桥连型核苷和使用其的核苷酸的制作方法
1.本发明涉及桥连型核苷和使用了该桥连型核苷的核苷酸。更详细而言涉及具有良好的核酸酶抗性的桥连型核苷和使用了该桥连型核苷的核苷酸。
背景技术:
2.对dna或rna具有优异的结合亲和性的人工核酸能够在基因诊断或核酸医药中应用,目前为止开发了各种各样类型的人工核酸。其中,将核酸糖部的构象通过桥连固定化为n型构象的2’,4’-bna(2’,4’-bridged nucleic acid;别名lna)对单链rna(ssrna)具有优异的结合亲和性,被期待作为能够进行反义法等各种应用的核酸医药(非专利文献1和2)。但是,2’,4’-bna缺乏酶耐性,存在容易诱发肝毒性的问题(非专利文献3)。
3.相对于此,核酸糖部被取代为吡喃糖环的己糖醇核酸(hna)具有模拟天然核酸的n型构象的结构,已知使对单链rna(ssrna)的结合亲和性提高(非专利文献4和5)。因此,近年来,报道了将hna及其类似物应用于反义医药的研究,得到了暗示优异的活性和毒性降低这样的发现(非专利文献6和7)。
4.这样,具有hna骨架的人工核酸在医药中的应用上具有吸引人的性质,作为改善2’,4’-bna所存在的问题的物质备受关注。
5.现有技术文献
6.非专利文献
7.非专利文献1:s.obika等,tetrahedron lett.,1997,38,8735-8738
8.非专利文献2:s.singh等,chem.commun.,1998,455-456
9.非专利文献3:e.swayze等,nucleic acids res.,2007,35,687-700
10.非专利文献4:a.aerschot等,angew.chem.int.ed.,1995,34,1338-1339
11.非专利文献5:p.herdewijn,p.chem.biodiversity,2010,7,1-59
12.非专利文献6:m.egli等,j.am.chem.soc.,2011,133,16642-16649
13.非专利文献7:b.t.le等,chem.commun.,2016,52,13467-13470
技术实现要素:
14.发明所要解决的技术问题
15.本发明是解决上述课题的发明,其目的在于提供具有良好的核酸酶抗性且具有hna骨架的桥连型核苷和使用了该桥连型核苷的核苷酸。
16.用于解决技术问题的技术方案
17.本发明为以下的式(i)所示的化合物或其盐:
[0018][0019]
(式中,
[0020]
base(碱基)表示可以具有1个以上选自α组中的任意取代基的嘌呤-9-基或2-氧代-1,2-二氢嘧啶-1-基,其中,该α组包括羟基、被核酸合成的保护基所保护的羟基、碳原子数1至6的直链烷基、碳原子数1至6的直链烷氧基、巯基、被核酸合成的保护基所保护的巯基、碳原子数1至6的直链烷硫基、氨基、碳原子数1至6的直链烷基氨基、被核酸合成的保护基所保护的氨基和卤原子,
[0021]
r2和r3分别独立地表示氢原子、核酸合成的羟基的保护基、可以形成支链或环的碳原子数1至7的烷基、可以形成支链或环的碳原子数2至7的烯基、可以具有1个以上选自该α组中的任意取代基且可以含有杂原子的碳原子数3至10的芳基、具有可以具有1个以上选自该α组中的任意取代基且可以含有杂原子的碳原子数3至12的芳基部分的芳烷基、可以具有1个以上选自该α组中的任意取代基的酰基、可以具有1个以上选自该α组中的任意取代基的甲硅烷基、可以具有1个以上选自该α组中的任意取代基的磷酸基、被核酸合成的保护基所保护的磷酸基、-p(r4)r5[式中,r4和r5分别独立地表示羟基、被核酸合成的保护基所保护的羟基、巯基、被核酸合成的保护基所保护的巯基、氨基、碳原子数1至6的烷氧基、碳原子数1至6的烷硫基、碳原子数1至6的氰基烷氧基或具有碳原子数1至6的烷基的二烷基氨基],
[0022]
x1为碳原子数1~5的亚烷基、碳原子数2~5的亚烯基或-y1-(ch2)n-、-(ch2)n-y1-或-(ch2)
l
-y1-(ch2)m-[其中,y1为磺酰基、磺酰胺基、酰胺基、酯基或羰基,n为1~5的整数,l和m为正的整数且l与m的合计为2~5],
[0023]
x2为氧原子、硫原子、-nh-或亚甲基)。
[0024]
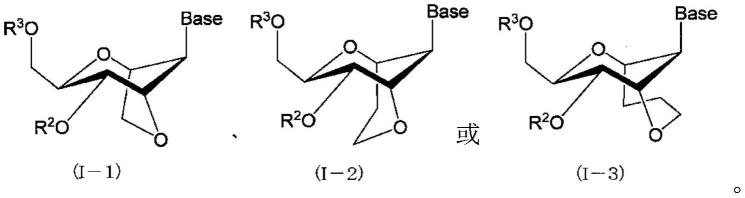
在一个实施方式中,上述式(i)由以下的式(i-1)~(i-3)的任意一个表示:
[0025][0026]
在一个实施方式中,上述base为6-氨基嘌呤-9-基、2,6-二氨基嘌呤-9-基、2-氨基-6-氯嘌呤-9-基、2-氨基-6-氟嘌呤-9-基、2-氨基-6-溴嘌呤-9-基、2-氨基-6-羟基嘌呤-9-基、6-氨基-2-甲氧基嘌呤-9-基、6-氨基-2-氯嘌呤-9-基、6-氨基-2-氟嘌呤-9-基、2,6-二甲氧基嘌呤-9-基、2,6-二氯嘌呤-9-基、6-巯基嘌呤-9-基、2-氧代-4-氨基-1,2-二氢嘧啶-1-基、4-氨基-2-氧代-5-氟-1,2-二氢嘧啶-1-基、4-氨基-2-氧代-5-氯-1,2-二氢嘧啶-1-基、2-氧代-4-甲氧基-1,2-二氢嘧啶-1-基、2-氧代-4-巯基-1,2-二氢嘧啶-1-基、2-氧代-4-羟基-1,2-二氢嘧啶-1-基、2-氧代-4-羟基-5-甲基-1,2-二氢嘧啶-1-基或4-氨基-5-甲基-2-氧代-1,2-二氢嘧啶-1-基。
[0027]
在一个实施方式中,上述base为以下的式:
[0028][0029]
所示的基团。
[0030]
本发明还为含有至少1个以下的式(ii)所示的核苷结构的寡核苷酸或其药理学上可接受的盐:
[0031][0032]
(式中,
[0033]
base表示可以具有1个以上选自α组中的任意取代基的嘌呤-9-基或2-氧代-1,2-二氢嘧啶-1-基,其中,该α组包括羟基、被核酸合成的保护基所保护的羟基、碳原子数1至6的直链烷基、碳原子数1至6的直链烷氧基、巯基、被核酸合成的保护基所保护的巯基、碳原子数1至6的直链烷硫基、氨基、碳原子数1至6的直链烷基氨基、被核酸合成的保护基所保护的氨基和卤原子,
[0034]
x1为碳原子数1~5的亚烷基、碳原子数2~5的亚烯基或-y1-(ch2)n-、-(ch2)n-y1-或-(ch2)
l
-y1-(ch2)m-[其中,y1为磺酰基、磺酰胺基、酰胺基、酯基或羰基,n为1~5的整数,l和m为正的整数且l与m的合计为2~5],
[0035]
x2为氧原子、硫原子、-nh-或亚甲基)。
[0036]
在一个实施方式中,上述式(ii)由以下的式(ii-1)~(ii-3)的任意一个表示:
[0037][0038]
在一个实施方式中,上述base为6-氨基嘌呤-9-基、2,6-二氨基嘌呤-9-基、2-氨基-6-氯嘌呤-9-基、2-氨基-6-氟嘌呤-9-基、2-氨基-6-溴嘌呤-9-基、2-氨基-6-羟基嘌呤-9-基、6-氨基-2-甲氧基嘌呤-9-基、6-氨基-2-氯嘌呤-9-基、6-氨基-2-氟嘌呤-9-基、2,6-二甲氧基嘌呤-9-基、2,6-二氯嘌呤-9-基、6-巯基嘌呤-9-基、2-氧代-4-氨基-1,2-二氢嘧啶-1-基、4-氨基-2-氧代-5-氟-1,2-二氢嘧啶-1-基、4-氨基-2-氧代-5-氯-1,2-二氢嘧啶-1-基、2-氧代-4-甲氧基-1,2-二氢嘧啶-1-基、2-氧代-4-巯基-1,2-二氢嘧啶-1-基、2-氧代-4-羟基-1,2-二氢嘧啶-1-基、2-氧代-4-羟基-5-甲基-1,2-
二氢嘧啶-1-基或4-氨基-5-甲基-2-氧代-1,2-二氢嘧啶-1-基。
[0039]
在一个实施方式中,上述base为以下的式:
[0040][0041]
所示的基团。
[0042]
本发明还为上述寡核苷酸或其药理学上可接受的盐的制造方法,其包括使用以下的式(i)所示的化合物或其药理学上可接受的盐合成寡核苷酸的工序,
[0043][0044]
(式中,
[0045]
base表示可以具有1个以上选自α组中的任意取代基的嘌呤-9-基或2-氧代-1,2-二氢嘧啶-1-基,其中,该α组包括羟基、被核酸合成的保护基所保护的羟基、碳原子数1至6的直链烷基、碳原子数1至6的直链烷氧基、巯基、被核酸合成的保护基所保护的巯基、碳原子数1至6的直链烷硫基、氨基、碳原子数1至6的直链烷基氨基、被核酸合成的保护基所保护的氨基和卤原子,
[0046]
r2和r3分别独立地表示氢原子、核酸合成的羟基的保护基、可以形成支链或环的碳原子数1至7的烷基、可以形成支链或环的碳原子数2至7的烯基、可以具有1个以上选自该α组中的任意取代基且可以含有杂原子的碳原子数3至10的芳基、具有可以具有1个以上选自该α组中的任意取代基且可以含有杂原子的碳原子数3至12的芳基部分的芳烷基、可以具有1个以上选自该α组中的任意取代基的酰基、可以具有1个以上选自该α组中的任意取代基的甲硅烷基、可以具有1个以上选自该α组中的任意取代基的磷酸基、被核酸合成的保护基所保护的磷酸基、-p(r4)r5[式中,r4和r5分别独立地表示羟基、被核酸合成的保护基所保护的羟基、巯基、被核酸合成的保护基所保护的巯基、氨基、碳原子数1至6的烷氧基、碳原子数1至6的烷硫基、碳原子数1至6的氰基烷氧基或具有碳原子数1至6的烷基的二烷基氨基],
[0047]
x1为碳原子数1~5的亚烷基、碳原子数2~5的亚烯基或-y1-(ch2)n-、-(ch2)n-y1-或-(ch2)
l
-y1-(ch2)m-[其中,y1为磺酰基、磺酰胺基、酰胺基、酯基或羰基,n为1~5的整数,l和m为正的整数且l与m的合计为2~5],
[0048]
x2为氧原子、硫原子、-nh-或亚甲基)。
[0049]
发明的效果
[0050]
根据本发明,提供使用了具有hna骨架的新型的桥连型核苷的核苷酸。本发明的桥连型核苷也可以作为存在在特定的脏器的蓄积等风险的硫代磷酸酯修饰核酸的替代。
附图说明
[0051]
图1是表示用3’-核酸外切酶对5’-tttttttttx-3’的序列的各种寡核苷酸进行处理时的未切断寡核苷酸的比例的经时变化的图表。
[0052]
图2是表示用3’-核酸外切酶对5’-ttttttttxt-3’的序列的各种寡核苷酸进行处理时的20分钟后的未切断寡核苷酸的比例的图表。
[0053]
图3是表示投予各种寡核苷酸时的小鼠的各种组织中的相对malat1表达水平的图表。
[0054]
图4是表示投予各种寡核苷酸时的小鼠的各种组织中的相对malat1表达水平的图表。
具体实施方式
[0055]
首先,定义本说明书中所使用的术语。
[0056]
在本说明书中,术语“碳原子数1至6的直链烷基”是指碳原子数1~6的任意的直链烷基,具体是指甲基、乙基、正丙基、正丁基、正戊基或正己基。另一方面,术语“碳原子数1至6的烷基”的情况是指碳原子数1~6的任意的直链、支链或环状的烷基。
[0057]
在本说明书中,术语“碳原子数1至6的直链烷氧基”包括具有碳原子数1~6的任意的直链烷基的烷氧基。例如可以列举甲氧基、乙氧基、正丙氧基等。另一方面,术语“碳原子数1至6的烷氧基”的情况是指碳原子数1~6的任意的直链、支链或环状的烷氧基。
[0058]
在本说明书中,术语“碳原子数1至6的氰基烷氧基”是指碳原子数1~6的任意的直链、支链或环状的烷氧基中的至少1个氢原子被氰基取代后的基团。
[0059]
在本说明书中,术语“碳原子数1至6的直链烷硫基”包括具有碳原子数1~6的任意的直链烷基的烷硫基。例如可以列举甲硫基、乙硫基、正丙硫基等。另一方面,术语“碳原子数1至6的烷硫基”的情况是指碳原子数1~6的任意的直链、支链或环状的烷硫基。
[0060]
在本说明书中,术语“碳原子数1至6的直链烷基氨基”包括具有1个或2个具有碳原子数1~6的任意的直链烷基的烷基氨基的烷基氨基。例如可以列举甲基氨基、二甲基氨基、乙基氨基、甲基乙基氨基、二乙基氨基等。
[0061]
在本说明书中,术语“可以形成支链或环的碳原子数1至7的烷基”包括碳原子数1~7的任意的直链烷基、碳原子数3~7的任意的支链烷基、和碳原子数3~7的任意的环状烷基。有时仅称为“低级烷基”。例如作为碳原子数1~7的任意的直链烷基,可以列举甲基、乙基、正丙基、正丁基、正戊基、正己基、和正庚基,作为碳原子数3~7的任意的支链烷基,可以列举异丙基、异丁基、叔丁基、异戊基等,并且作为碳原子数3~7的任意的环状烷基,可以列举环丁基、环戊基、环己基等。
[0062]
在本说明书中,术语“可以形成支链或环的碳原子数2至7的烯基”包括碳原子数2~7的任意的直链烯基、碳原子数3~7的任意的支链烯基、和碳原子数3~7的任意的环状烯基。有时也简称为“低级烯基”。例如,作为碳原子数2~7的任意的直链烯基,可以列举乙烯基、1-丙烯基、2-丙烯基、1-丁烯基、2-丁烯基、1-戊烯基、2-戊烯基、3-戊烯基、4-戊烯基、1-己烯基等,作为碳原子数3~7的任意的支链烯基,可以列举异丙烯基、1-甲基-1-丙烯基、1-甲基-2-丙烯基、2-甲基-1-丙烯基、2-甲基-2-丙烯基、1-甲基-2-丁烯基等,而且,作为碳原子数3~7的任意的环状烯基,可以列举环丁烯基、环戊烯
基、环己烯基等。
[0063]
在本说明书中,术语“可以含有杂原子的碳原子数3至10的芳基”包括仅由烃构成的碳原子数6~10的任意的芳基、和构成该芳基的环结构的至少1个碳原子被杂原子(例如、氮原子、氧原子和硫原子、以及它们的组合)取代的碳原子数3~12的任意的杂芳基。作为该碳原子数6~10的芳基,可以列举苯基、萘基、茚基、薁基等,而且作为该碳原子数3~12的任意的杂芳基,可以列举吡啶基、吡咯基、喹啉基、吲哚基、咪唑基、呋喃基、噻吩基等。
[0064]
在本说明书中,作为术语“具有可以含有杂原子的碳原子数3至12的芳基部分的芳烷基”的例子,可以列举苄基、苯乙基、萘基甲基、3-苯基丙基、2-苯基丙基、4-苯基丁基、2-苯基丁基、吡啶基甲基、吲哚基甲基、呋喃基甲基、噻吩基甲基、吡咯基甲基、2-吡啶基乙基、1-吡啶基乙基、3-噻吩基丙基等。
[0065]
在本说明书中,作为术语“酰基”的例子,可以列举脂肪族酰基和芳香族酰基。具体而言,作为脂肪族酰基的例子,可以列举:甲酰基、乙酰基、丙酰基、丁酰基、异丁酰基、戊酰基(pentanoyl group)、叔戊酰基(pivaloyl group)、戊酰基(valeryl group)、异戊酰基、辛酰基、壬酰基、癸酰基、3-甲基壬酰基、8-甲基壬酰基、3-乙基辛酰基、3,7-二甲基辛酰基、十一碳酰基、十二碳酰基、十三碳酰基、十四碳酰基、十五碳酰基、十六碳酰基、1-甲基十五碳酰基、14-甲基十五碳酰基、13,13-二甲基十四碳酰基、十七碳酰基、15-甲基十六碳酰基、十八碳酰基、1-甲基十七碳酰基、十九碳酰基、二十碳酰基和二十一碳酰基这样的烷基羰基;琥珀酰基、戊二酰基、己二酰基这样的羧基化烷基羰基;氯乙酰基、二氯乙酰基、三氯乙酰基、三氟乙酰基这样的卤代低级烷基羰基;甲氧基乙酰基这样的低级烷氧基低级烷基羰基;(e)-2-甲基-2-丁烯酰基这样的不饱和烷基羰基。另外,作为芳香族酰基的例子,可以列举苯甲酰基、α-萘甲酰基、β-萘甲酰基这样的芳基羰基;2-溴苯甲酰基、4-氯苯甲酰基这样的卤代芳基羰基;2,4,6-三甲基苯甲酰基、4-甲苯酰基这样的低级烷基化芳基羰基;4-甲氧苯甲酰基这样的低级烷氧基化芳基羰基;2-羧基苯甲酰基、3-羧基苯甲酰基、4-羧基苯甲酰基这样的羧基化芳基羰基;4-硝基苯甲酰基、2-硝基苯甲酰基这样的硝基化芳基羰基;2-(甲氧基羰基)苯甲酰基这样的低级烷氧基羰基化芳基羰基;4-苯基苯甲酰基这样的芳基化芳基羰基等。优选为甲酰基、乙酰基、丙酰基、丁酰基、异丁酰基、戊酰基(pentanoyl group)、戊酰基(valeryl group)、苯甲酰基。
[0066]
在本说明书中,作为术语“甲硅烷基”的例子,可以列举三甲基甲硅烷基、三乙基甲硅烷基、异丙基二甲基甲硅烷基、叔丁基二甲基甲硅烷基、甲基二异丙基甲硅烷基、甲基二叔丁基甲硅烷基、三异丙基甲硅烷基这样的三低级烷基甲硅烷基;二苯基甲基甲硅烷基、丁基二苯基丁基甲硅烷基、二苯基异丙基甲硅烷基、苯基二异丙基甲硅烷基这样的被取代有1~2个芳基的三低级烷基甲硅烷基等。优选为三甲基甲硅烷基、三乙基甲硅烷基、三异丙基甲硅烷基、叔丁基二甲基甲硅烷基、叔丁基二苯基甲硅烷基,更优选为三甲基甲硅烷基。
[0067]
在本说明书中,作为术语“卤原子”,例如可以列举氟原子、氯原子、溴原子或碘原子。优选为氟原子或氯原子。
[0068]
在本说明书中,术语“核酸合成的氨基的保护基”、“核酸合成的羟基的保护基”、“被核酸合成的保护基所保护的羟基”、“被核酸合成的保护基所保护的磷酸基”、“被核酸合成的保护基所保护的巯基”的“保护基”,只要是在核酸合成时稳定,能够保护氨基、羟基、磷酸基或巯基的基团就没有特别限制。具体是指在酸性或中性条件下稳定,可以通过氢解、水
解、电解和光解这样的化学方法开裂的保护基。作为这样的保护基,例如可以列举低级烷基、低级烯基、酰基、四氢吡喃基或四氢硫代吡喃基、四氢呋喃基或四氢硫代呋喃基、甲硅烷基、低级烷氧基甲基、低级烷氧基化低级烷氧基甲基、卤代低级烷氧基甲基、低级烷氧基化乙基、卤代乙基、由1~3个芳基所取代的甲基、“由芳基环取代有低级烷基、低级烷氧基、卤原子或氰基的1~3个芳基所取代的甲基”、低级烷氧基羰基、“由卤原子、低级烷氧基或硝基所取代的芳基”、“由卤原子或三低级烷基甲硅烷基所取代的低级烷氧基羰基”、烯氧基羰基、“芳基环可以取代有低级烷氧基或硝基的芳烷氧基羰基”等。
[0069]
进一步具体而言,作为四氢吡喃基或四氢硫代吡喃基,可以列举四氢吡喃-2-基、3-溴四氢吡喃-2-基、4-甲氧基四氢吡喃-4-基、四氢硫代吡喃-4-基、4-甲氧基四氢硫代吡喃-4-基等。作为四氢呋喃基或四氢硫代呋喃基,可以列举四氢呋喃-2-基、四氢硫代呋喃-2-基。作为低级烷氧基甲基,可以列举甲氧基甲基、1,1-二甲基-1-甲氧基甲基、乙氧基甲基、丙氧基甲基、异丙氧基甲基、丁氧基甲基、叔丁氧基甲基等。作为低级烷氧基化低级烷氧基甲基,可以列举2-甲氧基乙氧基甲基等。作为卤代低级烷氧基甲基,可以列举2,2,2-三氯乙氧基甲基、双(2-氯乙氧基)甲基等。作为低级烷氧基化乙基,可以列举1-乙氧基乙基、1-(异丙氧基)乙基等。作为卤化乙基,可以列举2,2,2-三氯乙基等。作为由1~3个芳基所取代的甲基,可以列举苄基、α-萘基甲基、β-萘基甲基、二苯基甲基、三苯基甲基、α-萘基二苯基甲基、9-蒽基甲基等。作为“由芳基环取代有低级烷基、低级烷氧基、卤原子或氰基的1~3个芳基所取代的甲基”,可以列举4-甲基苄基、2,4,6-三甲基苄基、3,4,5-三甲基苄基、4-甲氧基苄基、4-甲氧基苯基二苯基甲基、4,4’-二甲氧基三苯基甲基、2-硝基苄基、4-硝基苄基、4-氯苄基、4-溴苄基、4-氰基苄基等。作为低级烷氧基羰基,可以列举甲氧基羰基、乙氧基羰基、叔丁氧基羰基、异丁氧基羰基等。作为“由卤原子、低级烷氧基或硝基所取代的芳基”,可以列举4-氯苯基、2-氟苯基、4-甲氧基苯基、4-硝基苯基、2,4-二硝基苯基等。作为“由卤原子或三低级烷基甲硅烷基所取代的低级烷氧基羰基”,可以列举2,2,2-三氯乙氧基羰基、2-三甲基甲硅烷基乙氧基羰基等。作为烯氧基羰基,可以列举乙烯基氧羰基、芳基氧羰基等。作为“芳基环可以取代有低级烷氧基或硝基的芳烷氧基羰基”,可以列举苄氧基羰基、4-甲氧基苄氧基羰基、3,4-二甲氧基苄氧基羰基、2-硝基苄氧基羰基、4-硝基苄氧基羰基等。
[0070]
在一个实施方式中,作为“核酸合成的羟基的保护基”,例如可以列举脂肪族酰基、芳香族酰基、由1~3个芳基所取代的甲基、“由芳基环取代有低级烷基、低级烷氧基、卤素、氰基的1~3个芳基所取代的甲基”和甲硅烷基。或者,在一个实施方式中,作为“核酸合成的羟基的保护基”,例如可以列举乙酰基、苯甲酰基、苄基、对甲氧基苯甲酰基、二甲氧基三苯甲基、单甲氧基三苯甲基、叔丁基二苯基甲硅烷基、叔丁基二甲基甲硅烷基(tbdms)基、[(三异丙基甲硅烷基)氧基]甲基(tom)基、[(2-硝基苄基)氧基]甲基(nbom)基、双(乙酰氧基乙氧基)甲基醚(ace)基、四氢-4-甲氧基-2h-吡喃-2-(mthp)基、1-(2-氰基乙氧基)乙基(cee)基、2-氰基乙氧基甲基(cem)基、叔丁基二硫代甲(dtm)基、2-(4-甲苯基磺酰基)乙氧基甲基(tem)基、和4-(n-二氯乙酰基-n-甲基氨基)苄氧基甲基(4-mabom)基。
[0071]
在一个实施方式中,作为“被核酸合成的保护基所保护的羟基”的保护基,例如可以列举脂肪族酰基、芳香族酰基、“由1~3个芳基所取代的甲基”、“由卤原子、低级烷氧基或硝基所取代的芳基”、低级烷基、和低级烯基。或者,在一个实施方式中,作为“被核酸合成的
保护基所保护的羟基”的保护基,例如可以列举苯甲酰基、苄基、2-氯苯基、4-氯苯基和2-丙烯基。
[0072]
在一个实施方式中,作为“核酸合成的氨基的保护基”,例如可以列举酰基,优选苯甲酰基。
[0073]
在一个实施方式中,作为“被核酸合成的保护基所保护的磷酸基”的“保护基”,例如可以列举低级烷基、由氰基所取代的低级烷基、芳烷基、“芳基环取代有硝基或卤原子的芳烷基”、和“由低级烷基、卤原子或硝基所取代的芳基”。或者,在一个实施方式中,作为“被核酸合成的保护基所保护的磷酸基”的“保护基”,例如可以列举2-氰乙基、2,2,2-三氯乙基、苄基、2-氯苯基和4-氯苯基。
[0074]
在一个实施方式中,作为“被核酸合成的保护基所保护的巯基”的“保护基”,例如可以列举脂肪族酰基和芳香族酰基,优选列举苯甲酰基。
[0075]
在本说明书中,在-p(r4)r5[式中,r4和r5分别独立地表示羟基、被核酸合成的保护基所保护的羟基、巯基、被核酸合成的保护基所保护的巯基、氨基、碳原子数1至6的烷氧基、碳原子数1至6的烷硫基、碳原子数1至6的氰基烷氧基或具有碳原子数1至6的烷基的二烷基氨基]所示的基团中,r4为or
4a
且r5为nr
5a
的基团称为“亚磷酰胺基”(phosphoramidite group)(其中,r
4a
例如为碳原子数1至6的氰基烷氧基,并且r
5a
例如为碳原子数1至6的烷基)。作为亚磷酰胺基,优选列举式-p(oc2h4cn)(n(ipr)2)所示的基团或式-p(och3)(n(ipr)2)所示的基团。其中,ipr表示异丙基。
[0076]
在本说明书中,“碳原子数1~5的亚烷基”是指-(ch2)n-(其中,n为1~5的整数)所示的基团、即亚甲基(-ch2-)以及由2个~5个亚甲基构成的2价亚烷基(亚乙基、亚丙基、亚丁基和亚戊基)。
[0077]
在本说明书中,“碳原子数2~5的亚烯基”是指由包含1个双键的碳原子数2~5的直链构成的二价基团,作为具体例,可以列举-ch=ch-、-ch2-ch=ch-、-ch=ch-ch2-、-ch2-ch2-ch=ch-、-ch2-ch=ch-ch2-、-ch=ch-ch2-ch2-、-ch2-ch2-ch2-ch=ch-、-ch2-ch2-ch=ch-ch2-、-ch2-ch=ch-ch2-ch2-和-ch=ch-ch2-ch2-ch2-。
[0078]
在本说明书中,术语“核苷”和“核苷类似物”是指嘌呤或嘧啶碱基与糖结合而成的“核苷”中的非天然型的核苷、以及嘌呤和嘧啶以外的能够替代嘌呤或嘧啶碱基的芳香族杂环和芳香族烃环与糖结合得到的核苷类似物。
[0079]
在本说明书中,术语“人工寡核苷酸”和“寡核苷酸类似物”是指例如2~50个相同或不同的“核苷”或“核苷类似物”通过磷酸二酯键结合得到的“寡核苷酸”的非天然型衍生物。作为那样的类似物,优选列举糖部分被修饰的糖衍生物;磷酸二酯部分被硫酯化的硫酯衍生物;末端的磷酸部分被酯化的酯体;嘌呤碱基上的氨基被酰胺化的酰胺体。
[0080]
在本说明书中,术语“其盐”是指本发明的式(i)所示的化合物的盐。作为这样的盐,例如可以列举钠盐、钾盐、锂盐这样的碱金属盐、钙盐、镁盐这样的碱土金属盐、铝盐、铁盐、锌盐、铜盐、镍盐、钴盐等的金属盐;铵盐这样的无机盐、叔辛基胺盐、二苄基胺盐、吗啉盐、葡萄糖胺(glucosamine)盐、苯基甘氨酸烷基酯盐、乙二胺盐、n-甲基葡糖胺(glucamine)盐、胍盐、二乙胺盐、三乙胺盐、二环己胺盐、n,n’-二苄基乙二胺盐、氯普鲁卡因盐、普鲁卡因盐、二乙醇胺盐、n-苄基-苯乙基胺盐、哌嗪盐、四甲基铵盐、三(羟甲基)氨
基甲烷盐这样的有机盐等的胺盐;氢氟酸盐、盐酸盐、氢溴酸盐、氢碘酸盐这样的氢卤酸盐、硝酸盐、高氯酸盐、硫酸盐、磷酸盐等的无机酸盐;甲磺酸盐、三氟甲磺酸盐、乙磺酸盐这样的低级烷烃磺酸盐、苯磺酸盐、对甲苯磺酸盐这样的芳基磺酸盐、乙酸盐、苹果酸盐、富马酸盐、琥珀酸盐、柠檬酸盐、酒石酸盐、草酸盐、马来酸盐等的有机酸盐;以及甘氨酸盐、赖氨酸盐、精氨酸盐、鸟氨酸盐、谷氨酸盐、天冬氨酸盐这样的氨基酸盐。
[0081]
在本说明书中,作为术语“其药理学上可接受的盐”,是指含有至少1个本发明的式(ii)所示的核苷结构的寡核苷酸类似物的盐。作为那样的盐,例如可以列举钠盐、钾盐、锂盐这样的碱金属盐、钙盐、镁盐这样的碱土金属盐、铝盐、铁盐、锌盐、铜盐、镍盐、钴盐等的金属盐;铵盐这样的无机盐、叔辛基胺盐、二苄基胺盐、吗啉盐、葡萄糖胺盐、苯基甘氨酸烷基酯盐、乙二胺盐、n-甲基葡糖胺盐、胍盐、二乙胺盐、三乙胺盐、二环己胺盐、n,n’-二苄基乙二胺盐、氯普鲁卡因盐、普鲁卡因盐、二乙醇胺盐、n-苄基-苯乙基胺盐、哌嗪盐、四甲基铵盐、三(羟甲基)氨基甲烷盐这样的有机盐等的胺盐;氢氟酸盐、盐酸盐、氢溴酸盐、氢碘酸盐这样的氢卤酸盐、硝酸盐、高氯酸盐、硫酸盐、磷酸盐等的无机酸盐;甲磺酸盐、三氟甲磺酸盐、乙磺酸盐这样的低级烷烃磺酸盐、苯磺酸盐、对甲苯磺酸盐这样的芳基磺酸盐、乙酸盐、苹果酸盐、富马酸盐、琥珀酸盐、柠檬酸盐、酒石酸盐、草酸盐、马来酸盐等的有机酸盐;以及甘氨酸盐、赖氨酸盐、精氨酸盐、鸟氨酸盐、谷氨酸盐、天冬氨酸盐这样的氨基酸盐。
[0082]
以下,详细说明本发明。
[0083]
(桥连型核苷)
[0084]
本发明的桥连型核苷为以下的式(i)所示的化合物或其盐:
[0085][0086]
(式中,
[0087]
base表示可以具有1个以上选自α组中的任意取代基的嘌呤-9-基或2-氧代-1,2-二氢嘧啶-1-基,其中,该α组包括羟基、被核酸合成的保护基所保护的羟基、碳原子数1至6的直链烷基、碳原子数1至6的直链烷氧基、巯基、被核酸合成的保护基所保护的巯基、碳原子数1至6的直链烷硫基、氨基、碳原子数1至6的直链烷基氨基、被核酸合成的保护基所保护的氨基和卤原子,
[0088]
r2和r3分别独立地表示氢原子、核酸合成的羟基的保护基、可以形成支链或环的碳原子数1至7的烷基、可以形成支链或环的碳原子数2至7的烯基、可以具有1个以上选自该α组中的任意取代基且可以含有杂原子的碳原子数3至10的芳基、具有可以具有1个以上选自该α组中的任意取代基且可以含有杂原子的碳原子数3至12的芳基部分的芳烷基、可以具有1个以上选自该α组中的任意取代基的酰基、可以具有1个以上选自该α组中的任意取代基的甲硅烷基、可以具有1个以上选自该α组中的任意取代基的磷酸基、被核酸合成的保护基所保护的磷酸基、-p(r4)r5[式中,r4和r5分别独立地表示羟基、被核酸合成的保护基所保护的羟基、巯基、被核酸合成的保护基所保护的巯基、氨基、碳原子数1至6的烷氧基、碳原子数1至6的烷硫基、碳原子数1至6的氰基烷氧基或具有碳原子数1至6的烷基的二烷基氨基],
[0089]
x1为碳原子数1~5的亚烷基、碳原子数2~5的亚烯基或-y1-(ch2)n-、-(ch2)n-y1-或-(ch2)
l
-y1-(ch2)m-[其中,y1为磺酰基、磺酰胺基、酰胺基、酯基或羰基,n为1~5的整数,l和m为正的整数且l与m的合计为2~5],
[0090]
x2为氧原子、硫原子、-nh-或亚甲基)。
[0091]
在上述式(i)中,“base(碱基)”例如为嘌呤碱基(即、嘌呤-9-基)或嘧啶碱基(即、2-氧代-1,2-二氢嘧啶-1-基)。这些碱基可以具有1个以上的选自包括羟基、碳原子数1至6的直链烷基、碳原子数1至6的直链烷氧基、巯基、碳原子数1至6的直链烷硫基、氨基、碳原子数1至6的直链烷基氨基、和卤原子的α组中的任意取代基。
[0092]
作为上述“base”的具体例,可以列举腺嘌呤基、鸟嘌呤基、胞嘧啶基、尿嘧啶基、和胸腺嘧啶基、以及6-氨基嘌呤-9-基、2,6-二氨基嘌呤-9-基、2-氨基-6-氯嘌呤-9-基、2-氨基-6-氟嘌呤-9-基、2-氨基-6-溴嘌呤-9-基、2-氨基-6-羟基嘌呤-9-基、6-氨基-2-甲氧基嘌呤-9-基、6-氨基-2-氯嘌呤-9-基、6-氨基-2-氟嘌呤-9-基、2,6-二甲氧基嘌呤-9-基、2,6-二氯嘌呤-9-基、6-巯基嘌呤-9-基、2-氧代-4-氨基-1,2-二氢嘧啶-1-基、4-氨基-2-氧代-5-氟-1,2-二氢嘧啶-1-基、4-氨基-2-氧代-5-氯-1,2-二氢嘧啶-1-基、2-氧代-4-甲氧基-1,2-二氢嘧啶-1-基、2-氧代-4-巯基-1,2-二氢嘧啶-1-基、2-氧代-4-羟基-1,2-二氢嘧啶-1-基、2-氧代-4-羟基-5-甲基-1,2-二氢嘧啶-1-基、和4-氨基-5-甲基-2-氧代-1,2-二氢嘧啶-1-基。
[0093]
或者,“base”从导入核酸医药的观点出发,优选分别由以下的结构式:
[0094][0095]
所示的基团、以及2-氧代-4-羟基-5-甲基-1,2-二氢嘧啶-1-基、2-氧代-4-氨基-1,2-二氢嘧啶-1-基、6-氨基嘌呤-9-基、2-氨基-6-羟基嘌呤-9-基、4-氨基-5-甲基-2-氧代-1,2-二氢嘧啶-1-基、和2-氧代-4-羟基-1,2-二氢嘧啶-1-基。“base”还优选在合成寡核苷酸时,构成上述基团的羟基和氨基被保护基保护。
[0096]
如式(i)所示,本发明的桥连型核苷酸具有核酸糖部由吡喃糖环构成的己糖醇核酸(hna)骨架,在该hna骨架的1’位与3’位之间导入有桥连结构(-x1-x2-)。
[0097]
这里,如果关注该桥连结构,则在一个实施方式中,作为式(i)所示的化合物的一例,可以列举以下的式(i-a)~(i-d)所示的化合物:
[0098][0099]
(式(i-a)~(i-d)中,base、r2、r3和x1的意义与上述式(i)中定义的基团相同)。
[0100]
或者,在一个实施方式中,作为式(i)所示的化合物的其他例子,可以列举以下的式(i-e)~(i-h)所示的化合物:
[0101][0102]
(式(i-e)~(i-g)中,base、r2、r3、x2、y1、l、m和n的意义与上述式(i)中定义的相同)。
[0103]
作为这样的式(i)所示的化合物的具体例,并没有限定,可以列举以下的式(i-1)~(i-3)所示的化合物:
[0104][0105]
(式(i-1)~(i-3)中、base、r2和r3的意义与上述式(i)中定义的相同)。
[0106]
本发明的桥连型核苷从上述式(i)可知具有类似于天然核酸的n型构象的hna骨架。由此,后述的寡核苷酸对单链rna(ssrna)具有良好的结合亲和性。
[0107]
(寡核苷酸)
[0108]
在本发明中,寡核苷酸能够使用这样的式(i)的桥连型核苷,例如经过该领域中公知的的亚酰胺法或m.kuwahara等、nucleic acids res.,2008年,36卷,13号,4257-4265页所记载的三磷酸化而容易地制造。
[0109]
本发明的寡核苷酸或其药理学上可接受的盐(以下,有时将这些总称为“本发明的寡核苷酸”)至少包含1个以下的式(ii)所示的核苷结构:
[0110][0111]
(式中,
[0112]
base表示可以具有1个以上选自α组中的任意取代基的嘌呤-9-基或2-氧代-1,2-二氢嘧啶-1-基,其中,该α组包括羟基、被核酸合成的保护基所保护的羟基、碳原子数1至6的直链烷基、碳原子数1至6的直链烷氧基、巯基、被核酸合成的保护基所保护的巯基、碳原子数1至6的直链烷硫基、氨基、碳原子数1至6的直链烷基氨基、被核酸合成的保护基所保护的氨基和卤原子,
[0113]
x1为碳原子数1~5的亚烷基、碳原子数2~5的亚烯基或-y1-(ch2)n-、-(ch2)n-y1-或-(ch2)
l
-y1-(ch2)m-[其中,y1为磺酰基、磺酰胺基、酰胺基、酯基或羰基,n为1~5的整数,l和m为正的整数且l与m的合计为2~5],
[0114]
x2为氧原子、硫原子、-nh-或亚甲基)。
[0115]
在一个实施方式中,作为本发明的寡核苷酸所含的式(ii)的核苷结构的一例,可以列举以下的式(ii-a)~(ii-d)所示的结构:
[0116][0117]
(式(ii-a)~(ii-d)中,base和x1的意义与上述式(ii)中定义的相同)。
[0118]
或者,在一个实施方式中,作为本发明的寡核苷酸所含的式(ii)的核苷结构的其他例子,可以列举以下的式(ii-e)~(ii-h)所示的结构:
[0119][0120]
(式(ii-e)~(ii-g)中、base、x2、y1、l、m和n的意义与上述式(ii)中定义的相同)。
[0121]
作为这样的本发明的寡核苷酸所含的式(ii)的核苷结构的具体例,并没有限定,可以列举以下的式(ii-1)~(ii-3)所示的结构:
[0122][0123]
(式(ii-1)~(ii-3)中,base的意义与上述式(i)中定义的相同)。
[0124]
本发明的寡核苷酸在任意位置至少具有1个上述核苷结构。其位置和数量没有特别限定,可以根据目的适当设计。
[0125]
包含这样的核苷结构的寡核苷酸(反义分子)与使用以往的2’,4’-bna/lna的情况相比,核酸酶抗性飞跃性地提高。另外,具有与公知的2’,4’-bna/lna相匹敌的ssrna结合亲和性。本发明的寡核苷酸还具有凌驾于现在市售的众多核酸医药品所含的硫代磷酸酯修饰核酸(以下有时称为ps修饰核酸)的酶耐性能。
[0126]
由此,可以期待使用本发明的桥连型核苷所合成的寡核苷酸作为以抗肿瘤剂、抗病毒剂为代表的抑制或恢复特定的基因的作用来治疗疾病的医药品(反义分子)的有用性。
[0127]
特别是在反义法中,需要对互补正义链rna的结合亲和性和生物体内dna分解酶的耐性这两者。一般而言,已知核酸在单链状态下,糖部的结构不断在近似于dna双链的形态和近似于dna-rna双链或近似于rna双链的形态之间摇摆。因此,通过对核酸糖部的构象以本发明那样预先规定的样式进行化学修饰,能够使对靶标ssrna的结合亲和性大幅提高。另外,核酸分解酶切断寡核酸的磷酸二酯部分,但在本发明的桥连型核苷的情况下,由于在糖部分配置有大体积的取代基,所以通过空间位阻能够抑制寡核酸的分解。进而,在本发明的桥连型核苷中,如上所述在hna骨架的1’位与3’位之间导入有桥连结构(-x1-x2-)。通过该hna骨架和桥连结构,本发明的桥连型核苷对这些靶标ssrna的结合亲和性和抗酶性能这两者都提高。
[0128]
本发明的寡核苷酸例如能够配合赋形剂、粘合剂、防腐剂、氧化稳定剂、崩解剂、润滑剂、矫味剂等医药的制剂技术领域中通常使用的辅助剂,制成非口服给药制剂或脂质体制剂。另外,例如能够配合该技术领域中通常使用的医药用载体,配置液剂、霜剂、软膏剂等局部用的制剂。
[0129]
实施例
[0130]
以下,基于实施例进一步详细地说明本发明,但本发明不受这些实施例限定。
[0131]
(实施例1:桥连型核苷的合成(1))
[0132][0133]
各工序的试剂和条件:(a)双(三亚甲基甲硅烷基)乙炔,sncl4,dcm,-20℃,0.5小时;(b)naome,meoh,室温,1小时,94%(2个工序);(c)mcpba,dcm,室温,24小时;(d)茴香醛二甲缩醛,csa,mecn,回流下,1.5小时,62%(2个工序);(e)pd/pei,h2,meoh/1,4-二噁烷,室温,1.5小时;(f)o3,dcm/meoh,-78℃,1小时;nabh4,室温,1小时,82%(2个工序);(g)胸腺嘧啶,dbu,mecn,85℃(mw),48小时;(h)tscl,tea,dmap,dcm,室温,2.5小时,52%(2个工序);(i)nah,dmf,室温,1小时,97%;(j)pd(oh)2/c,h2,meoh,室温,1小时;(k)dmtrcl,嘧啶,室温,3小时,75%(2个工序);(l)2-氰乙基n,n-二异丙基氯亚磷酰胺,dipea,nmi,mecn,室温,1小时,79%。
[0134]
(1-1)化合物2的合成
[0135][0136]
在化合物1(3.00g,11.02mmol)和双三甲基甲硅烷基乙炔(3.76g,22.04mmol)的无水二氯甲烷溶液(50ml)中在氮气流下以-20℃添加1.0m的sncl4·
二氯甲烷溶液(16.53ml,16.53mmol),在该温度搅拌0.5小时。反应结束后,将反应溶液加入到饱和碳酸氢钠水/饱和罗谢尔盐水溶液(=1:1(容量比),200ml),在0℃搅拌30分钟。然后,将混合物用二氯甲烷提取,用水和饱和食盐水清洗,用无水硫酸钠使其干燥,将溶剂在减压下蒸馏除去,由此作为粗产物得到化合物2。将该化合物2不作纯化而直接用于后续反应。
[0137]
(1-2)化合物3的合成
[0138][0139]
在上述得到的化合物2的甲醇溶液(50ml)中在冰冷下添加5m的甲氧基钠的甲醇溶液(2.20ml,11.02mmol),在氮气流下室温搅拌1小时。反应结束后,在反应溶液中加入强酸性阳离子交换树脂(富士胶片和光纯药株式会社制dowex 50
×
8 200-400网)搅拌30分钟,进行中和。然后将混合物过滤,浓缩滤液。将所得到的残渣通过硅胶柱色谱(sio2,甲醇/chcl3=5%)进行纯化,作为无色油状物质得到化合物3(1.60g,94%,从化合物1起2个工序)。
[0140]
将所得到的化合物3的物性数据表示于表1。
[0141]
[表1]
[0142][0143]
(1-3)化合物4的合成
[0144][0145]
在上述得到的化合物3(5.36g,34.8mmol)的二氯甲烷溶液(150ml)中,在冰冷下添加间氯苯甲酸(mcpba)(纯度70%,17.1g,69.5mmol),在室温搅拌24小时。反应结束后,将反应溶液直接载置在硅胶柱色谱上,简易地纯化(sio2,己烷/乙酸乙酯=1:1至0/1),作为无色油状物质得到环氧二醇的立体异构体混合物(5.62g)。然后,将该环氧二醇(5.62g)、茴香醛二甲缩醛(11.8ml,69.5mmol)和(
±
)-樟脑磺酸(807.6mg,3.48mmol)的无水乙腈溶液(150ml)在氮气流下加热回流1.5小时。反应结束后,将反应溶液用三乙胺(1ml)中和,缓慢进行冰冷。将生成的白色固体通过过滤回收,将滤液减压蒸馏除去,用甲醇清洗。将生成的白色固体通过再次过滤回收,合并得到化合物4(6.23g,62%,从化合物3起2个工序)。
[0146]
将所得到的化合物4的物性数据表示于表2。
[0147]
[表2]
[0148][0149]
(1-4)化合物5的合成
[0150][0151]
在上述得到的化合物4(300mg,1.04mmol)的甲醇/1,4-二噁烷混合溶液(10ml,甲醇/1,4-二噁烷=1:4(容量比))中添加钯/聚乙烯亚胺(pd/pei)(30mg,10重量%),在氢气流下以室温搅拌1.5小时。反应结束后,将混合物载持于快速硅胶柱色谱进行清洗(chcl3/甲醇=14/1),由此作为粗产物得到化合物5。在该粗产物(化合物5)中,过度还原的1-乙基体难以分离,因此不进行进一步纯化而直接用于后续反应。
[0152]
(1-5)化合物6的合成
[0153][0154]
使上述得到的化合物5(298mg)的甲醇/二氯甲烷混合溶液(10ml,dcm/meoh=4:1(容量比))在-78℃与臭氧反应。1小时后,在该温度加入硼氢化钠(157mg,4.16mmol),缓慢使其升温到室温。搅拌1小时后,加入饱和氯化铵水溶液和乙酸乙酯,进行提取。将有机层用饱和碳酸氢钠水和饱和食盐水清洗后,用无水硫酸钠干燥,使溶剂减压蒸馏除去。将所得到的残渣通过硅胶柱色谱(sio2,丙酮/chcl3=20%至27%)纯化,作为白色固体得到化合物6(250mg,82%,从化合物4起2个工序)。
[0155]
将所得到的化合物6的物性数据表示于表3。
[0156]
[表3]
[0157][0158]
(1-6)化合物7的合成
[0159][0160]
将上述得到的化合物6(353.1mg,1.20mmol)、胸腺嘧啶(302.6mg,2.40mmol)和二氮杂双环十一碳烯(dbu)(717.7μl,4.80mmol)的无水乙腈溶液(12ml)在微波的照射下以85℃加热48小时。反应结束后,将生成的白色固体通过过滤回收,作为粗产物得到化合物7。将该化合物7不作纯化而直接用于后续反应。
[0161]
(1-7)化合物8的合成
[0162][0163]
在上述得到的化合物7(516.5mg)、三乙胺(334.5μl,2.40mmol)和4,4-二甲基氨基吡啶(14.7mg,0.120mmol)的无水二氯甲烷溶液(12ml)中在冰冷下添加对甲苯磺酰氯(tscl)(274.5mg,1.44mmol),在氮气流下以室温搅拌2.5小时。反应结束后,将反应溶液加入到饱和碳酸氢钠水中,用二氯甲烷提取。将有机层用饱和食盐水清洗后,用无水硫酸钠使其干燥,将溶剂减压蒸馏除去。将所得到的残渣通过硅胶柱色谱(sio2,乙酸乙酯/chcl3=30%至80%)纯化,作为白色固体得到化合物8(360mg,52%,从化合物6起2个工序)。
[0164]
将所得到的化合物8的物性数据表示于表4。
[0165]
[表4]
[0166][0167]
(1-8)化合物9的合成
[0168][0169]
在上述得到的化合物8(360mg,0.626mmol)的无水dmf溶液(6.0ml)中加入60%油性氢化钠(62.7mg,1.57mmol),在氮气流下以室温搅拌1小时。反应结束后,加入饱和氯化铵水溶液和乙酸乙酯进行提取。将有机层用饱和碳酸氢钠水和饱和食盐水清洗后,用无水硫
酸钠使其干燥,将溶剂减压蒸馏除去。将所得到的残渣通过硅胶柱色谱(sio2,乙酸乙酯/己烷=60%至90%)纯化,作为白色固体得到化合物9(244.6mg,97%)。
[0170]
将所得到的化合物9的物性数据表示于表5。
[0171]
[表5]
[0172][0173]
(1-9)化合物10的合成
[0174][0175]
在上述得到的化合物9(28.7mg,0.0713mmol)的甲醇溶液(1.0ml)中加入pd(oh)2/c(7.1mg),在氢气流下以室温搅拌1小时。反应结束后,将混合物过滤,用甲醇清洗后,将滤液减压蒸馏除去,作为粗产物得到化合物10。将该化合物10不经纯化直接用于后续反应。
[0176]
(1-10)化合物11的合成
[0177][0178]
在上述得到的化合物10的无水吡啶溶液(1.0ml)中加入4,4’-二甲氧基三苯甲基氯(36.2mg,0.107mmol),在氮气流下以室温搅拌3小时。反应结束后,在反应溶液中加入甲醇,将溶剂减压蒸馏除去。在所得到的残渣中加入乙酸乙酯和饱和碳酸氢钠水,进行提取。将有机层用饱和碳酸氢钠水和饱和食盐水清洗后,用无水硫酸钠使其干燥,将溶剂减压蒸馏除去。将所得到的残渣通过硅胶柱色谱(sio2,乙酸乙酯/己烷=70%至100%)纯化,作为白色固体得到化合物11(31.2mg,75%,从化合物9起2个工序)。
[0179]
将所得到的化合物11的物性数据表示于表6。
[0180]
[表6]
[0181][0182]
(1-11)化合物12的合成
[0183][0184]
在上述得到的化合物11(321.9mg,0.549mmol)、n,n-二异丙基乙胺(286.7μl,1.65mmol)和1-甲基咪唑(13.2μl,0.165mmol)的无水乙腈溶液(5.5ml)中在冰冷下加入2-氰乙基-n,n-二异丙基磷酰氯(183.6μl,0.823mmol),在氮气流下以室温搅拌1小时。反应结束后,加入饱和碳酸氢钠水和乙酸乙酯进行提取。将有机层用饱和碳酸氢钠水和饱和食盐水清洗后,用无水硫酸钠使其干燥,将溶剂减压蒸馏除去。将所得到的残渣用硅胶柱色谱(sio2,乙酸乙酯/己烷=65%至95%)纯化,作为白色固体得到化合物12(341.4mg,79%)。
[0185]
将所得到的化合物12的物性数据表示于表7。
[0186]
[表7]
[0187][0188]
(实施例2:桥连型核苷的合成(2))
[0189][0190]
各工序的试剂和条件:(a)pd/pei,h2,meoh/1,4-二噁烷,室温,1.5小时;(b)9-bbn,thf,室温,1小时;nabo3·
4h2o,h2o,室温,1小时,85%(2个工序);(c)胸腺嘧啶,dbu,mecn,100℃(mw),24小时;(d)tscl,tea,dmap,dcm,室温,6小时,51%(2个工序);(e)nah,dmf,90℃,48小时,63%;(f)pd(oh)2/c,h2,meoh,室温,1.5小时;(g)dmtrcl,吡啶,室温,3小时,61%(2个工序);(h)2-氰乙基n,n-二异丙基氯亚磷酰胺,dipea,nmi,mecn,室温,1小时,84%。
[0191]
(2-1)化合物13的合成
[0192][0193]
首先,与实施例1同样得到化合物4至化合物5。然后,在所得到的化合物5(1.03g)的无水四氢呋喃溶液(6.0ml)中加入0.5m的9-硼双环[3.3.1]壬烷(9-bbn)的四氢呋喃溶液(13.9ml,6.94mmol),在氮气流下以室温搅拌1小时。原料消失后,在反应溶液中在冰冷下加入水(20ml)和过硼酸钠
·
四水合物(5.33g,34.7mmol),在室温再搅拌1小时。然后,过滤混合物,将滤液用乙酸乙酯提取。将有机层用饱和食盐水清洗后,用无水硫酸钠使其干燥,将溶剂减压蒸馏除去。将所得到的残渣通过硅胶柱色谱(sio2,甲醇/chcl3=2%至5%)纯化,作为白色固体得到化合物13(910mg,85%,从化合物4起2个工序)。
[0194]
将所得到的化合物13的物性数据表示于表8。
[0195]
[表8]
[0196][0197]
(2-2)化合物14的合成
[0198][0199]
将上述得到的化合物13(1.10g,3.57mmol)、胸腺嘧啶(900mg,7.14mmol)和二氮杂双环十一碳烯(dbu)(2.13ml,14.3mmol)的无水乙腈溶液(17.8ml)在微波照射下以100℃加热24小时。反应结束后,将溶剂减压蒸馏除去,加入二氯甲烷和饱和碳酸氢钠水进行提取。将有机层用饱和食盐水清洗后,用无水硫酸钠干燥,将溶剂减压蒸馏除去,作为粗产物得到化合物14。将该化合物14不作纯化而直接用于后续反应。
[0200]
(2-3)化合物15的合成
[0201][0202]
在上述得到的化合物14(1.58g)、三乙胺(1.24ml,8.92mmol)和4,4-二甲基氨基吡啶(43.6mg,0.357mmol)的无水二氯甲烷溶液(36ml)中在冰冷下加入对甲苯磺酰氯(tscl)(1.02g,5.35mmol),在氮气流下以室温搅拌6小时。反应结束后,将反应溶液加入到饱和碳酸氢钠水中,用二氯甲烷提取。将有机层用饱和食盐水清洗后,用无水硫酸钠使其干燥,将溶剂减压蒸馏除去。将所得到的残渣通过硅胶柱色谱(sio2,乙酸乙酯/chcl3=30%至80%)纯化,作为白色固体得到化合物15(1.08g,51%,从化合物13起2个工序)。
[0203]
将所得到的化合物15的物性数据表示于表9。
[0204]
[表9]
[0205][0206]
(2-4)化合物16的合成
[0207][0208]
在上述得到的化合物15(1.08g,1.84mmol)的无水dmf溶液(18ml)中加入60%油性氢化钠(183.5mg,4.59mmol),在氮气流下以90℃搅拌48小时。反应结束后,加入饱和氯化铵水溶液和乙酸乙酯进行提取。将有机层用饱和碳酸氢钠水和饱和食盐水清洗后,用无水硫酸钠使其干燥,将溶剂减压蒸馏除去。将所得到的残渣用硅胶柱色谱(sio2,乙酸乙酯/己烷=60%至100%)纯化,作为白色固体得到化合物16(480mg,63%)。
[0209]
将所得到的化合物16的物性数据表示于表10。
[0210]
[表10]
[0211][0212]
(2-5)化合物17的合成
[0213][0214]
在上述得到的化合物16(47.1mg,0.113mmol)的甲醇溶液(1.0ml)中加入pd(oh)2/c(11.3mg),在氢气流下以室温搅拌1小时。反应结束后,将混合物过滤,用甲醇清洗后,将滤
液减压蒸馏除去,由此作为粗产物得到化合物17。将该化合物17不作纯化而直接用于后续反应。
[0215]
(2-6)化合物18的合成
[0216][0217]
在上述得到的化合物17的无水吡啶溶液(1.0ml)中加入4,4’-二甲氧基三苯甲基氯(57.5mg,0.170mmol),在氮气流下以室温搅拌3小时。反应结束后,在反应溶液中加入甲醇,将溶剂减压蒸馏除去。在所得到的残渣中加入乙酸乙酯和饱和碳酸氢钠水进行提取。将有机层用饱和碳酸氢钠水和饱和食盐水清洗后,用无水硫酸钠使其干燥,将溶剂减压蒸馏除去。将所得到的残渣通过硅胶柱色谱(sio2,乙酸乙酯/己烷=70%至100%)纯化,作为白色固体得到化合物18(41.5mg,61%,从化合物16起2个工序)。
[0218]
将所得到的化合物18的物性数据表示于表11。
[0219]
[表11]
[0220][0221]
(2-7)化合物19的合成
[0222][0223]
在上述得到的化合物18(282.0mg,0.469mmol)、n,n-二异丙基乙胺(245.3μl,1.41mmol)和1-甲基咪唑(11.3μl,0.141mmol)的无水乙腈溶液(4.7ml)中在冰冷下加入2-氰乙基-n,n-二异丙基磷酰氯(157.1μl,0.704mmol),在氮气流下以室温搅拌1小时。反应结束后,加入饱和碳酸氢钠水和乙酸乙酯进行提取。将有机层用饱和碳酸氢钠水和饱和食盐水清洗后,用无水硫酸钠使其干燥将溶剂减压蒸馏除去。将所得到的残渣通过硅胶柱色谱(sio2,乙酸乙酯/己烷=65%至95%)纯化,作为白色固体得到化合物19(317.3mg,84%)。
[0224]
将所得到的化合物19的物性数据表示于表12。
[0225]
[表12]
[0226][0227]
(实施例3:桥连型核苷的合成(3))
[0228][0229]
各工序的试剂和条件:(a)pd/pei,h2,meoh/1,4-二噁烷,室温,3小时;(b)胸腺嘧啶,dbu,mecn,100℃(mw),24小时;(c)烯丙基溴,nah,thf,室温,4天;(d)第二代格拉布(grubbs)催化剂,甲苯,50℃,5.5小时,33%(4个工序);(e)pd(oh)2/c,h2,meoh,室温,3小时;(f)dmtrcl,吡啶,室温,2.5小时,71%(2个工序);(g)2-氰乙基n,n-二异丙基氯亚磷酰胺,dipea,nmi,mecn,室温,1小时,78%。
[0230]
(3-1)化合物20的合成
[0231][0232]
首先,与实施例1同样得到化合物4至化合物5。接着,将所得到的化合物5(1.15g)、胸腺嘧啶(1.01g,8.00mmol)和二氮杂双环十一碳烯(dbu)(2.39ml,16.0mmol)的无水乙腈溶液(18ml)在微波照射下以100℃加热24小时。反应结束后,将溶剂减压蒸馏除去,加入二氯甲烷和饱和碳酸氢钠水进行提取。将有机层用饱和食盐水清洗后,用无水硫酸钠使其干燥,将溶剂减压蒸馏除去。将所得到的残渣通过硅胶柱色谱(sio2,丙酮/chcl3=20%至40%)纯化,作为淡黄色固体得到化合物20和1-乙基体(副产物)的混合物(1.17g,化合物
20:1-乙基体=1:0.26)。化合物20和1-乙基体的分离困难,因此将它们不作纯化而直接用于后续反应。
[0233]
将所得到的化合物20的物性数据表示于表13。此外,关于nmr,以副产物(1-乙基体)与化合物20的混合物的状态测定,仅表示所得到的化合物20的信号的归属。
[0234]
[表13]
[0235][0236]
(3-2)化合物21的合成
[0237][0238]
在上述得到的化合物20和1-乙基体的混合物(1.17g,约2.81mmol)的无水四氢呋喃溶液(4.7ml)中加入60%油性氢化钠(336.8mg,8.42mmol),在氮气流下以室温搅拌1小时。接着,在反应溶液中加入烯丙基溴(308.9μl,3.65mmol),在氮气流下以室温搅拌4天。反应结束后,加入饱和氯化铵水溶液和乙酸乙酯进行提取。将有机层用饱和碳酸氢钠水和饱和食盐水清洗后,用无水硫酸钠使其干燥,将溶剂减压蒸馏除去。将所得到的残渣用硅胶柱色谱(sio2,乙酸乙酯/己烷=40%至70%)纯化,作为黄色固体得到化合物21和1-乙基体(副产物)的混合物(900mg,化合物21:1-乙基体=1:0.26)。
[0239]
将所得到的化合物21的物性数据表示于表14。此外,关于nmr,以副产物(1-乙基体)与化合物21的混合物的状态测定,仅表示所得到的化合物21的信号的归属。
[0240]
[表14]
[0241][0242]
(3-3)化合物22的合成
[0243]
[0244]
在上述得到的化合物21和1-乙基体的混合物(900mg,约1.56mmol)的脱氧甲苯溶液(31ml)中在氮气流下以室温加入第二代格拉布催化剂(66.3mg,0.078mmol),以50℃搅拌5.5小时。然后,将反应溶液减压蒸馏除去,将所得到的残渣用硅胶柱色谱(sio2,乙酸乙酯/己烷=60%至100%)纯化,作为淡黄色固体得到化合物22(564.1mg,从化合物4的33%)。
[0245]
将所得到的化合物22的物性数据表示于表15。
[0246]
[表15]
[0247][0248]
(3-4)化合物23的合成
[0249][0250]
在上述得到的化合物22(522.9mg,1.22mmol)的甲醇溶液(12.0ml)中加入pd(oh)2/c(122.0mg),在氢气流下以室温搅拌3小时。反应结束后,将混合物过滤并用甲醇清洗,将滤液减压蒸馏除去,由此作为粗产物得到化合物23。将该化合物23不作纯化而直接用于后续反应。
[0251]
(3-5)化合物24的合成
[0252][0253]
在上述得到的化合物23的无水吡啶溶液(12.0ml)中加入4,4’-二甲氧基三苯甲基氯(620.3mg,1.83mmol),在氮气流下以室温搅拌2.5小时。反应结束后,在反应溶液中加入甲醇,将溶剂减压蒸馏除去。在所得到的残渣中加入乙酸乙酯和饱和碳酸氢钠水进行提取。将有机层用饱和碳酸氢钠水和饱和食盐水清洗后,用无水硫酸钠使其干燥,将溶剂减压蒸馏除去。将所得到的残渣通过硅胶柱色谱(sio2,丙酮/氯仿=15%至40%)纯化,作为白色固体得到化合物24(530.1mg,71%,从化合物22起2个工序)。
[0254]
将所得到的化合物24的物性数据表示于表16。
[0255]
[表16]
[0256][0257]
(3-6)化合物25的合成
[0258][0259]
在上述得到的化合物24(227.2mg,0.370mmol)、n,n-二异丙基乙胺(193.2μl,1.12mmol)和1-甲基咪唑(8.90μl,0.111mmol)的无水乙腈溶液(3.7ml)中在冰冷下加入2-氰乙基-n,n-二异丙基磷酰氯(123.7μl,0.554mmol),在氮气流下以室温搅拌1小时。反应结束后,加入饱和碳酸氢钠水和乙酸乙酯进行提取。将有机层用饱和碳酸氢钠水和饱和食盐水清洗后,用无水硫酸钠使其干燥,将溶剂减压蒸馏除去。将所得到的残渣通过硅胶柱色谱(sio2,乙酸乙酯/己烷=40%至80%)纯化,作为白色固体得到化合物25(235.4mg,78%)。
[0260]
将所得到的化合物25的物性数据表示于表17。
[0261]
[表17]
[0262][0263]
(实施例4:桥连型核苷的合成(4))
[0264][0265]
各工序的试剂和条件:(a)tescl、吡啶、室温、4小时、96%;(b)1,2,4-三唑,三乙胺,磷酰氯、乙腈、室温、45分钟;(c)氢氧化铵、1,4-二噁烷、室温、3小时、99%(2个工序);(d)苯甲酸酐、吡啶、40℃、6小时、79%;(e)四正丁基氟化铵、thf、室温、4小时、95%;(f)2-氰乙基n,n-二异丙基氯亚磷酰胺,dipea,n-甲基咪唑,mecn,室温,4小时,84%。
[0266]
(4-1)化合物26的合成
[0267][0268]
在上述得到的化合物24(484.7mg,0.79mmol)的吡啶溶液(8ml)中滴加三乙基氯硅烷(650μl,3.9mmol),在氮气流下以室温搅拌4小时。反应结束后,加入饱和碳酸氢钠水和乙酸乙酯进行提取。将有机层用水和饱和食盐水清洗后,用无水硫酸钠使其干燥,将溶剂减压蒸馏除去。将所得到的残渣通过硅胶柱色谱(sio2,己烷/乙酸乙酯=60/40至40/60)纯化,作为白色固体得到化合物26(551.2mg,96%)。
[0269]
将所得到的化合物26的物性数据表示于表18。
[0270]
[表18]
[0271][0272]
(4-2)化合物27的合成
[0273][0274]
在上述得到的化合物26(514.3mg,0.71mmol)、三乙胺(1.5ml,10.8mmol)和1,2,4-三唑(714.4mg,10.3mmol)的无水乙腈溶液(7ml)中滴加磷酰氯(200μl,2.15mmol),在氮气流下以室温搅拌45分钟。反应结束后,加入饱和碳酸氢钠水和乙酸乙酯进行提取。将有机层用水和饱和食盐水清洗后,用无水硫酸钠使其干燥,将溶剂减压蒸馏除去。加入1,4-二噁烷(7ml)、28%铵水溶液(1.2ml,9.9mmol),以室温搅拌3小时。反应完结后,将溶剂减压蒸馏除去。将所得到的残渣通过硅胶柱色谱(sio2,乙酸乙酯/甲醇=95/5至90/10)纯化,作为白色固体得到化合物27(511.9mg,99%)。
[0275]
将所得到的化合物27的物性数据表示于表19。
[0276]
[表19]
[0277][0278]
(4-3)化合物28的合成
[0279][0280]
在上述得到的化合物27(702.8mg,0.97mmol)的吡啶溶液(10ml)中加入无水苯甲酸(328.4mg,1.45mmol),在氮气流下以40℃搅拌6小时。反应结束后,加入饱和碳酸氢钠水和乙酸乙酯进行提取。将有机层用水和饱和食盐水清洗后,用无水硫酸钠使其干燥,将溶剂减压蒸馏除去。将所得到的残渣通过硅胶柱色谱(sio2,氯仿/甲醇=99/1至97/3)纯化,作为白色固体得到化合物28(634.8mg,79%)。
[0281]
将所得到的化合物28的物性数据表示于表20。
[0282]
[表20]
[0283][0284]
(4-4)化合物29的合成
[0285][0286]
在上述得到的化合物28(43.5mg,0.05mmol)的四氢呋喃(thf)溶液(1.0ml)中滴加四丁基氟化铵(1m thf溶液、157μl,0.16mmol),在室温搅拌4小时。反应结束后,将溶剂减压蒸馏除去。将所得到的残渣通过硅胶柱色谱(sio2,己烷/乙酸乙酯=60/40至40/60)纯化,作为白色固体得到化合物29(35.6mg,95%)。
[0287]
将所得到的化合物29的物性数据表示于表21。
[0288]
[表21]
[0289][0290]
(4-5)化合物30的合成
[0291][0292]
在上述得到的化合物29(31,0mg,0.04mmol)的无水乙腈溶液(1ml)中加入n,n-二异丙基乙胺(23μl,0.13mmol)、1-甲基咪唑(1μl,0.013mmol)和2-氰乙基-n,n-二异丙基磷酰氯(15μl,0.067mmol),在氮气流下以室温搅拌4小时。反应结束后,加入甲醇、饱和碳酸氢钠水和乙酸乙酯进行提取。将有机层用水和饱和食盐水清洗后,用无水硫酸钠使其干燥,将溶剂减压蒸馏除去。将所得到的残渣通过硅胶柱色谱(sio2,己烷/乙酸乙酯=65/35至45/55)纯化,作为白色固体得到化合物30(34.3mg,84%)。
[0293]
将所得到的化合物30的物性数据表示于表22。
[0294]
[表22]
[0295][0296]
(实施例5:桥连型核苷的合成(5))
[0297][0298]
各工序的试剂和条件:(b)腺嘌呤,dbu,dmf,150℃(mw),2小时;(c)n,n-二甲基甲酰胺二甲基缩醛,thf,室温,16小时;(d)烯丙基溴、氢化钠,碘化钠,dmf,-30℃,3小时;(e)naoh水溶液,meoh,40℃,1小时;(f)第二代hoveyda-grubbs催化剂、1,4-苯醌、甲苯、50℃,20小时,53%(从化合物4起6个工序);(g)pd(oh)2/c,meoh,thf,60℃,7小时,70%;(h)四甲基甲硅烷基氯,苯甲酰氯、氨水、吡啶、室温、23小时、55%。
[0299]
(5-1)化合物31的合成
[0300][0301]
将上述得到的化合物5(1.27g,4.36mmol)、腺嘌呤(650mg,4.81mmol)和二氮杂双环十一碳烯(dbu)(976μl,6.54mmol)的无水二甲基甲酰胺溶液(11.0ml)在微波照射下以150℃加热2小时。放置冷却后,加入乙酸乙酯和水进行提取。将有机层用饱和食盐水清洗后,用无水硫酸钠使其干燥,将溶剂减压蒸馏除去。将所得到的残渣通过硅胶柱色谱(sio2,乙酸乙酯/甲醇=95/5至85/15)纯化,作为淡黄色固体得到化合物31和1-乙基体的混合物(1.69g)。由于化合物31和1-乙基体的分离困难,所以将它们不作纯化而直接用于后续反应。
[0302]
将所得到的化合物31的物性数据表示于表23。此外,关于nmr,以副产物(1-乙基体)和化合物31的混合物的状态进行测定,仅表示所得到的化合物31的信号的归属。
[0303]
[表23]
[0304][0305]
(5-2)化合物32的合成
[0306][0307]
在上述得到的化合物31和1-乙基体的混合物(423mg,约0.10mmol)的无水四氢呋喃溶液(10ml)中加入n,n-二甲基甲酰胺二甲基缩醛(400μl,2.99mmol),在氮气流下以室温搅拌16小时。反应结束后,将溶剂减压蒸馏除去,加入无水二甲基甲酰胺(10ml)、60%油性氢化钠(60.1mg,1.50mmol),在氮气流下以-30℃搅拌1小时。然后在反应溶液中加入烯丙基溴(100μl,1.19mmol)、碘化钠(30.1mg,0.20mmol),在氩气流下以-30℃搅拌3小时。反应结束后,加入甲醇(1ml)以-20℃搅拌20分钟后,加入饱和氯化铵水溶液,用乙酸乙酯提取。将有机层用饱和碳酸氢钠水、水和饱和食盐水清洗后,用无水硫酸钠使其干燥,将溶剂减压蒸馏除去。将所得到的残渣通过硅胶柱色谱(sio2,乙酸乙酯/甲醇=98/2至93/7)纯化,作为白色固体得到化合物32和1-乙基体的混合物(426.0mg)。
[0308]
将所得到的化合物32的物性数据表示于表24。此外,关于nmr,以副产物(1-乙基体)与化合物32的混合物的状态进行测定,仅表示所得到的化合物32的信号的归属。
[0309]
[表24]
[0310][0311]
(5-3)化合物33的合成
[0312]
[0313]
在上述得到的化合物32和1-乙基体的混合物(254.8mg,约0.49mmol)的甲醇溶液(4.9ml)中加入2n的氢氧化钠水溶液(1.46ml,2.92mmol)中以40℃搅拌1小时。反应结束后,滤取所析出的白色固体。将滤液减压蒸馏除去,将所得到的残渣用甲醇清洗,将产生的白色固体再次通过过滤回收,将其合并作为白色固体得到化合物33和1-乙基体的混合物(194.6mg)。
[0314]
将所得到的化合物33的物性数据表示于表25。此外,关于nmr,以副产物(1-乙基体)与化合物33的混合物的状态进行测定,仅表示所得到的化合物33的信号的归属。
[0315]
[表25]
[0316][0317]
(5-4)化合物34的合成
[0318][0319]
在上述得到的化合物33和1-乙基体的混合物(23.5mg,约0.045mmol)的脱氧甲苯溶液(4.5ml)中将第二代hoveyda-grubbs催化剂(3.2mg,0.005mmol)、对苯醌(0.7mg,0.006mmol)在氮气流下以室温加入,以50℃搅拌20小时。然后,将反应溶液减压蒸馏除去,将所得到的残渣通过硅胶柱色谱(sio2,氯仿/丙酮=95/5至70/30)纯化,作为白色固体得到化合物34(18.2mg,53%,从化合物4起6个工序)。
[0320]
将所得到的化合物34的物性数据表示于表26。
[0321]
[表26]
[0322][0323]
(5-5)化合物35的合成
[0324][0325]
在上述得到的化合物34(57.8mg,0.13mmol)的甲醇/thf混合溶剂(3.9ml,甲醇/thf=2/1)中添加乙酸(116μl),加入氢氧化钯/碳(约50%水湿润、48.1mg)后,在氢气流下以60℃搅拌7小时。反应结束后,将混合物过滤,用乙酸乙酯清洗后,将滤液减压蒸馏除去,作为白色固体得到化合物35(29.5mg,70%)。
[0326]
将所得到的化合物35的物性数据表示于表27。
[0327]
[表27]
[0328][0329]
(5-6)化合物36的合成
[0330][0331]
在上述得到的化合物35(25.2mg,0.078mmol)的吡啶溶液(1ml)中加入四甲基甲硅烷基氯(40μl,0.32mmol),在氮气流下以室温搅拌2小时后,滴加苯甲酰氯(35μl,0.30mmol),在氮气流下以室温搅拌18小时。反应结束后,加入氨水(680μl),5小时后,将反应液浓缩。在残渣中加入吡啶(1ml)、氨水,在室温搅拌3小时后,将反应液浓缩。将所得到的残渣通过硅胶柱色谱(sio2,乙酸乙酯/甲醇=100/0至93/7)纯化,作为白色固体得到化合物36(18.2mg,55%)。
[0332]
将所得到的化合物36的物性数据表示于表28。
[0333]
[表28]
[0334][0335]
(5-7)化合物37的合成
[0336]
在上述得到的化合物36的无水吡啶溶液中加入4,4’-二甲氧基三苯甲基氯,在氮
气流下进行搅拌。反应结束后,在反应溶液中加入甲醇,将溶剂减压蒸馏除去。在所得到的残渣中加入乙酸乙酯和饱和碳酸氢钠水进行提取。将有机层用饱和碳酸氢钠水和饱和食盐水清洗后,用无水硫酸钠使其干燥,将溶剂减压蒸馏除去。将所得到的残渣通过硅胶柱色谱纯化,得到化合物37。
[0337]
(5-8)化合物38的合成
[0338]
在上述得到的化合物37、n,n-二异丙基乙胺和1-甲基咪唑的无水乙腈溶液中在冰冷下加入2-氰乙基-n,n-二异丙基磷酰氯,在氮气流下进行搅拌。反应结束后,加入饱和碳酸氢钠水和乙酸乙酯进行提取。将有机层用饱和碳酸氢钠水和饱和食盐水清洗后,用无水硫酸钠使其干燥,将溶剂减压蒸馏除去。将所得到的残渣通过硅胶柱色谱纯化,由此得到化合物38。
[0339]
(实施例6:桥连型核苷的合成(6))
[0340][0341]
各工序的试剂和条件:(a)2-氨基-6氯嘌呤,碳酸钾,18-冠醚-6,hmpa,室温,14小时;(b)n,n-二甲基甲酰胺二甲基缩醛,dmf,50℃,19小时。
[0342]
(6-1)化合物39的合成
[0343][0344]
将上述得到的化合物5(170.6mg,0.59mmol)、2-氨基-6-氯嘌呤(199.3mg,1.18mmol)和碳酸钾(448.0mg,3.24mmol)、18-冠醚-6(387.7mg,1.47mmol)的hmpa(2ml)溶液以室温在氮气流下搅拌14小时。反应结束后,倒入冰水中反应液搅拌1小时。滤取所析出的白色固体,用冰水和二乙基醚清洗。将所得到的残渣通过硅胶柱色谱(sio2,氯仿/甲醇=100/0至95/5)纯化,作为白色固体得到化合物39和1-乙基体的混合物(75.4mg)。
[0345]
将所得到的化合物39的物性数据表示于表29。此外,关于nmr,以副产物(1-乙基体)与化合物39的混合物的状态测定,仅表示所得到的化合物39的信号的归属。
[0346]
[表29]
[0347][0348]
(6-2)化合物40的合成
[0349][0350]
在上述得到的化合物39和1-乙基体的混合物(72.0mg,约0.16mmol)的无水二甲基甲酰胺(16ml)溶液中加入n,n-二甲基甲酰胺二甲基缩醛(199.3mg,1.18mmol),以50℃在氮气流下搅拌19小时。反应结束后,将反应液浓缩。将所得到的残渣通过硅胶柱色谱(sio2,乙酸乙酯)纯化,作为白色固体得到化合物40和1-乙基体的混合物(50.1mg)。
[0351]
将所得到的化合物40的物性数据表示于表30。此外,关于nmr,以副产物(1-乙基体)与化合物40的混合物的状态测定,仅表示所得到的化合物40的信号的归属。
[0352]
[表30]
[0353][0354]
(6-8)化合物48的合成
[0355]
使用上述得到的化合物40,通过与实施例5同样的方法(从化合物31至化合物38的各合成方法)合成化合物40至化合物48。
[0356]
(实施例7:桥连型核苷的合成(7))
[0357][0358]
各工序的试剂和条件:(a)烯丙基三甲基硅烷、tmsotf,mecn,0℃,1小时;(b)naome,meoh,室温,1小时,87%(2个工序);(c)mcpba,甲苯,0℃,1小时;(d)茴香醛二甲缩醛,csa,mecn,50℃,16小时,26%(2个工序);(e)胸腺嘧啶,dbu,mecn,100℃(mw),41.5小时,90%;(f)烯丙基溴,nah,nai,thf,50℃,7.5小时,63%;(g)第二代hoveyda-grubbs催化剂,甲苯,70℃,2.5小时;(h)pd(oh)2/c,h2,meoh,室温,10分钟(i)4,4’-二甲氧基三苯甲基氯,吡啶,室温,1.5小时,38%(3个工序);(j)2-氰乙基二异丙基氯磷酰胺,二异丙基乙基胺,1-甲基咪唑,mecn,室温,1小时,84%。
[0359]
(7-1)化合物51的合成
[0360][0361]
在化合物1(1.03g,3.77mmol)的无水乙腈(14.7ml)溶液中在冰冷下加入烯丙基三甲基硅烷(0.705ml,4.44mmol)后,滴加三甲基甲硅烷基三氟甲磺酸酯(0.71ml,3.93mmol),在氮气流下以室温搅拌1小时。反应结束后,在反应溶液加入饱和碳酸氢钠水进行搅拌。然后,将混合物用乙酸乙酯提取。用水和饱和食盐水清洗后,用无水硫酸钠使其干燥,将溶剂减压蒸馏除去。将所得到的粗产物不进一步不作纯化而直接用于后续反应。
[0362]
(7-2)化合物52的合成
[0363][0364]
在上述得到的化合物51的甲醇(14.4ml)溶液中在冰冷下加入5m的naome(甲醇溶液,0.75ml,3.75mmol),在氮气流下以室温搅拌1小时。反应结束后,在反应溶液中加入dowex 50
×
8 200mesh进行搅拌、中和。然后过滤混合物,将滤液浓缩,作为黄褐色油状物质得到化合物52(0.56g,87%,从化合物1起2个工序)。此外,该化合物52记载于mallikharjuna r.lambu等、j.med.chem.,2013,56,6123-6124。
[0365]
(7-3)化合物54的合成
[0366][0367]
在上述得到的化合物52(201.1mg,1.18mmol)的甲苯(4.70ml)溶液中在冰冷下加入mcpba(纯度70%,866mg,3.51mmol),在0℃搅拌1小时。反应结束后,在反应溶液中加入环己烯进行搅拌。然后过滤混合物,将滤液浓缩。接着,将所得到的环氧二醇用甲苯共沸后,加入无水乙腈(5.0ml)和茴香醛二甲缩醛(0.40ml,2.35mmol)以及(
±
)-樟脑磺酸(27.3mg,0.118mmol),在氮气流下以50℃搅拌16小时。反应结束后,将反应溶液在冰冷下用三乙胺(0.25ml)中和。将生成的混合溶液通过硅胶柱色谱(sio2,己烷/乙酸乙酯=80/20至20/80))纯化,作为黄色固体得到化合物54(92.1mg,26%,从化合物52起2个工序)。
[0368]
将所得到的化合物54的物性数据表示于表31。
[0369]
[表31]
[0370][0371]
(7-4)化合物55的合成
[0372][0373]
将上述得到的化合物54(574.8mg,1.89mmol)、胸腺嘧啶(480mg,3.81mmol)和二氮杂双环十一碳烯(dbu)(1.13ml,7.57mmol)的无水乙腈(9.4ml)溶液在微波照射下以100℃加热24.5小时。在反应溶液中再加入胸腺嘧啶(477.2mg,3.78mmol)和dbu(1.13ml,7.57mmol)以100℃反应17小时。反应结束后,将生成的混合溶液通过硅胶柱色谱(sio2,己烷/乙酸乙酯=40/60)纯化。使所得到的化合物溶解于二氯甲烷,用饱和碳酸氢钠水和饱和食盐水清洗后。然后,将有机层用无水硫酸钠干燥,将溶剂减压蒸馏除去,结果得到白色固体55(732.7mg,90%)。
[0374]
将所得到的化合物55的物性数据表示于表32。
[0375]
[表32]
[0376][0377]
(7-5)化合物56的合成
[0378][0379]
在上述得到的化合物55(254.4mg,0.59mmol)的无水thf(6.0ml)溶液中在冰冷下
加入氢化钠(60%油性,61.8mg,1.55mmol),在氮气流下以室温搅拌1小时。然后,在溶液中滴加烯丙基溴(60μl,0.71mmol),加入碘化钠(27.1mg,0.18mmol)以50℃搅拌9小时。反应结束后,加入饱和氯化铵水,用乙酸乙酯提取。将有机层用饱和碳酸氢钠水和饱和食盐水清洗,用无水硫酸钠使其干燥,将溶剂减压蒸馏除去。将生成的残渣通过硅胶柱色谱(sio2,氯仿/甲醇=7/1)纯化,作为白色固体得到化合物56(55.5mg,63%)。
[0380]
将所得到的化合物56的物性数据表示于表33。
[0381]
[表33]
[0382][0383]
(7-6)化合物57的合成
[0384][0385]
在上述得到的化合物56(234.7mg,0.50mmol)的脱氧二氯乙烷溶液(50ml)中在氮气流下以室温加入1,4-苯醌(5.2mg,0.048mmol)和第二代hoveyda-grubbs催化剂(17.0mg,0.027mmol),以70℃搅拌1.5小时。然后,将反应溶液减压蒸馏除去,将所得到的残渣通过硅胶柱色谱(sio2,己烷/乙酸乙酯=33/67)纯化。将所得到的粗产物不进一步纯化而直接用于后续反应。
[0386]
(7-7)化合物58的合成
[0387][0388]
在上述得到的化合物57的粗产物(128.5mg,约0.291mmol)的甲醇溶液(2.9ml)中加入pd(oh)2/c(31.1mg,约50%水湿润),在氢气流下以室温搅拌10分钟。反应结束后,将混合物过滤,用甲醇清洗后,将滤液减压蒸馏除去。将所得到的粗产物58不进一步纯化而直接用于后续反应。
[0389]
(7-8)化合物59的合成
[0390][0391]
在上述得到的化合物58的无水吡啶溶液(3.0ml)中加入4,4’-二甲氧基三苯甲基氯(142.7mg,0.42mmol),在氮气流下以室温搅拌1.5小时。反应结束后,在反应溶液中加入碳酸氢钠水和乙酸乙酯进行提取。将有机层用饱和碳酸氢钠水和饱和食盐水清洗后,用无水硫酸钠使其干燥,将溶剂减压蒸馏除去。将所得到的残渣通过硅胶柱色谱(sio2,己烷/乙酸乙酯=25/75)纯化,作为白色固体得到化合物59(118.2mg,38%,从化合物56起3个工序)。
[0392]
将所得到的化合物59的物性数据表示于表34。
[0393]
[表34]
[0394][0395]
(7-9)化合物60的合成
[0396][0397]
在上述得到的化合物59(34.6mg,0.055mmol)、n,n-二异丙基乙胺(30μl,0.176mmol)的无水乙腈溶液(1.0ml)中加入10%的1-甲基咪唑(无水乙腈溶液,13.0μl,0.0163mmol)后,在冰冷下加入2-氰乙基-,n-二异丙基磷酰氯(20.0μl,0.0897mmol),在氩气流下以室温搅拌1小时。反应结束后,加入饱和碳酸氢钠水和乙酸乙酯进行提取。将有机层用饱和碳酸氢钠水和饱和食盐水清洗后,用无水硫酸钠使其干燥,将溶剂减压蒸馏除去。将所得到的残渣通过硅胶柱色谱(sio2,己烷/乙酸乙酯=33/67)纯化,作为白色固体得到化合物60(38.3mg,84%)。
[0398]
将所得到的化合物60的物性数据表示于表35。
[0399]
[表35]
[0400][0401]
(实施例8:寡核苷酸的合成和纯化)
[0402]
使用实施例1~3中制作得到的化合物12、19和25作为亚酰胺砌块,如下所述合成寡核苷酸。构成寡核苷酸的化合物12、19和25以外的化合物只要没有特别说明均从proligo公司购入。
[0403]
由实施例1~3中制作得到的化合物12、19和25分别制成0.1m的无水乙腈溶液,装入genedesign公司制ns-8oligonucleotides synthesizer。在trityl off条件下进行各合成。活化剂使用activator-42(注册商标)(proligo公司制),将化合物12、19和25的缩合时间延长到120秒
×
5。关于其他操作按照通常的亚磷酰胺法进行合成。
[0404]
合成结束后,将产物使用28%氨水溶液在室温下进行1.5小时处理,从柱载体上的切出,接着在55℃静置15小时,由此进行碱基部分和磷酸部分的脱保护。接着,将所得到的粗寡核苷酸用反相hplc纯化。
[0405]
其中,该hplc的条件如下所述。
[0406]
洗脱液
[0407]
·
a液:0.1m乙酸三乙铵缓冲液(ph7.0)
[0408]
·
b液:乙腈
[0409]
梯度
[0410]
·
b液浓度:6~12%(20分钟)
[0411]
色谱柱
[0412]
·
waters公司制xbridgetm ost c18 2.5μm(10
×
50mm)(制备)
[0413]
·
waters公司制xbridgetm ost c18 2.5μm(4.6
×
50mm)(分析)
[0414]
流速
[0415]
·
4.0ml/分钟(制备)
[0416]
·
1.0ml/分钟(分析)
[0417]
柱温
[0418]
·
50℃
[0419]
检测
[0420]
·
uv(260nm)
[0421]
通过maldi-tof-ms测定来确定纯化得到的寡核苷酸的组成。在该测定时,首先,
在干燥有3-羟基吡啶二羧酸水溶液(10mg/ml)与柠檬酸二铵水溶液(1mg/ml)以1:1的容量比混合得到的基质(1μl)的anchor chip上,载置寡核苷酸水溶液(50μm,1μl)并再次使其干燥,然后进行maldi-tof-ms测定。以负离子模式进行分子量的测定,使用寡胸苷酸(7mer、15mer和23mer)作为外部标准。另外,使用吸光度测定装置(株式会社岛津制作所制shimadzu uv-1800)测定260nm的紫外部分吸收来进行所合成的寡核苷酸的定量。
[0422]
(实施例9:双链形成能力的评价)
[0423]
如实施例8所记载的那样,合成和纯化以下的表所示的序列的寡核苷酸。
[0424]
(1)5’-d(gcgttttttgct)-3’(序列编号1)
[0425]
(2)5’-d(gcgtthtttgct)-3’(序列编号2)
[0426]
(3)5’-d(gcgtt1tttgct)-3’(序列编号3)
[0427]
(4)5’-d(gcgtt2tttgct)-3’(序列编号4)
[0428]
(5)5’-d(gcgtt3tttgct)-3’(序列编号5)
[0429]
(6)5’-d(gcghththtgct)-3’(序列编号6)
[0430]
(7)5’-d(gcg1t1t1tgct)-3’(序列编号7)
[0431]
(8)5’-d(gcg2t2t2tgct)-3’(序列编号8)
[0432]
(9)5’-d(gcg3t3t3tgct)-3’(序列编号9)
[0433]
(10)5’-d(gcgtthhhtgct)-3’(序列编号10)
[0434]
(11)5’-d(gcgtt111tgct)-3’(序列编号11)
[0435]
(12)5’-d(gcgtt222tgct)-3’(序列编号12)
[0436]
(13)5’-d(gcgtt333tgct)-3’(序列编号13)
[0437]
(14)5’-d(gcghhhhhhgct)-3’(序列编号14)
[0438]
(15)5’-d(gcg111111gct)-3’(序列编号15)
[0439]
(16)5’-d(gcg222222gct)-3’(序列编号16)
[0440]
(17)5’-d(gcg333333gct)-3’(序列编号17)
[0441]
上述的h、1、2和3表示如下:
[0442]
h=hna-t(非专利文献4的化合物)
[0443]
1=化合物12(bana-t1)
[0444]
2=化合物19(bana-t2)
[0445]
3=化合物25(bana-t3)
[0446]
作为靶标链,使用单链寡rna5’-r(agcaaaaaacgc)-3’(序列编号18)和单链寡dna5’-d(agcaaaaaacgc)-3’(序列编号19),如下调查双链形成能力(结合亲和性)。
[0447]
寡核苷酸的双链形成能通过将各种寡核苷酸和靶标链进行退火处理使其形成双链后,测定tm值来调查。更详细而言,将各寡核苷酸(最终浓度4μm)与氯化钠(最终浓度100mm)的磷酸缓冲液(10mm,ph7.2,130μl)的混合液置于沸水浴中,缓慢冷却到室温。然后,在氮气流下冷却到5℃,开始测定。以0.5℃/分钟升温到90℃,以0.5℃间隔绘制260nm的吸光度。利用中线法或微分法(序列编号17)算出tm值,作为独立的3次测定的平均值。
[0448]
在表36(序列(1)~(9))和表37(序列(1)和(10)~(17))中表示双链形成能力的结果。这些表中,将对于单链寡rna的结果表示为“ssrna”,将对于单链寡dna的结果表示为“ssdna”,并且表示各寡核苷酸的tm和人工修饰核酸每1个碱基的tm变动温度(“δtm/
mod.”)。
[0449]
[表36]
[0450][0451]
[表37]
[0452][0453]
相对于ssrna,tm值是bana-t3(化合物25)最高,接着依次为bana-t2(化合物19)、bana-t1(化合物12)。通过在bana-t1、bana-t2(化合物19)和bana-t3(化合物25)中分别导入多个寡核苷酸,显示出相对于ssrna的tm值上升,存在对于rna的双链形成能力提高的倾向。另外,通过在bana-t1、bana-t2(化合物19)和bana-t3(化合物25)中分别进行连续的导入,相对于rna的双链形成能力进一步提高,显示出比hna-t时更高的双链形成能力。
[0454]
(实施例10:碱基选择性的评价)
[0455]
关于上述的序列(1)~(5),作为靶标链使用单链寡rna5’-r(agcaaayaacgc)-3’和单链寡dna5’-d(agcaaayaacgc)-3’,同样地调查双链形成能力(结合亲和性)。
[0456]
就y而言,关于单链寡rna,为ra(序列编号18)、ru(序列编号20)、rg(序列编号21)
和rc(序列编号22)的任意一个,并且关于单链寡dna,设为da(序列编号19)、dt(序列编号23)、dg(序列编号24)和dc(序列编号25)的任意一个。
[0457]
将结果表示于以下的表38。表中、δtm是从错配的tm值减去配对(da或ra)的tm值求得的。
[0458]
[表38]
[0459][0460]
通过在bana-t1(化合物12)、bana-t2(化合物19)和bana-t3(化合物25)中分别导入寡核苷酸,与使用天然dna(dt)的情况(序列编号1)相比,抑制了与g的碱基对的形成。
[0461]
(实施例11:核酸酶抗性的评价)
[0462]
如实施例8所记载的那样,合成和纯化了以下的10mer的序列的寡核苷酸,作为受试寡核苷酸使用:
[0463]5’
-d(tttttttttx)-3’[0464]
x设为以下的任意一种:
[0465]
x=胸腺嘧啶核苷(dt)
[0466]
x=5’-硫代磷酸酯胸腺嘧啶核苷(ps)
[0467]
x=lna-t(lna)
[0468]
x=hna-t(hna)
[0469]
x=化合物12(bana-t1)
[0470]
x=化合物19(bana-t2)
[0471]
x=化合物25(bana-t3)
[0472]
在包含7.5μm受试寡核苷酸和10mm氯化镁的50mm tris盐酸缓冲液(ph8.0)中加入1.0μg/ml的3’-核酸外切酶(crotalus adamanteus venom phosphodiesterase,cavp),在37℃孵育。在孵育开始时(0分钟)、5分钟后、10分钟后、20分钟后、40分钟后分别各取出20μl试样,与milliq90μl合并成为110μl。其中将100μl用反相hplc解析,算出未切断的寡核苷酸的比例。另外,评价通过独立的3次测定来导出。
[0473]
将结果表示于图1。如图1所示,在x为lna-t(lna)或hna-t(hna)的情况下,与天然dna(dt)同样地利用核酸酶将寡核苷酸分解。即使在硫代磷酸酯化(ps)寡(x=5’-硫代磷酸酯胸腺嘧啶核苷(ps))的情况下,核酸酶处理40分钟后的未切断寡核苷酸的残存率也约为20%程度,而相对于此,在作为x分别使用化合物12(bana-t1)、化合物19(bana-t2)和化合物25(bana-t3)的情况下,即使在核酸酶处理40分钟后,也有约60%未切断而残存,难以被分解。
[0474]
(实施例12:核酸酶抗性的评价)
[0475]
如实施例8中记载那样,合成和纯化以下的10mer的序列的寡核苷酸,作为受试寡核苷酸使用:
[0476]5’
-d(ttttttttxt)-3’[0477]
x设为以下的任意一种:
[0478]
x=3’-硫代磷酸酯胸腺嘧啶核苷(ps)
[0479]
x=lna-t(lna)
[0480]
x=hna-t(hna)
[0481]
x=化合物12(bana-t1)
[0482]
x=化合物19(bana-t2)
[0483]
x=化合物25(bana-t3)
[0484]
在包含7.5μm受试寡核苷酸和10mm氯化镁的50mm tris盐酸缓冲液(ph8.0)中加入1.0μg/ml的3’-核酸外切酶(crotalus adamanteus venom phosphodiesterase,cavp)在37℃孵育。在孵育开始时20分钟后取出20μl试样,与milliq90μl合并成为110μl。其中将100μl用反相hplc解析,算出未切断的寡核苷酸的比例。
[0485]
将结果表示于图2。如图2可知,在x为lna-t(lna)或hna-t(hna)的情况下,通过核酸酶,寡核苷酸完全分解。对于硫代磷酸酯化(ps)寡(x=3’-硫代磷酸酯胸腺嘧啶核苷(ps)),核酸酶处理20分钟后的未切断寡核苷酸的残存率约为50%程度。在作为x使用化合物12(bana-t1)、化合物19(bana-t2)的情况下,核酸酶处理20分钟后残存率分别为2%、33%,相对于此在作为x使用化合物25(bana-t3)的情况下,95%残存,与其他修饰相比,压倒性地难以被分解。
[0486]
(实施例13:寡核苷酸的合成和纯化以及双链形成能力的评价)
[0487]
如实施例8中记载的那样,合成和纯化以下的序列的寡核苷酸:
[0488]
(18)5’-d(gcgttllltgct)-3’(序列编号26)
[0489]
(19)5’-d(gcgttl33tgct)-3’(序列编号27)
[0490]
(20)5’-d(gcgtt3l3tgct)-3’(序列编号28)
[0491]
(21)5’-d(gcgtt33ltgct)-3’(序列编号29)
[0492]
上述的l和3表示如下:
[0493]
l=lna-t(lna)
[0494]
3=化合物25(bana-t3)
[0495]
将各寡核苷酸的产率和maldi-tof ms测定的结果表示于表39。
[0496]
[表39]
[0497][0498]
进而,如实施例9所记载的那样,对各寡核苷酸,评价对rna的双链形成能力。作为靶标链,使用单链寡rna5’-r(agcaaaaaacgc)-3’(序列编号18)。将结果表示于表38。表38表示形成双链时的各寡核苷酸的解链温度tm与序列(18)的tm的差异(“δt
m”)。如表40所示,可知包含化合物25(bana-t3)的寡核苷酸具有与包含lna的寡核苷酸同等的高双链形性能。
[0499]
[表40]
[0500][0501]
(实施例14:寡核苷酸的合成和纯化以及双链形成能力的评价)
[0502]
使用化合物25(bana-t3)和化合物30(bana-mc3),如实施例8所记载的那样,合成和纯化表41所示的序列(22)~(25)的寡核苷酸(分别也表示于序列编号30~33)。表41中的寡核苷酸为相对于mmalat1的反义寡核苷酸。
[0503]
使用0.05m((二甲基氨基-亚甲基)氨基)-3h-1,2,4-二噻唑-3-硫酮(ddtt)(吡啶/乙腈(3:2)溶液、glen research公司),按照该试剂的制造公司推荐的方法进行硫代磷酸酯(ps)化。
[0504]
表41还表示这些寡核苷酸的产率和maldi-tof ms测定的结果。
[0505]
[表41]
[0506][0507]
进而,与实施例9同样对各寡核苷酸评价了对于rna的双链形成能力。作为对照,使用以下的表42所示的2个序列(分别也表示于序列编号34和35)。
[0508]
[表42]
[0509][0510]
作为靶标链,使用单链寡rna5’-r(gcauucagugaacuag)-3’(序列编号36)。将结果表示于表43。表43表示形成双链时的各寡核苷酸的解链温度tm与序列(26)的tm的差异(“δt
m”)。如表41所示,可知包含化合物30(bana-mc3)和化合物25(bana-t3)的寡核苷酸具有与包含lna的寡核苷酸同等的高双链形性能。
[0511]
[表43]
[0512][0513]
(实施例15:寡核苷酸的合成和纯化以及毒性降低效果的评价)
[0514]
如实施例8中记载的那样,合成和纯化表44所示的序列(28)的寡核苷酸(序列编号37)。与实施例14同样进行硫代磷酸酯(ps)化。表44还表示该寡核苷酸的产率和maldi-tof ms测定的结果。
[0515]
[表44]
[0516][0517]
在6周龄的小鼠(c57bl/6j、雄)的腹腔内投予表45所示的受试寡核苷酸(20mg/kg)(5只/组)。96小时后,在吸入麻醉下(异氟烷)进行采血,放血安乐死。然后,利用自动分析装置(fujifilm制富士drichem 4000v)测定血清中的天冬氨酸转氨酶(ast)和丙氨酸转氨酶(alt)的活性。
[0518]
[表45]
[0519]
受试寡核苷酸
[0520][0521]
表46表示投予受试寡核苷酸的情况和投予生理盐水的情况下的血液中的天冬氨酸转氨酶(ast)和丙氨酸转氨酶(alt)的活性。在投予了已知显示肝毒性的序列(29)的组中,小鼠5只全部死亡。而将序列(29)的寡核苷酸的序列的一部分用bana取代的序列(28)的寡核苷酸基本不显示alt和ast的上升,确认到了毒性的降低效果。
[0522]
[表46]
[0523][0524]
alt/ast值:平均
±
sd.
[0525]
(实施例16:小鼠体内的反义寡核苷酸的表达抑制效果的评价)
[0526]
如实施例8中记载的那样,合成和纯化表47所示的序列(22)~(25)和(30)的寡核苷酸(分别也表示于序列编号30~33和39)。与实施例14同样进行硫代磷酸酯(ps)化。表47中的寡核苷酸为相对于mmalat1的反义寡核苷酸。
[0527]
[表47]
[0528][0529]
在6周龄的小鼠(balb/canncrlcrlj、雌)的尾静脉投予受试寡核苷酸(20nmol:100μm生理盐水溶液200μl)(5只/组)。72小时后,在吸入麻醉下(异氟烷)进行采血,放血安乐死。然后,采集各组织,进行rna提取(使用试剂盒:rneasy)。通过实时pcr(使用试剂盒:one step tb green(注册商标)primescript
tm rt-pcrkit(perfect real time)、takara bio株式会社制)测定各组织中的malat1的mrna表达量。实时pcr中使用了以下的引物:
[0530]
malat1 forward:acattccttgaggtcggcaa(序列编号40)
[0531]
malat1 reverse:cacccgcaaaggcctacata(序列编号41)
[0532]
gapdh forward:tcaccaccatggagaaggc(序列编号42)
[0533]
gapdh reverse:gctaagcagttggtggtgca(序列编号43)
[0534]
将结果表示于图3和图4。图3和图4表示投予各种寡核苷酸时的小鼠的各种组织中的相对的malat1表达水平(图3:肝脏、心脏、肾脏、胰脏、骨骼肌、肺和胃、以及图4:脾脏、皮肤、大肠、脑、乳腺、眼球和软骨)。“相对的malat1表达水平”是以仅投予生理盐水(无寡核苷酸)时的表达水平作为1的情况下的相对值表示。在图3和图4中,将序列(22)~(25)的寡核苷酸分别表示为on22~on25。在图3和图4中,表示结果的棒的标记在作为包含化合物25(bana-t3)和化合物30(bana-mc3)的寡核苷酸的序列(22)~(25)的寡核苷酸(“on22”~“on25”)、代替化合物25和化合物30而包含lna的寡核苷酸即序列(30)的寡核苷酸(“on30”)、以及对照(仅投予生理盐水)(“生理盐水”)之间互相区分。
[0535]
包含化合物25和化合物30这样的bana的序列(22)~(25)的寡核苷酸在大量组织中,与代替bana而包含lna的序列(30)的寡核苷酸相比,显示出同等或其以上的高靶标基因抑制效果。
[0536]
(实施例17:寡核苷酸的合成和纯化)
[0537]
使用化合物60(bana-t4),如实施例8所记载的那样,合成和纯化以下所示的序列(31)的寡核苷酸。
[0538]
(31)5’-d(gcgtt4tttgct)-3’(序列编号44)
[0539]
4=化合物60(bana-t4)
[0540]
如实施例8中记载的那样利用maldi-tof-ms测定来确定纯化的寡核苷酸的组成。合成的寡核苷酸利用吸光度测定装置来定量。
[0541]
[表48]
[0542][0543]
产业上的可利用性
[0544]
根据本发明,可以提供能够作为硫代磷酸酯修饰核酸的替代的新型的桥连型核苷和使用了该桥连型核苷的核苷酸。使用本发明的桥连型核苷得到的寡核苷酸例如作为核酸医药中的原材料有用。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1