为乳 酸、磷酸、乙酸和酒石酸,最优选的缓冲剂是柠檬酸。通过包含合适 的碱来调节控制制剂的ph,优选的碱包括胆碱、葡甲胺、苄星、二 乙胺、三(羟基甲基)氨基甲烷、二乙醇胺、哌嗪,更优选的碱是氢氧 化钾、氢氧化镁和氢氧化钙,且最优选的碱是氢氧化钠。
[0616]
还可以包括填充剂、张度调节剂或水清除赋形剂,其中优选的赋 形剂包括糖、多元醇、聚合物和氨基酸,更优选的赋形剂包括葡聚糖、 聚乙烯基吡咯烷酮和甘氨酸,且最优选的赋形剂包括海藻糖、蔗糖、 乳糖、甘露醇、聚乙二醇400和聚乙二醇3350。此外,制剂可以包括 增溶剂,其中优选的赋形剂包括表面活性剂和络合剂(例如环糊精), 更优选的赋形剂是聚山梨酯20、cremophor el、kolliphor hs-15、 羟丙基-β-环糊精、磺丁基醚-β环糊精、γ环糊精,和最优选聚山梨酯 80。
[0617]
在重构之后磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯的浓度 优选在10-300mg/ml的范围内,更优选25-150mg/ml,和最优选 在50-125mg/ml的范围内。可以将制剂在无菌注射用水、0.9%w/v 氯化钠(生理盐水)或5%w/v右旋糖溶液中重构和稀释。
[0618]
pf-07304814制剂实施例的一般方法
[0619]
含水量的karl fischer评估
[0620]
使用配有连接至分析天平(mettler toledo xp56)的双针铂电极 dm143-sc的karl fisher(kf)滴定计(mettler toledo c30)通过库仑 测定方法确定冷冻干燥的样品的含水量。hydranal coulomat ad (fluka)用作kf容器溶液。调节仪器直到背景漂移低于20mg/min。 根据指南,在通过系统适用性标准的初始水检查后,对样品进行分析。 简单地说,将100mg样品置于试管中并置于分析天平上。然后对天 平进行去皮,并将样品快速转移到kf容器中并塞上塞子。再次在天 平上称量空管,以检查是否有残余样品。自动滴定样品,并将实验结 果打印为样品大小和含水量。一式两份地测量样品,并将%含水量报 告为平均值。
[0621]
纯度的超高效液相色谱法(uplc)评估
[0622]
使用具有紫外检测的梯度uplc方法确定pf-07304814的测定和 纯度。用于分析的柱具有用tms封端的五氟苯基固定相。通过将甲 酸铵水溶液和甲酸铵在甲醇中的溶液混合制备流动相。根据 pf-07304814峰,通过它们的相对保留时间(rrt)确定杂质。通过 将样品溶液色谱图的相应峰面积与已知浓度标准溶液的pf-07304814 峰的峰面积值进行比较,定量测定。通过将杂质峰面积与总峰面积 (pf-07304814和杂质的峰面积之和)进行比较,计算每个杂质峰的 面积百分比(%)。
[0623]
调制的示差扫描量热法(mdsc)表征
[0624]
使用ta instruments dsc q1000仪器对冷冻干燥的样品进行分 析。用于分析的软件是universal analysis 2000(版本4.5a,构造4.5.0.5)。简单地说,将样品密封在铝盘
中,并使温度以2℃/min的 速率从-20℃上升到200℃,每100秒调制
±
0.53℃。对tg和其它热事 件进行了分析。
[0625]
粉末x-射线衍射(pxrd)表征
[0626]
将冷冻干燥的样品通过pxrd进行分析,以使用rigaku miniflex600衍射仪评估亲液物的结构。衍射仪与40kv/15ma管电源和闪烁 探测器一起使用。所使用的狭缝条件为可变+固定系统,ihs的入射光 束路径设置为5.0
°
、10.0mm,ds为1.25
°
,且rs的衍射光束设置为 0.3mm。在分步模式下分析样品,从2
°
的2θ开始,在40
°
结束,分步 为0.02
°
,持续1秒。使用rigaku pdxl软件(版本1.8.0.3)处理原 始数据。
[0627]
磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯(pf-07304814)的制剂实 施例:pf-07304814制剂的ph、缓冲液和pf-07304814浓度
[0628]
制剂ph
[0629]
为了评价pf-07304814制剂的可能ph范围,在ph 1至ph 7制 备pf-07304814制剂,并通过hplc评价随时间变化的化学稳定性。
[0630]
将约1.35mg pf-07304814称量进20ml闪烁瓶中,每个ph值 重复2次。向每个小瓶中添加7ml纯化水,然后添加预测量的0.01m、 0.1m或1m hcl或naoh。测量每种制剂的ph,如果偏离超过
±
0.2 单位,则使用0.01m hcl或naoh调整ph,直到达到目标ph。然 后将纯化水添加至10ml的目标体积。然后将小瓶加盖、混合,并在 室温下稳定放置。在1、3和6天后,取出每种制剂的150μl等分试 样,并转移到hplc小瓶中进行纯度分析。
[0631]
根据下面ph稳定性表中的实验数据,优选的ph范围是约ph 2 至约ph 6,且最优选的ph范围是约ph 3至约ph 5。该优选ph适 用于冷冻干燥前的溶液、冷冻干燥后的重构溶液和用于静脉内施用的 稀释溶液。
[0632]
ph稳定性表:在室温下6天内,评价约0.13mg/ml pf-07304814 溶液的化学稳定性随ph的变化,以了解最佳ph范围。报告了通过 色谱纯度的uplc测定所确定的总杂质。
[0633]
ph稳定性表
[0634][0635]
缓冲液组成
[0636]
为了评价pf-07304814制剂的可能缓冲液组成,在无缓冲液、有 柠檬酸盐缓冲液和有乳酸盐缓冲液的情况下制备pf-07304814制剂。 对于这些制剂中的每一种,通过hplc
评价随时间的化学稳定性和 ph。
[0637]
在250ml容量瓶中制备浓储备溶液,以便在添加pf-07304814 至约25mg/ml浓度后,最终ph为3、4或5。对于无缓冲液的储备 溶液,添加规定量的1m naoh,然后用纯化水将烧瓶填充至体积。 对于柠檬酸盐缓冲液,向每个烧瓶中添加约1471mg二水合柠檬酸钠 以及规定量的1m naoh,然后用纯化水将烧瓶填充至体积。对于乳 酸盐缓冲液,将约1868mg乳酸钠(60%w/w)与规定量的1m naoh 一起添加到每个烧瓶中,然后用纯化水将烧瓶填充至体积。
[0638]
称取约75mg pf-07304814至3ml玻璃小瓶中。向每个小瓶中 添加1.2ml纯化水,然后添加1.5ml浓储备溶液。然后测量每种制 剂的ph,如果偏离超过
±
0.2单位,则使用0.1m hcl或naoh调整 ph,直到达到目标ph。然后将纯化水添加至3ml的目标体积。所 得制剂应具有无缓冲液、10mm柠檬酸盐缓冲液或20mm乳酸盐缓 冲液,每个制剂的最终ph为3、4和5。然后将小瓶塞住,加盖,混 合,并在25℃下稳定放置。4天后,测量溶液ph,并将每种制剂的 50μl等分试样转移到hplc小瓶中以进行纯度分析。
[0639]
根据以下缓冲液组成表中的实验数据,在特定ph下,不同制剂 中的总杂质没有显著差异,这表明受试缓冲液与pf-07304814之间没 有化学相容性问题。形成的定性杂质分布没有随使用的缓冲液变化的 明显趋势(数据未显示)。但是,与没有缓冲液的样品相比,10mm 的柠檬酸盐缓冲液的包含似乎能够更好地在约3和4的ph值控制ph。 因此,为了使药物产品保持在目标ph范围内,进而限制不希望的ph 依赖性降解,在4.5:1的pf-07304814与缓冲液的摩尔比选择柠檬酸 盐缓冲液。该优选缓冲液组成适用于冷冻干燥前的溶液、冷冻干燥的 粉末、冷冻干燥后的重构溶液和用于静脉内施用的稀释溶液。
[0640]
缓冲液组成表:在无缓冲液、有10mm柠檬酸盐缓冲液和有20 mm乳酸盐缓冲液的情况下,在3种不同ph水平(3、4和5)评价 25mg/ml pf-07304814制剂在45mm下的化学稳定性,以了解最佳 缓冲液组成。将总杂质的增加报告为初始总杂质与25℃下4天后测得 的总杂质之间的差异。
[0641]
缓冲液组成表
[0642]
[0643]
pf-07304814浓度
[0644]
为了评价冷冻干燥前的溶液和重构的亲液物溶液中可能存在的 pf-07304814浓度,在保持4.5:1的pf-07304814与柠檬酸盐缓冲液的 固定比例的同时,以约50、约100和约200mg/ml的浓度制备 pf-07304814制剂。对于这些制剂中的每一种,通过hplc随时间评 价化学稳定性和ph。
[0645]
首先在250ml容量瓶中制备浓缓冲溶液,以便在添加 pf-07304814至约50mg/ml、约100mg/ml或约200mg/ml的浓度 后,最终ph为4。对于50mg/ml pf-07304814制剂,通过添加2.94 g二水合柠檬酸钠和27.4ml 1n naoh并用纯化水稀释至体积,制备 40mm柠檬酸盐缓冲液。对于100mg/ml pf-07304814制剂,通过添 加5.88g二水柠檬酸钠和55.5ml 1n naoh并用纯化水稀释至体积, 制备80mm柠檬酸盐缓冲液。对于200mg/ml pf-07304814制剂, 通过添加11.76g二水柠檬酸钠和112.5ml 1n naoh并用纯化水稀 释至体积,制备160mm柠檬酸盐缓冲液。
[0646]
将约200mg、约400mg或约800mg pf-07304814称重至10ml 玻璃小瓶中。向每个小瓶中添加2ml适当的浓缓冲溶液,然后添加2 ml纯化水。然后测量每种制剂的ph,如果偏离超过
±
0.2单位,则使 用0.1m hcl或naoh调整ph,直到达到目标ph。得到的制剂的 pf-07304814浓度应为约50mg/ml、约100mg/ml或约200mg/ml, 分别含有20mm、40mm或80mm柠檬酸盐缓冲液。然后将小瓶塞 住,加盖,混合,并在25℃下保持稳定。3、6和13天后,测量溶液 ph,并将每种制剂的等分试样转移到hplc小瓶中进行纯度分析。
[0647]
根据以下制剂化学稳定性表中的实验数据,pf-07304814制剂的 化学稳定性在50mg/ml至200mg/ml的pf-07304814浓度范围内是 相当的。测试的浓度表现出可比性。此优选pf-07304814浓度范围支 持在冷冻干燥前的可能溶液和在冷冻干燥后的重构溶液。具有较低 pf-07304814浓度(约1mg/ml至约25mg/ml)的制剂也具有可接 受的化学稳定性,如上文ph稳定性表(error!reference source notfound)和缓冲液组成表中的示例所示,这可能进一步涵盖用于静脉内 施用的可能稀释溶液。
[0648]
制剂化学稳定性表:用4.5:1的pf-07304814与柠檬酸盐缓冲液 固定摩尔比,在3种不同的pf-07304814浓度(50mg/ml、100mg/ml 和200mg/ml)评价pf-07304814制剂的化学稳定性。将总杂质的增加 报告为初始总杂质与25℃下3、6或13天后测得的总杂质之间的差异。
[0649]
制剂化学稳定性表
[0650][0651]
制剂实施例1:100mg/ml磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲 哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁 酯在柠檬酸盐缓冲液中的溶液的制备
[0652]
步骤1:80mm柠檬酸钠缓冲液的250ml制备物(preparation) 的制备
[(3s)-2-氧代吡咯烷-3-基]丁酯形式1水合物加入烧杯以形成溶液和混合5分钟。将1.12ml1n氢氧化钠溶液加入烧杯和混合5分钟。将0.556g磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯形式1水合物加入烧杯以形成溶液和混合5分钟。使用0.29ml1n氢氧化钠溶液将溶液滴定至4的目标ph。用纯化水将溶液在容量瓶中稀释至目标体积或基于密度稀释至目标质量并倒转混合直到均匀。将溶液穿过0.2umpvdf过滤器进行注射器式过滤。制剂的最终组成是10mlph4溶液,其含有~100mg/ml磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯形式1水合物和40mm柠檬酸盐缓冲液(4.5:1的pf-07304814与柠檬酸盐的摩尔比)。
[0662]
药物产品制剂1e
[0663]
含有5mg/ml聚山梨酯80的100mg/ml磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯(pf-07304814)溶液的制备
[0664]
为了防止难溶性的pf-07304814相关杂质或降解物的沉淀,我们研究了含有增溶赋形剂、具体而言含有聚山梨酯80的制剂的制备。制剂的组成与实施例1a-1d一致,即具有100mg/mlpf-07304814、ph4.0、40mm柠檬酸盐缓冲液(约4.5:1的pf-07304814与柠檬酸盐的摩尔比),和约1.3:1的钠与pf-07304814的大致比率。该溶液还包括5mg/ml聚山梨酯80。为了进一步确认此类溶液可以在不发生显著降解的情况下进行冻干以产生亲液物,进行了冷冻干燥循环开发。冷冻干燥的粉末中聚山梨酯80的含量为约4%w/w。
[0665]
80mm柠檬酸钠缓冲液的制备
[0666]
将5.89g二水合柠檬酸钠(美国药典级)加入250ml容量瓶。将125ml纯化水加入容量瓶,随后加入55.5ml1n氢氧化钠溶液。将溶液用纯化水稀释至目标体积并倒转混合直到均匀。将溶液穿过0.2μm尼龙过滤器真空过滤。
[0667]
250mg/ml聚山梨酯80溶液的制备
[0668]
将2.50g聚山梨酯80(nfgrade,spectrum)加入10ml容量瓶。将溶液用纯化水稀释至目标体积并倒转混合直到均匀。
[0669]
药物产品制剂的制备
–
含有5mg/ml聚山梨酯80的100mg/mlpf-07304814溶液
[0670]
将5.0ml冷冻的80mm柠檬酸钠缓冲溶液加入具有磁棒的20ml烧杯。将烧杯放入控制至2-8℃的水浴。将约1.04g磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯形式1水合物加入烧杯以形成溶液和混合约25分钟。将3.0ml冷冻的纯化水加入烧杯和混合5分钟。将溶液用纯化水稀释至10.35g的目标质量,并通过搅拌棒混合直到均匀。将溶液穿过0.2μmpvdf过滤器进行注射器式过滤。制剂的最终组成是约10mlph4溶液,其含有约100mg/ml磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯形式1水合物,40mm柠檬酸盐缓冲液,和5mg/ml聚山梨酯80。(4.5:1的pf-07304814与柠檬酸盐的摩尔比)。
[0671]
药物产品制剂的稀释
[0672]
为了证实增溶赋形剂的存在有助于防止难溶杂质或降解物的沉淀,制备了含有或不含有聚山梨酯80的制剂,并以与用于静脉内施用时的制备一致的方式进行稀释,并监
测可见和亚可见微粒(sub-visibleparticulate)的形成。重要的是,药物组合物中的少量聚山梨酯80(5 mg/ml)(重构之前和之后)可显著减少用于静脉内施用的稀释溶液中 的微粒形成。
[0673]
含有0和5mg/ml聚山梨酯80的制剂分别按照药物产品制剂1d 和1e中的上述方法制备。然后,将制剂在0.9%w/v氯化钠注射液(usp, b.braun)或5%w/v右旋糖注射液(usp,b.braun)中稀释至25mg/ml 磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨 基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯。对于25mg/ml的磷酸二 氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧 代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯稀释液,向小瓶中添加3.0ml0.9%w/v氯化钠注射液或5%w/v右旋糖注射液,然后添加1.0ml制 剂。将所有溶液加塞(stopped),倒置混合,并在室温下储存两天。 然后,在0、1和2天后,通过usp《790》分析所得稀释液中的可见微 粒,并通过动态流动成像分析亚可见微粒。
[0674]
可见微粒分析
[0675]
对稀释制剂样品和稀释安慰剂进行目视检查,以监测可见微粒的 形成。当按照usp《790》方法进行测试时,含有聚山梨酯80的样品在 2天内“基本上不含微粒”。不含聚山梨酯80的样品在稀释后立即具 有》10个可见微粒。微粒与api相关的杂质有关。
[0676]
亚可见微粒分析
[0677]
通过动态流动成像在两天内对25mg/ml稀释制剂样品和稀释安 慰剂进行分析。使用带有校准过的10倍物镜的flowcam 8100,从每 个小瓶中取样1.00ml溶液并使其流经干净的液体流动池。在图像采 集过程中,应用4-100μm等效球直径(esd)预滤器,以与流动池定 义的尺寸约束对齐。将采集结果从flowcam 8100导出到 lumetics link软件进行进一步处理。使用lumetics link,将每次运 行的整个颗粒(particle)群过滤到亚可见尺寸范围(4-10μm、10-25μm、 25-50μm和50-100μm)。通过该分析,发现观察到大多数微粒的大小 在4-10μm之间。利用esd超过90%的微粒尺寸小于25μm。重要的 是,数据表明,与不含聚山梨酯80的药品相比,含有聚山梨酯80的 药品样品中的微粒明显较少。采用聚山梨酯80时微粒的减少与目视检 查结果一致,并证明聚山梨酯80可溶解造成可见和亚可见微粒形成的 api相关杂质。亚可见微粒计数与安慰剂相当。在微粒数据表中报告 了第0天的结果,但在第1天和第2天观察到一致的趋势。
[0678]
微粒数据表:含有0或5mg/ml聚山梨酯80的磷酸二氢 (3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁酯的100mg/ml制剂的每ml亚可见微 粒计数,所述制剂已在盐水或右旋糖中稀释至25mg/ml的磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁酯浓度。通过动态流动成像进行分析。 在第0天报告了数据,在几小时稀释内.
[0679][0680]
制剂实施例2:100mg/ml磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲 哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁 酯形式1水合物溶液的冷冻干燥
[0681]
将10ml过滤的磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯形式1 水合物溶液的100mg/ml溶液(如在上面制剂实施例1d中制备)填充 到具有20mm颈直径的20ml小瓶中,并部分加塞。将填充和部分塞 好的小瓶放在托盘上,并将托盘加载进冻干机(lyostar)。将冻干机密 封并将存放温度以0.5℃/分钟的速率冷却至-45℃并保持1小时。将真 空压设定至150毫托并将冻干机保持1小时。然后将存放温度以0.5℃/ 分钟的速率加热至25℃并保持27小时。在初次干燥结束时,给腔室 回填氮并将存放温度冷却至5℃。将样品在冻干机中加塞,释放真空, 并移出样品,以在小瓶中得到作为亲液物的产物。
[0682]
pf-07304814亲液物的表征
[0683]
在冷冻干燥以后,样品呈现为饼状至粉末状,具有白色至灰白色/ 黄色/棕色颜色。冷冻干燥的样品显示回熔、塌陷和皱缩的微小迹象。 通过karl fischer测量,冷冻干燥的粉末的含水量为约0.6%w/w。通 过uplc测量,样品的色谱纯度在冷冻干燥之前和之后变化了约 0.1%。通过mdsc观察到单个tg,温度为109.4℃。通过pxrd测量, 冷冻干燥的样品的结构似乎主要为无定形,在约3.0
°
的2θ处观察到一 个宽峰(参见图22)。
[0684]
制剂实施例2a:在10.9ml填充体积含有5mg/ml聚山梨酯80 的100mg/ml pf-07304814溶液的冷冻干燥
[0685]
为了防止难溶性的pf-07304814相关杂质或降解物的沉淀,我们 研究了含有增溶赋形剂、具体而言含有聚山梨酯80的制剂的制备。制 剂的组成与实施例2一致,即含有100mg/ml pf-07304814,ph为 4.0,柠檬酸盐缓冲液为40mm(约4.5:1的pf-07304814与柠檬酸盐 的摩尔比),钠与pf-07304814的大致比率为约1.3:1。该溶液还包括5 mg/ml聚山梨酯
80。为了进一步确认此类溶液可以在不发生显著降解的情况下进行冷冻干燥以产生亲液物,进行了冷冻干燥循环开发。冷冻干燥的粉末中聚山梨酯80的含量为约4%w/w。
[0686]
在10.9ml填充体积含有5mg/ml聚山梨酯80的100mg/mlpf-07304814溶液的冷冻干燥
[0687]
将10.9ml过滤的100mg/mlpf-07304814溶液(来自以上制剂1e)填充进具有20mm颈直径的20ml小瓶,并部分加塞。将填充并部分加塞的小瓶放在托盘上并将托盘加载进冻干机。将冻干机密封并将存放温度以0.5℃/分钟的速率冷却至-45℃并保持1小时。将真空压设定至150毫托并将冻干机保持1小时。然后将存放温度以0.5℃/分钟的速率加热至25℃并保持27小时。在第二次干燥结束时,给腔室回填氮并将存放温度冷却至5℃。将样品在冻干机中加塞,释放真空,并移出样品。
[0688]
在10.9ml填充体积用5mg/ml聚山梨酯80制备的pf-07304814亲液物的表征
[0689]
在冷冻干燥以后,样品呈现为饼状至粉末状,具有白色至灰白色/黄色/棕色颜色。冷冻干燥的样品显示回熔、塌陷和皱缩的微小迹象。通过karlfischer测量,冷冻干燥的粉末的含水量为约0.5%w/w。通过uplc测量,样品的色谱纯度在冷冻干燥之前和之后变化了约0.1%。通过mdsc观察到单个tg,具有101.2℃的温度。
[0690]
通过pxrd测量,冷冻干燥的样品的结构似乎主要为无定形,在约3.0
°
的2θ处观察到一个宽峰(参见图23(error!referencesourcenotfound.3))。
[0691]
制剂实施例2b:在5.45ml填充体积含有5mg/ml聚山梨酯80的100mg/mlpf-07304814溶液的冷冻干燥
[0692]
将5.45ml过滤的100mg/mlpf-07304814溶液填充进具有20mm颈直径的20ml小瓶,并部分加塞。将填充并部分加塞的小瓶放在托盘上并将托盘加载进冻干机。将冻干机密封并将存放温度以0.5℃/分钟的速率冷却至-45℃并保持1.5小时。将真空压设定至150毫托并将冻干机保持1小时。然后将存放温度以0.5℃/分钟的速率加热至25℃并保持16.7小时。然后将存放温度以0.2℃/分钟的速率加热至40℃并保持6.7小时。在第二次干燥结束时,给腔室回填氮并将存放温度冷却至5℃。将样品在冻干机中加塞,释放真空,并移出样品。
[0693]
制剂实施例3:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯亲液物的重构和得到的溶液的稀释
[0694]
将9.2ml无菌注射用水穿过塞子注入上述制剂实施例2的冷冻干燥的小瓶中,以将药物产品重构为10ml目标体积的水溶液。然后将小瓶倒置以混合,直到亲液物完全重构,所需时间不到1分钟。重构溶液的ph是在冷冻干燥前ph的
±
0.2单位内。
[0695]
重构后,然后从小瓶中取出溶液,并用0.9%w/v氯化钠(生理盐水)或5%w/v右旋糖溶液稀释以提供所需浓度的磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯溶液(例如,浓度为25mg/ml)。所得稀释溶液然后可用于胃肠外施用,诸如静脉内施用,尤其是静脉内输注。
[0696]
制剂实施例3a:在10.9ml填充体积用5mg/ml聚山梨酯80制备的pf-07304814亲液物的重构
[0697]
将10.0ml无菌注射用水穿过塞子注入上述实施例2a中的冷冻干燥的小瓶中,以
将药物产品重构至10.9ml的目标体积。然后将小 瓶倒置以混合,直到亲液物完全重构,这需要大约2至3分钟。重构 溶液的ph是在冷冻干燥前ph的
±
0.2单位内。
[0698]
制剂实施例4:含有钾抗衡离子的100mg/ml pf-07304814溶液 的制备和冷冻干燥
[0699]
为了研究制备具有替代抗衡离子的溶液的可行性,将含钠的赋形 剂从溶液制备中移除或者用不含抗衡离子或含有钾作为抗衡离子的赋 形剂替代。具体地说,柠檬酸代替二水合柠檬酸钠,氢氧化钾代替 naoh。制剂的所得组成为100mg/ml pf-07304814,ph为4.0,柠 檬酸盐缓冲液为40mm,钾与pf-07304814的大致比例为约1.3:1。 为了进一步确认此类溶液可以在不发生显著降解的情况下进行冻干以 产生亲液物,进行了冷冻干燥循环开发。得到的亲液物的表征表明可 接受的外观和结构。
[0700]
含有钾抗衡离子的80mm柠檬酸缓冲液的制备
[0701]
将1.54g无水柠檬酸加入100ml容量瓶。将约50ml纯化水加 入容量瓶,随后加入5.17ml 50%w/v氢氧化钾溶液。将溶液用纯化 水稀释至目标体积并倒转混合直到均匀。将溶液穿过0.2μm pvdf过 滤器真空过滤。
[0702]
含有钾抗衡离子的100mg/ml pf-07304814溶液的制备
[0703]
将15.0ml冷冻的80mm柠檬酸缓冲溶液加入具有磁棒的50ml 烧杯。将烧杯放入控制至2-8℃的水浴。将约3.12g pf-07304814加入 烧杯以形成溶液和混合约25分钟。将9.0ml冷冻的纯化水加入烧杯 和混合5分钟。将溶液用纯化水稀释至31.05g的目标质量并用搅拌棒 混合直到均匀。将溶液穿过0.2μm pvdf过滤器进行注射器式过滤。 制剂的最终组成是约30ml ph 4.0溶液,含有约100mg/mlpf-07304814和40mm柠檬酸盐缓冲液(4.5:1的pf-07304814与柠檬 酸盐的摩尔比)。由于用于制备柠檬酸盐缓冲液的氢氧化钾,该制剂含 有钾作为抗衡离子。
[0704]
含有钾抗衡离子的100mg/ml pf-07304814溶液的冷冻干燥
[0705]
将5ml过滤的100mg/ml pf-07304814溶液填充进具有20mm 颈直径的6ml小瓶,并部分加塞。将填充并部分加塞的小瓶放在托 盘上并将托盘加载进冻干机。将冻干机密封并将存放温度以0.5℃/分 钟的速率冷却至-45℃并保持1小时。将真空压设定至150毫托并将冻 干机保持1小时。然后将存放温度以0.5℃/分钟的速率加热至25℃并 保持20小时。在第二次干燥结束时,给腔室回填氮并将存放温度冷却 至5℃。将样品在冻干机中加塞,释放真空,并移出样品。
[0706]
用钾抗衡离子制备的pf-07304814亲液物的表征
[0707]
在冷冻干燥以后,样品呈现为饼状至粉末状,具有白色至灰白色/ 黄色/棕色颜色。冷冻干燥的样品显示收缩或皱缩的微小迹象。通过 karl fischer测量,冷冻干燥的粉末的含水量为约0.5%w/w。通过 uplc测量,样品的色谱纯度在冷冻干燥之前和之后变化了约0.1%。 通过mdsc观察到单个tg,具有108.9℃的温度。通过pxrd测量, 冷冻干燥的样品的结构似乎主要为无定形,观察到在约3.0
°
的2θ的一 个宽峰(参见图24)。
[0708]
制剂实施例5:含有哌嗪抗衡离子的100mg/ml pf-07304814溶 液的制备和冷冻干燥
[0709]
为了研究pf-07304814抗衡离子对制剂的化学稳定性的影响,将 含钠的赋形剂除去或替换为不含抗衡离子或含哌嗪作为抗衡离子的赋 形剂。具体而言,柠檬酸代替二水合柠檬酸钠,和哌嗪代替naoh。制 剂的所得组成为100mg/ml pf-07304814,ph为4.0,40mm柠
檬酸 盐缓冲液(4.5:1的pf-07304814与柠檬酸盐的摩尔比)和约0.6:1的哌 嗪与pf-07304814的大致比率。为了进一步确认此类溶液可以在不发 生显著降解的情况下进行冻干以产生亲液物,进行了冷冻干燥循环开 发。随后将冷冻干燥的样品置于加速稳定性试验,令人惊讶的是,观 察向降解产物1的降解的显著减少。
[0710]
160mm柠檬酸缓冲液的制备
[0711]
将3.07g无水柠檬酸和4.03g哌嗪加入100ml容量瓶。将溶液 用纯化水稀释至目标体积并倒转混合直到均匀。将溶液穿过0.2μmpvdf过滤器真空过滤。
[0712]
含有哌嗪抗衡离子的100mg/ml pf-07304814溶液的制备
[0713]
将25ml冷冻的纯化水加入具有磁棒的100ml烧杯并用搅拌棒 混合。将烧杯放入控制至2-8℃的水浴。将约6.24g pf-07304814加 入烧杯以形成悬浮液和混合约5分钟。将15ml冷冻的160mm柠檬 酸缓冲溶液(含有哌嗪)加入烧杯并用搅拌棒混合直到均匀。将溶液用 纯化水稀释至60ml的目标体积并用搅拌棒混合直到均匀。检查溶液 的ph。使用1n hcl将ph调至目标。将溶液穿过0.2μm pvdf过 滤器进行注射器式过滤。制剂的最终组成是约60ml ph 4.0溶液,含 有约100mg/ml pf-07304814和40mm柠檬酸盐缓冲液(4.5:1的 pf-07304814与柠檬酸盐的摩尔比)。由于用于制备柠檬酸盐缓冲液的 哌嗪,该溶液含有哌嗪作为抗衡离子。
[0714]
含有哌嗪抗衡离子的100mg/ml pf-07304814溶液的冷冻干燥
[0715]
将5ml过滤的含有哌嗪抗衡离子的100mg/ml pf-07304814溶 液填充进具有20mm颈直径的6ml小瓶,并部分加塞。将填充并部 分加塞的小瓶放在托盘上并将托盘加载进冻干机(lyostar)。将冻干机 密封并将存放温度以0.5℃/分钟的速率冷却至-45℃并保持1小时。将 真空压设定至150毫托并将冻干机保持1小时。然后将存放温度以 0.5℃/分钟的速率加热至25℃并保持20小时。然后将存放温度以0.2℃/分钟的速率加热至40℃并保持10小时。在第二次干燥结束时, 给腔室回填氮并将存放温度冷却至5℃。将样品在冻干机中加塞,释 放真空,并移出样品。
[0716]
用哌嗪抗衡离子制备的pf-07304814亲液物的表征
[0717]
在冷冻干燥以后,样品呈现为饼状至粉末状,具有白色至灰白色/ 黄色/棕色颜色。冷冻干燥的样品显示收缩或皱缩的微小迹象。通过 karl fischer测量,冷冻干燥的粉末的含水量为约0.3%w/w。通过 uplc测量,样品的色谱纯度在冷冻干燥之前和之后变化了约0.3%。 通过mdsc观察到单个tg,具有102.7℃的温度。
[0718]
通过pxrd测量,冷冻干燥的样品的结构似乎主要为无定形,观 察到在约3.0
°
的2θ的一个宽峰(参见图24(error!reference source notfound))。
[0719]
用哌嗪抗衡离子制备的pf-07304814亲液物的加速稳定性
[0720]
将如制剂实施例3(钠)和制剂实施例5(哌嗪)中所述用不同抗衡 离子制备的冷冻干燥的制剂在40℃/75%相对湿度(rh)下进行加速稳 定性试验。在储存0周和4周后,对每种制剂的两个小瓶进行测试。 第一个小瓶保持为固体,第二个小瓶用4.6ml水重构。下面的加速稳 定性表中显示了此加速稳定性研究的数据。该数据表明,使用哌嗪抗 衡离子可减少在加速稳定性试验中的总降解和向降解物1(磷酸酯 (phosphate)切割的化合物即母体化合物n-((1s)-1-{[((1s)-3-羟基-2
‑ꢀ
氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁 基)-4-甲氧基-1h-吲哚-2-甲酰胺)的降解(4周后)。
[0721]
加速稳定性表:将使用钠或哌嗪抗衡离子制备的pf-07304814冷冻干燥的制剂置于加速稳定性试验。在加速稳定性研究开始时报告了结果,以证明冷冻干燥的药物产品在含水量和重构后ph方面具有可比性。报告了通过hplc测量的总杂质和降解物1的变化。
[0722][0723]
制剂实施例6:含有聚乙二醇(peg)的100mg/mlpf-07304814溶液的制备和冷冻干燥
[0724]
为了提高冷冻干燥的制剂的化学稳定性,我们研究了含有稳定赋形剂,具体而言peg400和peg3350的制剂的制备。制剂的组成与制剂实施例1d一致,含有100mg/mlpf-07304814,ph为4.0,40mm的柠檬酸盐缓冲液(4.5:1的pf-07304814与柠檬酸盐的摩尔比),钠与pf-07304814的近似比为约1.3:1。为了进一步确认此类溶液可以在不发生显著降解的情况下进行冷冻干燥以产生亲液物,进行了冷冻干燥循环开发。冷冻干燥的粉末的peg400或peg3350含量为约8%w/w(单独包含时),当包含二者时冷冻干燥的粉末的peg总含量为约15%w/w。随后将冷冻干燥的样品置于加速稳定性试验,令人惊讶的是,观察到总降解和向降解物1的降解的显著减少。
[0725]
160mm柠檬酸盐缓冲液的制备
[0726]
将11.77g二水合柠檬酸钠和111ml1nnaoh加入250ml容量瓶。将溶液用纯化水稀释至目标体积并倒转混合直到均匀。将溶液穿过0.2μmpvdf过滤器真空过滤。
[0727]
批量制剂(bulkformulation)的制备
[0728]
将280ml冷冻的纯化水加入具有磁棒的500ml烧杯。将烧杯放入控制至2-8℃的水浴。将约68.6gpf-07304814加入烧杯以形成悬浮液和混合约5分钟。将165ml160mm柠檬酸缓冲溶液加入烧杯并用搅拌棒混合直到均匀。
[0729]
含有10mg/mlpeg400的100mg/mlpf-07304814溶液的制备
[0730]
将52ml批量制剂加入具有磁棒的100ml烧杯。按质量加入约700mgpeg400并混合直到均匀。将溶液稀释至70ml的总体积并穿过0.2μmpvdf过滤器进行注射器式过滤。最终组合物是约70mlph4.0溶液,含有约100mg/mlpf-07304814、40mm柠檬酸盐缓冲液(4.5:1的pf-07304814与柠檬酸盐的摩尔比)和10mg/mlpeg400。
[0731]
含有10mg/mlpeg3350的100mg/mlpf-07304814溶液的制备
[0732]
将52ml批量制剂加入具有磁棒的100ml烧杯。按质量加入约700mgpeg3350并混合直到均匀。将溶液稀释至70ml的总体积并穿过0.2μmpvdf过滤器进行注射器式过滤。最终组合物是约70mlph4.0溶液,含有约100mg/mlpf-07304814、40mm柠檬酸盐缓冲液(4.5:1的pf-07304814与柠檬酸盐的摩尔比)和10mg/mlpeg3350。
[0733]
含有10mg/mlpeg400和10mg/mlpeg3350的100mg/mlpf-07304814溶液的制备
[0734]
将52ml批量制剂加入具有磁棒的100ml烧杯。按质量加入约700mgpeg400和约700mgpeg3350并混合直到均匀。将溶液稀释至70ml的总体积并穿过0.2μmpvdf过滤器进行注射器式过滤。最终组合物是约70mlph4.0溶液,含有约100mg/mlpf-07304814、
40mm柠檬酸盐缓冲液(4.5:1的pf-07304814与柠檬酸盐的摩尔比)、 10mg/ml peg400和10mg/ml peg3350。
[0735]
用peg制备的100mg/ml pf-07304814溶液的冷冻干燥
[0736]
将5ml过滤的含有peg的100mg/ml pf-07304814溶液填充进 具有20mm颈直径的6ml小瓶,并部分加塞。将填充并部分加塞的 小瓶放在托盘上并将托盘加载进冻干机(lyostar)。将冻干机密封并将 存放温度以0.5℃/分钟的速率冷却至-45℃并保持1小时。将真空压设 定至150毫托并将冻干机保持1小时。然后将存放温度以0.5℃/分钟 的速率加热至25℃并保持20小时。然后将存放温度以0.2℃/分钟的速 率加热至40℃并保持10小时。在第二次干燥结束时,给腔室回填氮 并将存放温度冷却至5℃。将样品在冻干机中加塞,释放真空,并移 出样品。
[0737]
用peg制备的100mg/ml pf-07304814亲液物的表征
[0738]
在冷冻干燥以后,所有样品呈饼状至粉末状,具有白色至灰白色/ 黄色/棕色颜色。通过karl fischer测量,所有三个样品的冷冻干燥的 粉末的含水量为约0.2%w/w。通过uplc测量,所有三个样品的色谱 纯度在冷冻干燥之前和之后变化了约0.1%。含有10mg/ml peg400、 10mg/ml peg3350和10mg/ml peg400/10mg/ml peg3350的样品 分别在91.8、92.4和76.3℃所有三个样品通过mdsc观察到单个tg。 通过pxrd测量,冷冻干燥的样品的结构似乎主要为无定形,观察到 在约3.0
°
的2θ的一个宽峰(参见图26)。
[0739]
用peg制备的pf-07304814亲液物的加速稳定性
[0740]
将如实施例3(ps80)和实施例7(peg)所述用peg400、peg3350 和ps80的不同组合制备的冷冻干燥的制剂在40℃/75%相对湿度(rh) 下置于加速稳定性试验。在储存0周和4周后,对每种制剂的两个小 瓶进行测试。第一个小瓶保持为固体,第二个小瓶用4.6毫升水重构。 本加速稳定性研究的数据如以下peg加速稳定性表所示。实验数据表 明,peg的加入可在稳定性方面减少总降解和向降解物1降解(4周 后),最大的好处来自peg400和peg3350的同时加入。将用ps80、 peg400或peg3350制备的pf-07304814冷冻干燥的制剂置于加速稳 定性试验。在加速稳定性研究开始时报告了结果,以证明冷冻干燥的 药物产品在ph和含水量方面具有可比性。通过hplc测量的总杂质 和降解物1的变化报告为两瓶测量值的平均值。
[0741]
peg加速稳定性表
[0742][0743]
制剂实施例7:使用通过二甲亚砜(dmso)溶剂化物的重结晶纯 化的pf-07304814制备和冷冻干燥100mg/ml pf-07304814溶液
[0744]
为了研究使用从替代纯化方案衍生的pf-07304814作为起始材料 的影响,使用pf-07304814水合物制备以下起始api表所示的药物产 品制剂,所述pf-07304814水合物通过pf-07304814dmso溶剂化物 (pf-07304814批1)向与水合物同构的dmso溶剂化物(pf-07304814 批2)的转化获得,或来自未转化的pf-07304814dmso溶剂化物 (pf-07304814批3)。制剂的组成与药物产品制剂1e一致,具有100 mg/ml pf-07304814,ph为4.0,40mm的柠檬酸盐缓冲液(4.5:1的 pf-07304814与柠檬酸盐的摩尔比),钠与pf-07304814的近似比例为 约1.3:1,和约5mg/ml聚山梨酯80。为了确认dmso的存在是否影 响生产冷冻干燥的制剂的能力,进行了冷冻干燥循环开发。冷冻干燥 的粉末中聚山梨酯80的含量为约4%w/w。
[0745]
起始api表:从dmso纯化方法衍生出的pf-07304814批的描 述.
[0746][0747]
160mm柠檬酸盐缓冲液的制备
[0748]
将约47.04g二水合柠檬酸钠、44.4ml 10n naoh和700ml纯 化水加入容器并混合直到所有成分溶解。用纯化水使溶液达到1.0l 的最终体积,并将溶液穿过0.2μm pvdf过滤器真空过滤。
[0749]
100mg/ml聚山梨酯80溶液的制备
[0750]
将约10g聚山梨酯80加入容器,并使其最终体积达到100.0ml (按重量计)。混合液体,直到获得均质溶液。
[0751]
使用通过二甲亚砜(dmso)溶剂化物的重结晶纯化的 pf-07304814制备100mg/ml pf-07304814溶液
[0752]
以与药物产品实施例1c类似的方式制备药物产品制剂。具体而 言,将8.5ml冷冻的纯化水加入具有磁棒的20ml烧杯并混合。将 烧杯放入控制至2-8℃的水浴。对于每种制剂,向烧杯中添加约2.0g pf-07304814(关于dmso含量调整)并混合,直到获得均匀的湿悬 浮液。通过添加约5.0ml冷冻的160mm柠檬酸盐缓冲液,然后添加 约1.0ml 100mg/ml聚山梨酯80溶液,溶解各悬浮液中的湿的 pf-07304814。将溶液混合,用纯化水稀释至20ml的目标体积,混 合,并通过0.2μm pvdf注射器式滤器过滤。制剂的最终组成是约20 ml ph 4.0溶液,含有约100mg/ml pf-07304814、40mm柠檬酸盐 缓冲液(4.5:1的pf-07304814与柠檬酸盐的摩尔比)和5mg/ml聚山梨 酯80。
[0753]
使用通过dmso溶剂化物的重结晶纯化的pf-07304814冷冻干燥 100mg/ml pf-07304814溶液
[0754]
将5.45ml过滤的100mg/ml pf-07304814药物产品溶液(如上 所述)填充进具有20mm颈直径的20ml小瓶,并部分加塞。将填 充并部分加塞的小瓶放在托盘上并将托盘加载进冻干机(lyostar)。将 冻干机密封并将存放温度以0.5℃/分钟的速率冷却至-45℃并保
持90 分钟。将真空压设定至150毫托并将冻干机保持1小时。然后将存放 温度以0.5℃/分钟的速率加热至25℃并保持1000分钟。然后将存放温 度以0.2℃/分钟的速率加热至40℃并保持400分钟。在第二次干燥结 束时,给腔室回填氮并将存放温度冷却至5℃。将样品在冻干机中加 塞,释放真空,并移出样品。
[0755]
使用通过dmso溶剂化物的重结晶纯化pf-07304814制备的 pf-07304814亲液物的表征
[0756]
在冷冻干燥以后,样品呈现为饼状至粉末状,具有白色至灰白色/ 黄色/棕色颜色。冷冻干燥的样品显示收缩或皱缩的微小迹象。样品的 色谱纯度在冷冻干燥之前和之后变化了约0.1%分析。通过mdsc在 每个样品中观察到单个tg,如在下面tg和dmso水平表中所示。
[0757]
tg和dmso水平表:用从dmso纯化过程衍生出的pf-07304814 制备的冷冻干燥的药物产品tg.
[0758][0759]
通过pxrd测量,冷冻干燥的样品的结构似乎主要为无定形,观 察到在约3.0
°
的2θ的一个宽峰(参见图27)。
[0760]
实施例50:磷酸(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基二甲酯
[0761]
lcms m/z 581.5[m+h]
+
。保留时间:2.36分钟(分析条件:柱: waters atlantis dc18,4.6x 50mm,5μm;流动相a:含有0.05%三 氟乙酸的水(v/v);流动相b:含有0.05%三氟乙酸的乙腈(v/v);梯度: 5.0%至95%b,线性历时4.0分钟,然后95%b保持1.0分钟;流速: 2ml/分钟)。
[0762]
实施例51:磷酸(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮 氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁基二丙烷-2-基酯
[0763]
实施例52:磷酸(3s)-4-[(3s)-1-乙酰基-2-氧代吡咯烷-3
‑ꢀ
基]-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代丁 基二甲酯
[0764]
实施例53:4-甲氧基-n-[(2s)-4-甲基-1-({(2s)-4-[(2-氧-4-苯基
ꢀ‑
1,3,2-二氧杂磷杂环己烷-2-基)氧基]-3-氧代-1-[(3s)-2-氧代吡咯烷-3
‑ꢀ
基]丁-2-基}氨基)-1-氧代戊烷-2-基]-1h-吲哚-2-甲酰胺
[0765]
实施例54:磷酸二乙基(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯
[0766]
lcms m/z 609.5[m+h]
+
。保留时间:2.53分钟(分析条件:柱: waters atlantis dc18,4.6x 50mm,5μm;流动相a:含有0.05%三氟 乙酸的水(v/v);流动相b:含有0.05%三
氟乙酸的乙腈(v/v);梯度: 5.0%至95%b,线性历时4.0分钟,然后95%b保持1.0分钟;流速: 2ml/分钟)。
[0767]
实施例55:(3s)-3-[(2s)-4-[(二甲氧基磷酰基)氧基]-2-({n-[(4-甲氧 基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-3-氧代丁基]-2-氧代吡咯烷
ꢀ‑
1-甲酸甲酯
[0768]
实施例56:碳酸(1s)-1-{(3s)-3-[(2s)-2-({n-[(4-甲氧基-1h-吲哚-2
‑ꢀ
基)羰基]-l-亮氨酰基}氨基)-2-(2-氧代-1,3-二氧杂环戊烯-4-基)乙 基]-2-氧代吡咯烷-1-基}乙基甲酯
[0769]
实施例57:4-甲氧基-n-[(2s)-4-甲基-1-氧代-1-({(1s)-1-(2-氧代
ꢀ‑
1,3-二氧杂环戊烯-4-基)-2-[(3s)-2-氧代吡咯烷-3-基]乙基}氨基)戊烷
ꢀ‑
2-基]-1h-吲哚-2-甲酰胺(57)
[0770][0771]
将碘甲烷(10.5μl,0.169mmol)加入1,1
’‑
羰基二咪唑(13.7mg, 84.5μmol)在1,2-二氯乙烷(0.84ml)中的溶液中。将得到的混合物搅拌 30分钟,此后加入c1(40mg,85μmol),随后加入4-甲基吗啉(18.5μl, 0.168mmol)。将反应混合物在室温搅拌,直到通过lcms分析确定 向活化酯的转化结束;然后将它在80℃加热过夜。与使用c1(20mg, 42μmol)进行的类似反应合并以后,将反应混合物在乙酸乙酯和10% 硫酸氢钾水溶液之间分配。将有机层经硫酸钠干燥,过滤,在真空中 浓缩,并通过反相hplc(柱:waters sunfire c18,19x 100mm,5μm; 流动相a:含有0.05%三氟乙酸的水;流动相b:含有0.05%三氟乙酸 的乙腈;梯度:25%至45%b历时8.5分钟,然后45%至95%b历时 0.5分钟,然后95%b保持1.0分钟;流速:25ml/分钟)纯化以得到 4-甲氧基-n-[(2s)-4-甲基-1-氧代-1-({(1s)-1-(2-氧代-1,3-二氧杂环戊烯
ꢀ‑
4-基)-2-[(3s)-2-氧代吡咯烷-3-基]乙基}氨基)戊烷-2-基]-1h-吲哚-2-甲 酰胺(57)。合并收率:3.5mg,7.0μmol,6%。lcms m/z 499.4[m+h]
+
。 保留时间:2.47分钟(分析条件:柱:waters atlantis dc18,4.6x 50mm, 5μm;流动相a:含有0.05%三氟乙酸的水(v/v);流动相b:含有0.05% 三氟乙酸的乙腈(v/v);梯度:5.0%至95%b,线性历时4.0分钟,然后 95%b保持1.0分钟;流速:2ml/分钟)。
[0772]
实施例58:n-[(2s)-1-{[(1s)-2-[(3s)-1-乙酰基-2-氧代吡咯烷-3
‑ꢀ
基]-1-(2-氧代-1,3-二氧杂环戊烯-4-基)乙基]氨基}-4-甲基-1-氧代戊烷
ꢀ‑
2-基]-4-甲氧基-1h-吲哚-2-甲酰胺
[0773]
实施例59:(3s)-3-[(2s)-2-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l
‑ꢀ
亮氨酰基}氨基)-2-(2-氧代-1,3-二氧杂环戊烯-4-基)乙基]-2-氧代吡咯烷
ꢀ‑
1-甲酸甲酯
[0774]
实施例60:碳酸(1s)-1-{(3s)-3-[(2s)-4-羟基-2-({n-[(4-甲氧基-1h
‑ꢀ
吲哚-2-基)羰基]-l-亮氨酰基}氨基)-3-氧代丁基]-2-氧代吡咯烷-1-基} 乙基甲酯
[0775]
实施例61:n-[(2s)-1-({(2s)-1-[(3s)-1-乙酰基-2-氧代吡咯烷-3
‑ꢀ
基]-4-羟基-3-氧代丁-2-基}氨基)-4-甲基-1-氧代戊烷-2-基]-4-甲氧基
ꢀ‑
1h-吲哚-2-甲酰胺
[0776]
实施例62:(3s)-3-[(2s)-4-羟基-2-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-3-氧代丁基]-2-氧代吡咯烷-1-甲酸甲酯
[0777]
实施例63:碳酸{(3s)-3-[(2s)-4-羟基-2-({n-[(4-甲氧基-1h-吲哚
ꢀ‑
2-基)羰基]-l-亮氨酰基}氨基)-3-氧代丁基]-2-氧代吡咯烷-1-基}甲基 甲基酯
[0778]
实施例64:碳酸苄基(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯(64)
[0779][0780]
将c1(15mg,32μmol)在四氢呋喃(0.32ml)中的0℃溶液用2,6
‑ꢀ
二甲基吡啶(4.4μl,38μmol)处理,随后用氯甲酸苄酯(4.98μl, 34.9μmol)处理。使反应混合物温热至室温和搅拌28小时,此后加入 额外当量的氯甲酸苄酯,并使温度增加至40℃。在已经将反应混合物 在40℃搅拌过夜以后,将它加热至60℃保持2小时,然后用二氯甲烷 稀释,并用10%硫酸氢钾水溶液洗涤。将有机层经硫酸钠干燥,过滤, 并在真空中浓缩;将残余物通过反相hplc(柱:waters sunfire c18, 19x 100mm,5μm;流动相a:含有0.05%三氟乙酸的水;流动相b:含 有0.05%三氟乙酸的乙腈;梯度:30%至70%b历时8.5分钟,然后 70%至95%b历时0.5分钟,然后95%b保持1.0分钟;流速:25ml/ 分钟)纯化,得到碳酸苄基(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯(64)。收 率:11.6mg,19.1μmol,60%。lcms m/z 607.5[m+h]
+
。保留时间:2.92 分钟(分析条件:柱:waters atlantis dc18,4.6x 50mm,5μm;流动相 a:含有0.05%三氟乙酸的水(v/v);流动相b:含有0.05%三氟乙酸的 乙腈(v/v);梯度:5.0%至95%b,线性历时4.0分钟,然后95%b保 持1.0分钟;流速:2ml/分钟)。
[0781]
实施例65:2-甲基吡咯烷-1-甲酸(3s)-3-({n-[(4-甲氧基-1h-吲哚
ꢀ‑
2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯 (65)
[0782][0783]
将碘甲烷(2.63μl,42.2μmol)加入1,1
’‑
羰基二咪唑(3.43mg, 21.2μmol)在二氯甲烷(0.21ml)中的溶液中。将得到的混合物已经搅拌 30分钟以后,加入c1(10mg,21μmol)。
在搅拌另外1小时以后,将 反应混合物用2-甲基吡咯烷(2.16μl,21.2μmol)处理并搅拌2.5小时, 此后将它在真空中浓缩。使用反相hplc(柱:waters sunfire c18,19 x 100mm,5μm;流动相a:含有0.05%三氟乙酸的水;流动相b:含 有0.05%三氟乙酸的乙腈;梯度:5%至95%b历时8.54分钟,然后 95%b保持1.46分钟;流速:25ml/分钟)纯化,得到2-甲基吡咯烷-1
‑ꢀ
甲酸(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2
‑ꢀ
氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯(65)。收率:7.6mg,13μmol, 61%。lcms m/z 584.6[m+h]
+
。保留时间:2.70分钟(分析条件:柱: waters atlantis dc18,4.6x 50mm,5μm;流动相a:含有0.05%三氟 乙酸的水(v/v);流动相b:含有0.05%三氟乙酸的乙腈(v/v);梯度: 5.0%至95%b,线性历时4.0分钟,然后95%b保持1.0分钟;流速: 2ml/分钟)。
[0784]
对接实验
[0785]
方法:
[0786]
同源性建模:sars和covid-19中3c样蛋白酶的序列可以在 来自rcsb(例如,3iwm)1和ncbi(例如,参照序列: yp_009725301.1ncbi)2的参考文献中找到。
[0787]
sars 3c蛋白酶序列(pdb 3iwm):
[0788][0789]
新冠状病毒sars-cov-2序列(相同部分,6y84):
[0790][0791]
使用的prime3从pfizer的数据库中sars 3c样蛋 白酶的晶体结构构建同源模型。在检查具有这些配体部分的其他 sars 3c样晶体结构的蛋白质构象后,使用与配体复合的同源模型的 最小化来消除与含有苯并噻唑酮或苄基侧链的配体的冲突。185-190 环、his41和met49中残基的松弛导致同源模型的三个不同的最小化 版本。催化性cys突变为gly(c145g)以促进agdock核心对接 (docking)和随后的评分,而不会与催化性cys发生冲突。
[0792]
对接:使用核心对接4与agdock5将化合物对接到同源模型中。 对接是在不形成蛋
白质-配体共价键的情况下进行的。相反,在配体中鉴定出包含内酰胺侧链和反应性酮的共同核心,并将其固定在晶体结构方向作为共价对接的模拟物(参见图2)。agdock核心对接的亲和力测量是htscore6。
[0793]
方法参考文献:
[0794]
1.http://www.rcsb.org/structure/3iwm
[0795]
2.https://www.ncbi.nlm.nih.gov/protein/yp_009725301.1
[0796]
3.release2019-1:prime,llc,newyork,ny,2019.
[0797]
4.danielk.gehlhaar,gennadym.verkhivker,paula.rejto,christopherj.sherman,davidr.fogel,lawrencej.fogel,stephant.freer,molecularrecognitionoftheinhibitorag-1343byhiv-1protease:conformationallyflexibledockingbyevolutionaryprograming,chemistry&biology,volume2,issue5,1995,pages317-324.
[0798]
5.danielk.gehlhaar,djamalbouzida,andpaula.rejto,reduceddimensionalityinligand—proteinstructureprediction:covalentinhibitorsofserineproteasesanddesignofsite-directedcombinatoriallibrariesrationaldrugdesign.july7,1999,292-311.
[0799]
6.tamij.marrone,brocka.luty,peterw.discoveringhigh-affinityligandsfromthecomputationallypredictedstructuresandaffinitiesofsmallmoleculesboundtoatarget:avirtualscreeningapproach.perspectivesindrugdiscoveryanddesign20,209-230(2000).
[0800]
结果:
[0801]
同源模型:sars-cov和sars-cov-2之间的序列同源性为96.1%。306个残基中有12个不同(图a中蓝绿色突出显示的t35v、a46s、s65n、l86v、r88k、s94a、h134f、k180n、l202v、a267s、t285a和i286l)转换为96.1%同一性。
[0802]
与用于构建同源模型的晶体结构相关的配体是化合物b,n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺。在sars3c样蛋白酶和sars-cov-23c样蛋白酶模型之间不同的、最接近化合物b即n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺的氨基酸残基是a46s,且从c
α
至配体的最小距离为其它残基离化合物b中的最近原子是在和之间。
[0803]
表1.sars-cov-2中的c
α
原子至化合物b即n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺的近似距离
[0804][0805]
图1描绘了sars-cov和sars-cov-2之间的残基差异。在 sars-cov-2同源模型的这个丝带描述中,用蓝绿色突出显示残基变 化。化合物b即n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯 烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰 胺位置以洋红色显示。sars-cov-2氨基酸残基的c-α和化合物b即 n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙 基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺中的最近原子 之间的近似距离如上面的表1所示。
[0806]
对接结果:
[0807]
sars-cov-2 3cl与sars-cov 3cl的约96%同源性和配体之 间的相似性允许在sars-cov(参见图2)中的晶体配体的肽主链和 sars-cov-2 3cl模型中的对接配体之间的rmsd的对比。核心对接 配体rmsd与肽主链之间的差异不超过0.32a(平均0.28a)。一个实 例如图2所示。对于化合物b即n-((1s)-1-{[((1s)-3-羟基-2-氧代
ꢀ‑
1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲 氧基-1h-吲哚-2-甲酰胺;整个分子的rmsd为0.37a。
[0808]
图2.sars-cov-2 3cl的同源模型与核心对接配体(化合物b即 n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙 基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺)的结合位点 存在(紫色碳、红色氧、蓝色氮)。使用化合物b即n-((1s)-1-{[((1s)-3
‑ꢀ
羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲 基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺(肽主链、内酰胺侧链和附接的酮) 的晶体结构的部分来测量不同配体与该主链的rmsd(灰色碳,厚棒)。 在球形表示和在嵌入化学结构中,用于核心对接的核心显示为11个重 原子(浅蓝色碳)。距离以埃表示。
[0809]
下表2中的对接结果指示,化合物具有预测的亲和力(δg
结合
, kcal/mol),通常与靶标识别和结合相称。有效效力可能不同于δg结 合项,这取决于几个因素,如细胞摄取、
流出、辅因子竞争或底物竞争。
[0810]
表2:
[0811][0812]
将上述化合物通过fret生化测定和使用细胞培养技术的体外病毒学测定进行分析。
[0813]
保护免于sars感染:中性红终点
[0814]
利用中性红染色作为终点,通过类似于borenfreund,e.,andpuerner,j.1985.toxicitydeterminedinvitrobymorphologicalalterationsandneutralredabsorptiontoxicologyletters.24:119-124中描述的细胞活力测定,测量化合物保护细胞免受sars冠状病毒感染的能力。简而言之,将含有适当浓度化合物的培养基或仅培养基添加到vero细胞中。将细胞用sars相关病毒感染或仅用培养基模拟感染。一到七天后,除去培养基并将含有中性红的培养基加入到测试板中。在37℃下温育两小时后,将细胞用pbs洗涤两次并加入50%etoh、1%乙酸溶液。将细胞摇动1到2分钟,并在37℃温育5-10分钟。在540nm处用分光光度法定量中性红的量。数据表示为化合物处理的细胞的孔中的中性红相对于未感染的、不含化合物的细胞的孔中的中性红的百分比。将50%有效浓度(ec50)计算为将感染的、化合物处理的细胞中的中性红产生的百分比增加到未感染的、不含化合物的细胞产生的中性红的百分比的50%的化合物浓度。将50%细胞毒性浓度(cc50)计算为将未感染的、化合物处理的细胞中产生的中性红的百分比降低到未感染的、不含化合物的细胞中产生的中性红百分比的50%的化合物浓度。通过将细胞毒性(cc50)除以抗病毒活性(ec50)来计算治疗指数。
[0815]
保护免于sars-cov-2感染:glo终点
[0816]
化合物保护细胞免受sars-cov-2冠状病毒感染的能力也可以通过细胞活力测定法来测量,该测定法利用荧光素酶测量细胞内atp作为终点。简而言之,将含有适当浓度化合物的培养基或仅培养基添加到vero细胞中。细胞被sars-cov-2病毒感染或仅用培养基模拟感染。1到7天后,除去培养基并根据promegatechnicalbulletinno.288:celltiter-luminescentcellviabilityassay(promega,madison,wi)测量细胞内atp的量。将celltiter-试剂加入测试板,在37℃下温育1.25小时后,使用光度计在490nm处量化信号量。将数据表示为来自化合物处理的细胞的孔的发光信号相对于来自未感染的、不含化合物的细胞的孔的发光信号的百分比。将50%有效浓度(ec50)计算为将来自感染的、化合物处理的细胞的发光信号百分比增加到来自未感染的、不含化合物的细胞的发光信号的50%的化合物浓度。将50%细胞毒性浓度(cc50)计算为将来自未感染的、化合物处理的细胞的发光信号的百分比降低至来自未感染的、不含化合物的细胞的发光信号的50%的化合物浓度。通过将细胞毒性(cc50)除以抗病毒活性(ec50),计算治疗
指数。
[0817]
细胞毒性
[0818]
利用甲臢(formazan)作为终点,通过类似于在weislow,o.s., kiser,r.,fine,d.l.,bader,j.,shoemaker,r.h.,和boyd,m.r.1989. new soluble-formazan assay for hiv-1cytopathic effects: application to high-flux screening of synthetic and natural productsfor aids-antiviral activity.journal of the national cancer institute81(08):577-586中描述的细胞活力测定法,测量化合物在细胞中引起 细胞毒性的能力。简而言之,将vero细胞重新悬浮在含有适当浓度 化合物的培养基或仅培养基中。1到7天后,将xtt和pms添加到 测试板中,然后在37℃下温育2小时,在540nm下通过分光光度计 法对产生的甲臢量进行定量。将数据表示为化合物处理的细胞中产生 的甲臢相对于不含化合物的细胞孔的中产生的甲臢的百分比。将50% 细胞毒性浓度(cc50)计算为将未感染的、化合物处理的细胞中产生的 甲臢的百分比降低到未感染的、不含化合物的细胞中产生的甲臢作百 分比的50%的化合物浓度。
[0819]
保护免于sars-cov-2冠状病毒感染
[0820]
利用甲臢作为终点,通过类似于weislow,o.s.,kiser,r.,fine, d.l.,bader,j.,shoemaker,r.h.,and boyd,m.r.1989.newsoluble-formazan assay for hiv-1cytopathic effects:application tohigh-flux screening of synthetic and natural products foraids-antiviral activity.journal of the national cancer institute81(08):577-586中描述的细胞活力测定,测量化合物保护细胞免于 sars-cov-2感染的能力。简而言之,将含有适当浓度化合物的培养 基或仅培养基添加到mrc-5细胞中。将细胞用人冠状病毒 sars-cov-2感染或仅用培养基模拟感染。1到7天后,将xti和pms 添加到测试板中,然后在37℃下温育两小时,在540nm处通过分光 光度法定量产生的甲臢量。数据表示为化合物处理的细胞孔中的甲臢 相对于未感染、不含化合物的细胞孔中的甲臢的百分比。50%有效浓 度(ec50)被计算为将感染的、化合物处理的细胞中甲臢产生的百分比 增加到未感染的、不含化合物的细胞产生的甲臢百分比的50%的化合 物浓度。50%细胞毒性浓度(cc50)计算为将未感染的、化合物处理的 细胞中产生的甲臢的百分比降低到未感染的、不含化合物的细胞中产 生的甲臢作百分比的50%的化合物浓度。通过将细胞毒性(cc50)除以 抗病毒活性(ec50)来计算治疗指数。
[0821]
sars-cov-2冠状病毒3c蛋白酶fret测定和分析
[0822]
使用连续荧光共振能量转移测定,测量sars-cov-2冠状病毒 3cl蛋白酶的蛋白水解活性。sars-cov-2 3cl
pro fret测定测量蛋 白酶催化的tamra-sitsavlqsgfrkmk-(dabcyl)-oh向 tamra-sitsavlq和sgfrkmk(dabcyl)-oh的切割。使用 tecan safire荧光平板读数器历时10min测量切割的tamra(激 发558nm/发射581nm)肽的荧光。典型反应溶液含有20mm hepes (ph 7.0),1mm edta,4.0um fret底物,4%dmso和0.005%吐温
ꢀ‑
20。通过加入25nm sars 3cl
pro
(sars冠状病毒完整基因组序列 (ncbi登录号ay278741)的urbani株的核苷酸序列9985-10902)开始 测定。在0.001mm抑制剂水平一式两份地确定抑制百分比。使用以下 方程式用非线性回归分析程序kalidagraph分析数据:
[0823]
fu=offset+(limit)(1-e-(kobs)
t)
[0824]
其中offset等于未切割肽底物的荧光信号,并且limit等于完全切 割的肽底物的
荧光。kobs是该反应的一阶速率常数,并在没有任何抑 制剂的情况下代表底物的利用率。在含有不可逆抑制剂的酶启动反应 中,并在计算的limit值小于理论最大limit的20%时,计算出的kobs 代表冠状病毒3c蛋白酶的灭活速率。kobs相对于[i]的图的斜率 (kobs/i)是抑制剂对酶的亲和力的量度。对于非常快的不可逆抑制剂, kobs/i从仅一个或两个[i]的观测值计算,而不是作为斜率。
[0825]
可替换地,使用下面的sars cov-2fret测定可以评估化合物。
[0826]
sars cov-2蛋白酶fret测定和分析
[0827]
使用连续荧光共振能量转移(fret)测定监测sars-cov-2的主 要蛋白酶3clpro的蛋白水解活性。sars-cov-2 3clpro测定测量全 长sars-cov-2 3cl蛋白酶的切割合成荧光底物肽的活性,所述肽的 序列为dabcyl-ktsavlq-sgfrkme-edans,模拟一致肽。切割的 edans肽的荧光(激发340nm/发射490nm)使用flexstation读数器 (molecular devices)上的荧光强度协议进行测量。在存在 n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙 基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺(sars-cov-2 3cl pro的有效抑制剂)的情况下,荧光信号降低。测定反应缓冲液 含有20mm tris-hcl(ph 7.3),100nm nacl,1mm edta,5mmtcep和25μm肽底物。通过添加15nm sars-cov-2 3cl蛋白酶启 动酶反应,并在23℃下进行60分钟。根据不含化合物(0%抑制/100% 活性)和含有对照化合物(100%抑制/0%活性)的对照孔计算抑制百 分比或活性。使用abase软件(idbs)使用四参数拟合模型生成ic
50
值。将ki值拟合morrison方程式,酶浓度参数固定为15nm,km参 数固定为14μm,且底物浓度参数固定为25um,使用activity base 软件(idbs)。
[0828]
当在以上测定中评价时,实施例49的化合物磷酸二氢 (3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁酯具有350nm的ic
50
(330nm至380 nm的95%置信区间,n=7)和137nm的ki(136nm至137nm的95% 置信区间,n=7)。
[0829]
母体化合物n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯 烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰 胺(其在施用磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l
‑ꢀ
亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯后在体内形 成)已经在不同的细胞测定中进行了评估,且已经发现其表现出对 sars-cov-2的抗病毒活性。采用a549-ace2(人肺)细胞和 usa-wa1/2020sars-cov-2(在nyu langone)、原代人气道上皮 (hae)(人肺)细胞和washington sars-cov-2(在nyu langone)和 hela-ace2(人宫颈)和usa-wa1/2020sars-cov-2(在scripps) 的细胞测定。
[0830]
使用高容量成像测定,通过用mab定量病毒n蛋白,在 a549-ace2细胞中评价n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧 代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚
ꢀ‑
2-甲酰胺和瑞德西韦对sars-cov-2的抗病毒活性。通过基于atp 的定量监测细胞活力,在未感染的细胞中评价了两种化合物的细胞毒 性。在a549-ace2细胞中,n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2
‑ꢀ
氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲 哚-2-甲酰胺抑制sars-cov-2病毒复制,在感染后24小时具有 0.221/0.734μm的ec50/ec90值,和在48小时后0.158/0.439μm的ec50/ec90值。n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷
ꢀ‑
3-基]甲
基]甲基}丙 基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺对一组(a panel of)人蛋白酶和hiv蛋白酶是无活性的。n-((1s)-1-{[((1s)-3-羟 基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基 丁基)-4-甲氧基-1h-吲哚-2-甲酰胺对人组织蛋白酶b表现出可检测的 活性,但是与3clpro相比具有1000倍界限(margin)(表4)。这些 数据共同地支持n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯 烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰 胺作为泛冠状病毒3cl蛋白酶抑制剂。
[0834]
表3.母体化合物n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代 吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2
‑ꢀ
甲酰胺对冠状病毒的3clpro的活性
[0835][0836]
表4:母体化合物n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代 吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2
‑ꢀ
甲酰胺对人蛋白酶和hiv蛋白酶的活性-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲 氧基-1h-吲哚-2-甲酰胺对sars-cov-2的抗病毒活性。将这些细胞系 分别用sars-cov-2washington株1或betacov ghb-03021/2020株 感染,它们具有相同的3clpro氨基酸序列。分别在39.7μm和88.9μm, n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙 基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺保护细胞免于 病毒cpe(ec
50
,表6)。但是,vero细胞表达高水平的流出(efflux) 转运蛋白p-gp(也被称作mdr1或abcb1),n-((1s)-1-{[((1s)-3-羟基
ꢀ‑
2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁 基)-4-甲氧基-1h-吲哚-2-甲酰胺是其已知底物。因此,在有p-gp流出 抑制剂cp-100356即4-(3,4-二氢-6,7-二甲氧基-2(1h)-异喹啉 基)-n-2[2-(3,4-二甲氧基苯基)乙基]-6,7-二甲氧基-2-喹唑啉胺存在下 重复测定。在有2μm p-gp抑制剂存在下,n-((1s)-1-{[((1s)-3-羟基-2
‑ꢀ
氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁 基)-4-甲氧基-1h-吲哚-2-甲酰胺表现出117-173倍的活性增加,在 veroe6-enace2细胞中具有0.23μm的ec
50
值和在veroe6-egfp细 胞中具有0.76μm的ec
50
值(表6)。单独的p-gp抑制剂在这些浓度不 具有抗病毒或细胞毒性活性,且在有蛋白酶抑制剂存在下不会造成细 胞毒性。对递增剂量的母体化合物n-((1s)-1-{[((1s)-3-羟基-2-氧代
ꢀ‑
1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲 氧基-1h-吲哚-2-甲酰胺存在陡峭应答(steep response),在两种细胞 类型中在ec
50
和ec
90
之间具有约2-3倍差异(在有p-gp抑制剂存在 下,在veroe6-enace2细胞中ec
90
=0.48um和在veroe6-egfp细 胞中ec90=1.6um)。当在有和没有p-gp抑制剂存在下测试肺细胞系 (a549-ace2和mrc5)的抗病毒效能时,没有观察到抗病毒效能的显 著差异(表6)。另外,在具有2um p-gp的两种veroe6细胞系中的ec
50
和ec
90
值类似于用不同的细胞类型使用不同的测定方法得到的那些, 包括通过在a549-ace2细胞中检测病毒蛋白以及在极化的人气道上 皮细胞(其中pg-p表达较低)中使用噬斑测定。
[0842]
表5:磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨 酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯的体外抗病毒活 性、细胞毒性和治疗指数(ti)
[0843][0844]
表6:母体化合物n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代 吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2
‑ꢀ
甲酰胺的体外抗病毒活性、细胞毒性和治疗指数(ti)
基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰 胺+阿奇霉素的组合产生协同作用,协同评分为3.76,ci为0.4。观察 到的协同作用不是由于细胞毒性,因为所有试验组合都没有明显的细 胞毒性。n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基] 甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺和瑞 德西韦的组合表现出累加性,协同评分为5.1,ci为0.21。观察到的 协同作用可能潜在地用于减少剂量,并从而增加抑制剂的安全界限以 实现体内治疗窗口。此外,组合治疗可用于最小化抗药性。
[0849]
进行了额外研究以进一步评价母体化合物n-((1s)-1-{[((1s)-3-羟 基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基 丁基)-4-甲氧基-1h-吲哚-2-甲酰胺与瑞地西韦的组合使用的抗病毒组 合益处的潜力。
[0850]
抗病毒剂的组合,特别是针对病毒复制周期中的不同步骤的那些 抗病毒剂的组合,是治疗病毒性疾病的常用治疗策略。由于 n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙 基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺和瑞德西韦 (一种核苷rna依赖性的rna聚合酶抑制剂)针对病毒复制周期的 不同步骤,使用hela-ace2细胞单独和组合评价两种化合物的抗病 毒活性。在该测定中,使用来自两位不同covid-19患者的恢复期人 多克隆血清检测病毒蛋白。单独的n-((1s)-1-{[((1s)-3-羟基-2-氧代
ꢀ‑
1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲 氧基-1h-吲哚-2-甲酰胺(在表6中命名化合物1)抑制sars-cov-2复 制,平均ec
50
为0.14μm,ec
90
为0.40μm;而瑞德西韦具有0.074μm 的平均ec
50
和0.17μm的ec
90
(表7)。
[0851]
表7:n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3
‑ꢀ
基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺(化 合物1)和瑞德西韦在hela-ace2细胞中的体外活性
[0852][0853]
使用药物测试矩阵进行组合研究,并使用参考模型(loewe,bliss, hsa)分析药物组合的数据,以将药物组合的效果分为累加、协同或拮 抗(等效线图解法、协同评分和组合指数)。一般而言,协同评分》1 且组合指数《1表明组合治疗具有协同作用(yeo等人,2015)。为了 评价在高抑制水平下是否可以实现协同作用,将等效线图解法水平设 置为0.9,以捕获具有90%病毒减少(相当于1log
10
减少)的有意义 协同作用。
[0854]
如表8所总结,母体化合物n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲 氧基-1h-吲哚-2-甲酰胺和瑞德西韦的组合在2个独立实验中表现出来 自患者#1血清的协同作用,在用患者#2血清的单个实验中表现出累加 性(表8)。不同的分类很可能是因为用作检测试剂的恢复期血清不 同。这些相同的抗病毒数据也使用synergyfinder程序进行了分析, 这也表明两种药物是累加
{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲氧 基-1h-吲哚-2-甲酰胺的血浆蛋白结合,显示与血浆蛋白的中等结合,各 物种的血浆游离分数(plasma free fraction)为0.26至0.46。
[0860]
将n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基} 丙基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺静脉内施用给 大鼠、狗和猴(1或2mg/kg),其在各物种中表现出中等血浆清除率 (35-60%肝血流)、低分布容积(《1l/kg)和短半衰期(《1.5h),与其中性 生理化学和亲脂性(sflogd
7.4
=1.7)保持一致。对大鼠(2mg/kg)和猴(5 mg/kg)口服施用后,n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡 咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲 酰胺表现出低生物利用度(《2%),可能是由于以下原因的组合:由于 其低渗透性(1.3x10-6 cm/sec的表观mdck-le渗透性)的低吸收,低 溶解度,p-gp和bcrp在肠道中主动外流的潜力,以及胃肠道中的消 化酶进行酰胺水解的潜力。在大鼠、狗和猴中,约10%的 n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙 基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺在尿液中以原 形消除,表明肾脏消除可能在人类的n-((1s)-1-{[((1s)-3-羟基-2-氧代
ꢀ‑
1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲 氧基-1h-吲哚-2-甲酰胺的清除中也起微小作用。
[0861]
适合用于静脉内施用的人药代动力学预测-考虑人体外代谢数据 以及在大鼠、狗和猴中的体内药代动力学(pk)数据,预计 n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙 基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺在人类中表现 出~6ml/min/kg的血浆清除率(cl
p
)(主要cyp,次要肾途径)、1l/kg 的稳态分布体积(v
dss
)和约2h的半衰期。由于有限的口服生物利用度、 短的消除半衰期,并且可能需要随着时间的推移保持全身游离浓度, 因此建议连续静脉内(iv)输注作为最佳施用途径和方案。
[0862]
实现目标ceff的有效目标浓度和可行的人体剂量预测 (projection)
[0863]
抑制性商(iq)已成为将临床前抗病毒效能转化为跨多种病毒性疾 病的临床的有用指标。iq定义为人c
min,u
未结合浓度除以抗病毒测定 中体外未结合的(血清调整的)ec
50,u
值(方程式1)。
[0864][0865]
一些抗病毒疗法已经在iq接近1时显示出显著的益处;但是, 快速控制病毒复制通常需要保持比体外ec
50
高至少10倍的暴露。当 考虑蛋白结合和作用暴露部位以1-100的iq值施用时,临床批准的蛋 白酶抑制剂已经有效降低病毒载量。重要的是,当以低于1的iq施 用时,一般的抗病毒药物,和特别是蛋白酶抑制剂,可能导致增加的 突变和额外的抗药性。
[0866]
需要多高的iq值取决于剂量响应曲线的斜率。通过方程式2,希 尔系数(m)和ec
50
与一系列浓度(c)下的体外抗病毒活性相关:
[0867][0868]
n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基} 丙
1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁 基)-4-甲氧基-1h-吲哚-2-甲酰胺的安全特性(safety profile)。在体外 研究中,磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰 基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯和 n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙 基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺在细菌反向突 变测定中为阴性,且不会诱发微核形成。所述磷酸酯和母体化合物在 临床相关暴露下都具有最小的二级(脱靶效应(off-target))药理学 潜力。磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯和n-((1s)-1-{[((1s)-3-羟 基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基 丁基)-4-甲氧基-1h-吲哚-2-甲酰胺在高达300μm都没有抑制herg电 流振幅(在预计的人有效剂量下,预计的未结合的人c
max
分别为0.17 和0.50μm,所述300μm的浓度分别为它们的1,770倍和600倍), 表明有利的心血管安全特性。在人血液的血液相容性测定中,两种化 合物对溶血或絮凝/浊度参数没有影响,表明与人体血液相容并支持静 脉内施用。
[0877]
在glp研究中通过连续静脉内输注将磷酸二氢(3s)-3-({n-[(4-甲 氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡 咯烷-3-基]丁酯施用给大鼠24小时。没有试验物相关的发现,也没有 发现靶器官毒性。在24小时连续静脉内输注大鼠研究中,通过功能观 察组(battery)评估,磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基) 羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯对神 经系统安全性药理学参数没有影响。无观察到的不良作用水平 (noael)为1000mg/kg。在一项非glp探索性毒性研究中,还将母 体化合物n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基] 甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺通过 连续静脉内输注施用给雄性大鼠4天,所述化合物的耐受为 246mg/kg/天,这是测试的最高可行剂量。本研究中的 n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙 基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺相关发现限于 对临床化学参数(包括较高的平均甘油三酯、胆固醇和磷)的最小非 不良作用,无任何微观相关性或相关功能改变。在任何研究中均未发 现试验物相关的不良作用。
[0878]
在对大鼠进行磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯 (pf-07304814)的24小时glp连续静脉内输注研究的noael,在0.5 g/天的预计最小人有效剂量,未结合的c
max
和auc
24
的预期暴露界限 对于磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基} 氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯而言为97和65倍,且对 于n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基} 丙基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺而言为25 和21倍。这表明有可能在临床试验期间在人类中安全地评价多倍ec
90
以了解暴露-应答关系和在需要时实现高水平的抑制。此外,磷酸二氢 (3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代
ꢀ‑
4-[(3s)-2-氧代吡咯烷-3-基]丁酯在人类的施用预计不会与目前用于护 理标准covid-19治疗中的药物产生重叠或累加毒性,使该化合物成 为组合疗法的有吸引力的搭配物(partner)。基于进行的一系列安全 性研究的结果,磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰基]-l
‑ꢀ
亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯表现出
令人鼓 舞的非临床安全特性。磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基) 羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯的预 测的人药代动力学提供了母体化合物n-((1s)-1-{[((1s)-3-羟基-2-氧代
ꢀ‑
1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲 氧基-1h-吲哚-2-甲酰胺达到0.5μm(ec
90
)的全身未结合浓度的能力, 这通过如下实现:递送500mg磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲 哚-2-基)羰基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁 酯作为连续输注历时24小时和输注体积《250ml。
[0879]
体内鼠感染研究
[0880]
在动物模型中药物效力的证明对于建立pk/pd关联以及为临床 剂量参数的选择提供支持性证据非常重要。使用cov-1感染的小鼠适 应(ma15)模型评价母体化合物n-((1s)-1-{[((1s)-3-羟基-2-氧代
ꢀ‑
1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲 氧基-1h-吲哚-2-甲酰胺(在施用实施例49的化合物后形成的活性部 分);(参见deming ra.,等人.a mouse-adapted sars-coronaviruscauses disease and mortality in balb/c mice.plos pathog.2007;3(1), e5和frieman m.,等人.molecular determinants of severe acuterespiratory syndrome coronavirus pathogenesis and virulence inyoung and aged mouse models of human disease.j virol.2012;86(2), 884-97)。通过皮下(s.c.)途径,用n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲基丁基)-4-甲 氧基-1h-吲哚-2-甲酰胺治疗ma15-cov感染的小鼠,100mg/kg,每 天2次(bid)。预计该剂量可实现≈500nm或约1xec
90
(在cov-1和 cov-2复制的体外测定中将病毒减少90%所需的化合物浓度)的c
min
游离药物暴露,这与我们临床上潜在的最小有效剂量一致。在一个实 验中,治疗在感染时(第0天)开始,或在感染后延迟1或2天。分 别从感染后第0、1和2天开始治疗,在感染后第4天的肺病毒滴度降 低≈2.0、1.5和1.0log10。特别是在第0天开始施用n-((1s)-1-{[((1s)-3
‑ꢀ
羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲 基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺时,体重减轻和疾病的组织病理 学征象减少。在第二个实验中,在第0天开始用n-((1s)-1-{[((1s)-3
‑ꢀ
羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙基)氨基]羰基}-3-甲 基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺治疗,并且药物剂量不同(30、 100和300mg/kg,bid,皮下)。在vero细胞上在病毒噬斑测定中 确定用n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲 基}丙基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺或媒介 物治疗的ma15感染的小鼠的肺病毒滴度,并以每μg肺组织的噬斑 形成单位(pfu)表示。每天测定动物的体重,并将其作为%起始重量 做图,我们观察到三种剂量的第4天肺病毒滴度的剂量依赖性下降: 在30mg/kg为≈1.5log10;在100mg/kg为≈3log10;和在300mg/kg 为≥3.5log10。在所有三种剂量下通过使用母体化合物 n-((1s)-1-{[((1s)-3-羟基-2-氧代-1-{[(3s)-2-氧代吡咯烷-3-基]甲基}丙 基)氨基]羰基}-3-甲基丁基)-4-甲氧基-1h-吲哚-2-甲酰胺治疗减少了病 毒造成的体重减轻。该数据支持下述预测,即历时24小时作为连续输 注施用的500mg磷酸二氢(3s)-3-({n-[(4-甲氧基-1h-吲哚-2-基)羰 基]-l-亮氨酰基}氨基)-2-氧代-4-[(3s)-2-氧代吡咯烷-3-基]丁酯可作为 在人类中治疗sars-cov-2的有效剂量。
[0881]
上文所述的所有专利和出版物通过引用整体并入。虽然已经以各 种优选实施方
案和具体实施例的方式描述了本发明,但是本发明应当 理解为不受前述详细描述限制,而是由所附权利要求及其等同方案定 义。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1