顺丁烯二酰亚胺树脂及其制造方法、顺丁烯二酰亚胺溶液、以及可硬化性树脂组合物及其硬化物与流程
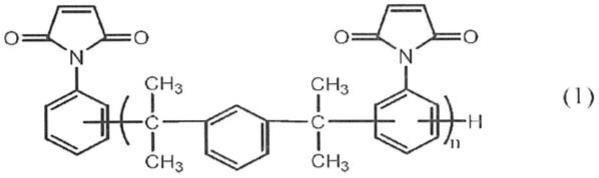
1.本发明涉及一种顺丁烯二酰亚胺树脂及其制造方法、顺丁烯二酰亚胺溶液、以及可硬化性树脂组合物及其硬化物。本发明可较佳地用于半导体密封材、印刷配线板、增层(
ビルドアップ
)积层板等电气-电子零件、碳纤维增强塑胶、玻璃纤维增强塑胶等轻量高强度材料。
背景技术:
2.近年来,搭载电气-电子零件的积层板由于其使用领域扩大,故所需特性范围变广且高度化。例如,现有半导体晶片主要搭载于金属制引线框架,而近年来cpu等具有高处理能力的半导体晶片大多搭载于以高分子材料制作的积层板。随着cpu等元件高速化的发展以及时钟频率的提高,信号传输延迟及传播损耗的问题亦增大,而对配线板要求低介电常数化及低介电损耗正切化。尤其,由于近年来5g的盛行,不仅是sub6,10ghz以上,特别是于28ghz以上的准毫米波、毫米波的电特性亦开始受到重视,从而要求可于这些范围稳定使用的材料。同时,由于随着元件的高速化,晶片发热会增加,因此亦需要提高耐热性。
3.又,由于行动电话等移动电子机器的普及,精密电子机器已于室外环境及极其靠近人体处使用、携带,因此需要对于外部环境(尤其是耐湿热环境)的耐性。进而,于汽车领域中,电子化迅速发展,精密电子机器有时亦配置于引擎附近,而以更高的水准要求耐热、耐湿性。又,由于精密电子机器被用于汽车及行动装置等,故阻燃性等安全性亦更为重要,但由于近年来环境问题意识的提高,而避免使用卤素系阻燃剂,因此,于不使用卤素的情况下赋予阻燃性的必要性增加。
4.以往,如专利文献1所述的使用将双酚a型氰酸酯化合物与双顺丁烯二酰亚胺化合物并用的树脂即bt树脂的配线板,其耐热性、耐化学品性、电特性等优异,已广泛地用作高性能配线板,但是在如上所述需要更高性能的状况下,需要对其进行改善。
5.又,近年来,出于节能的观点,正在推进飞机、汽车、列车、船舶等的轻量化。特别是于交通工具领域,正在研究用轻量且高强度的碳纤维复合材料取代先前使用金属材料者。例如,于波音787中,通过提高复合材料的比率来实现轻量化,从而大幅度改善燃料效率。于航空领域中,为了进一步实现轻量化,还有于引擎周围的构件亦导入碳纤维复合材的动向,自然需要高水准的耐热性。于汽车领域中,有一部分搭载有由复合材料制造的螺旋桨轴,且亦有针对高级车使用复合材料制作车体的动向。以往,于碳纤维复合材料的领域中,使用复合材料,该材料使用了环氧树脂的双酚a型二环氧丙基醚或四环氧丙基二氨基二苯基甲烷等、以及作为硬化剂的二氨基二苯甲烷、二氨基二苯砜等,但为了进一步推进轻量化、高耐热化,需要扩大复合材料的应用,关于用于该目的的材料,作为手段之一,正在研究顺丁烯二酰亚胺树脂。
6.于此情况下,可从市场获得的顺丁烯二酰亚胺化合物或双顺丁烯二酰亚胺树脂与先前使用的环氧树脂等相比,虽耐热性大幅度提高,欲将其用于如上所述的用途的需求强
烈,但介电特性,尤其是于高频区域的介电特性不够充分,无法满足高速通讯的需求。进而,存在由于吸收空气中的水分而使电特性大幅度劣化的问题,其应用范围有限。
7.对此,亦开发了如专利文献2、3的顺丁烯二酰亚胺树脂,但仍难谓充分。
8.现有技术文献
9.专利文献
10.专利文献1:日本特公昭54-30440号公报
11.专利文献2:日本特开平3-100016号公报
12.专利文献3:日本特开2009-1783号公报
13.专利文献4:日本特公平4-75222号公报
14.专利文献5:日本特公平6-37465号公报。
技术实现要素:
15.[发明所欲解决的课题]
[0016]
本发明通过提供一种溶剂溶解性优异、且即使于高频区域内电特性亦优异的顺丁烯二酰亚胺树脂,尤其有助于以5g为代表的高速通讯技术的发展。
[0017]
[解决课题的技术手段]
[0018]
本发明人等为解决上述课题而进行了努力研究,结果完成了本发明。
[0019]
即,本发明涉及以下[1]~[7]。
[0020]
[1]
[0021]
一种顺丁烯二酰亚胺树脂,其由下述式(1)表示,且
[0022]
上述顺丁烯二酰亚胺树脂中,n,n'-(亚苯基二(2,2-亚丙基)二亚苯基)双顺丁烯二酰亚胺的含量以gpc面积百分率计为98面积%以下,
[0023]
上述n,n'-(亚苯基二(2,2-亚丙基)二亚苯基)双顺丁烯二酰亚胺中,由下述式(2)表示的顺丁烯二酰亚胺化合物的含量以hplc面积百分率计为30面积%以上且未达60面积%。
[0024][0025]
(式(1)中,n为重复数,其平均值为1<n<5)
[0026][0027]
[2]
[0028]
如前项[1]的顺丁烯二酰亚胺树脂,其中,于上述n,n'-(亚苯基二(2,2-亚丙基)二亚苯基)双顺丁烯二酰亚胺中,通过hplc面积百分率得出的邻位位向与对位位向的面积比
(o/p)为100%以上且未达200%。
[0029]
[3]
[0030]
如前项[1]或[2]的顺丁烯二酰亚胺树脂,其中,于上述n,n'-(亚苯基二(2,2-亚丙基)二亚苯基)双顺丁烯二酰亚胺中,由下述式(3)表示的顺丁烯二酰亚胺化合物的含量以hplc面积百分率计为未达50面积%。
[0031][0032]
[4]
[0033]
一种顺丁烯二酰亚胺溶液,其包含前项[1]至[3]中任一项所述的顺丁烯二酰亚胺树脂及有机溶剂,且上述有机溶剂选自酮类、烃类、及酯类中的1种以上。
[0034]
[5]
[0035]
一种可硬化性树脂组合物,其包含前项[1]至[3]中任一项所述的顺丁烯二酰亚胺树脂、或前项[4]的顺丁烯二酰亚胺溶液,且进而含有硬化促进剂。
[0036]
[6]
[0037]
一种硬化物,其是使前项[1]至[3]中任一项所述的顺丁烯二酰亚胺树脂、或前项[5]的可硬化性树脂组合物进行硬化而获得。
[0038]
[7]
[0039]
一种前项[1]至[3]中任一项所述的顺丁烯二酰亚胺树脂的制造方法,其包括:向苯胺与二异丙烯苯或二(α-羟基异丙基)苯添加相对于苯胺总量为1~12重量%的质子酸,并于140~190℃反应,从而获得芳族胺树脂的步骤;及使上述芳族胺树脂进行顺丁烯二酰亚胺化的步骤。
[0040]
[发明的效果]
[0041]
本发明的顺丁烯二酰亚胺树脂不仅溶剂溶解性优异,其硬化物于高频区域的电特性亦优异,而可用于电子材料或结构材等各种用途。
附图说明
[0042]
[图1]为表示实施例15及比较例8~9的硬化物中的弹性模量根据温度的变化的曲线图。
具体实施方式
[0043]
以下,对本发明的一个实施方案进行详细说明。
[0044]
本实施方案的顺丁烯二酰亚胺树脂由下述式(1)表示。
[0045][0046]
(式(1)中,n为重复数,其平均值为1<n<5)
[0047]
式(1)中,n的值可根据通过顺丁烯二酰亚胺树脂的凝胶渗透层析法(gpc,检测器:ri)的测定所求出的数均分子量、或分离的峰的各自的面积比而算出。n通常为1<n<5,更优选为1<n<3。
[0048]
于平均值n为1的情形时,产生溶剂溶解性较低而使结晶析出的问题。另一方面,于平均值n为5以上的情形时,产生成形时的流动性变差而无法发挥出作为硬化物的特性的问题。
[0049]
本实施方案的顺丁烯二酰亚胺树脂中的式(1)中,作为n=1体的n,n'-(亚苯基二(2,2-亚丙基)二亚苯基)双顺丁烯二酰亚胺的通过gpc分析(ri)得出的含有率以gpc面积百分率计为98面积%以下,优选为20~90面积%,更优选为30~80面积%,进而优选为40~75面积%,最优选为40~70面积%的范围。当n=1体的含量为98面积%以下时,耐热性良好。又,结晶性降低,溶剂溶解性良好。另一方面,当n=1体的下限值为20面积%以上时,树脂溶液的粘度降低,含浸性良好。又,由于当作为固体取出时可于低温去除溶剂,因此不易发生自聚合而容易操作。
[0050]
上述面积%可通过gpc(凝胶渗透层析法)进行测定,于本实施方案中,上述面积%于以下条件进行测定。
[0051]
·
gpc分析
[0052]
gpc:dgu-20a3r、lc-20ad、sil-20aht、rid-20a、spd-20a、cto-20a、cbm-20a(均由岛津制作所制造)
[0053]
管柱:shodex kf-603、kf-602
×
2、kf-601
×2[0054]
连结洗脱液:四氢呋喃
[0055]
流速:0.5ml/min
[0056]
管柱温度:40℃
[0057]
检测:ri(示差折射率检测器)。
[0058]
本实施方案的顺丁烯二酰亚胺树脂的软化点优选为50℃~150℃,更优选为80℃~120℃,进而优选为90℃~120℃,尤其优选为95℃~120℃。又,150℃的熔融粘度为0.05~100pa
·
s,优选为0.1~40pa
·
s。但存在以下情况:由于在去除溶剂时的聚合中发生高分子量化而超过该值。该优选的范围是自溶液的状态固体化时n=1的gpc中的面积%的变化量为3面积%以下时的指标。
[0059]
本实施方案的顺丁烯二酰亚胺树脂的酸值优选为30mg koh/g,进而优选为1~15mg koh/g。若酸值较高,则未进行顺丁烯二酰亚胺化的分子较多,具有羧酸的结构过量,因此对电特性及耐水性造成影响。
[0060]
关于本实施方案的顺丁烯二酰亚胺树脂,作为上述式(1)中的n=1体的n,n'-(亚
苯基二(2,2-亚丙基)二亚苯基)双顺丁烯二酰亚胺,含有作为位置异构物的由下述式(2)~(4)表示的顺丁烯二酰亚胺化合物。
[0061][0062]
于n,n'-(亚苯基二(2,2-亚丙基)二亚苯基)双顺丁烯二酰亚胺中,由上述式(2)~(4)表示的顺丁烯二酰亚胺化合物的各自的含有率以hplc面积百分率计,优选为以下。
[0063]
关于由上述式(2)表示的顺丁烯二酰亚胺化合物,为30面积%以上且未达60面积%,优选为35面积%以上且未达55面积%,进而优选为40面积%以上且未达55面积%。通过使具有非对称结构的本化合物为30面积%以上,溶剂溶解性提高,此外介电特性提高。另一方面,由于需要例如通过晶析来去除由上述式(3)表示的顺丁烯二酰亚胺化合物等追加步骤以使含有率为60面积%以上,因此就制造成本上升及工业废弃物增加等方面而言不佳。
[0064]
关于由上述式(3)表示的顺丁烯二酰亚胺化合物,优选为未达50面积%,进而优选为2面积%以上且未达35面积%,优选为5面积%以上且未达30面积%。通过使含有率为未达50面积%,溶剂溶解性提高,且电特性亦良好。
[0065]
关于由上述式(4)表示的顺丁烯二酰亚胺化合物,优选为15面积%以上且未达60面积%,进而优选为25面积%以上且未达50面积%。通过使含有率为15面积%以上,介电特性良好,通过使含有率为未达60面积%,可硬化性及密接性良好,可抑制制作基板等时的不良情况。
[0066]
再者,由于由上述式(3)表示的顺丁烯二酰亚胺化合物对于结晶性的问题及电特性恶化的问题影响最大,因此由式(2)表示的顺丁烯二酰亚胺化合物与由式(4)表示的顺丁烯二酰亚胺化合物的合计比率于n,n'-(亚苯基二(2,2-亚丙基)二亚苯基)双顺丁烯二酰亚胺中,优选为50面积%以上,进而优选为60面积%以上,尤其优选为70面积%以上。
[0067]
又,于n,n'-(亚苯基二(2,2-亚丙基)二亚苯基)双顺丁烯二酰亚胺中,邻位位向与对位位向的面积比(o/p)优选为50%以上且未达300%,进而优选为100%以上且未达200%。
[0068]
当面积比(o/p)为50%以上时,溶剂溶解性及电特性良好。又,当面积比(o/p)为未
达300%时,不会产生制造成本上升及工业废弃物增加等问题。
[0069]
再者,邻位位向与对位位向的面积比(o/p)通过下述式算出。
[0070]
邻位位向与对位位向的面积比(o/p)=(由上述式(4)表示的顺丁烯二酰亚胺化合物的面积%)
×
2+(由上述式(2)表示的顺丁烯二酰亚胺化合物的面积%)/(由上述式(3)表示的顺丁烯二酰亚胺化合物的面积%)
×
2+(由上述式(2)表示的顺丁烯二酰亚胺化合物的面积%)。
[0071]
上述含有率可通过hplc(高效液相层析法)进行测定,于本实施方案中,于以下条件下进行测定。
[0072]
·
hplc分析
[0073]
岛津制作所股份有限公司制造的输液单元lc-20ad
[0074]
岛津制作所股份有限公司制造的光二极体阵列检测器spd-m20a
[0075]
岛津制作所股份有限公司制造的管柱烘箱cto-20a
[0076]
管柱:intersil ods-2.5μm,4.6
×
250mm 40℃
[0077]
流动相a:乙腈(an)
[0078]
流动相b:水(w)
[0079]
时间程式:
[0080]
0-28min.an/w=30%/70%
→
100%/0%
[0081]
28-40min.an/w=100%/0%
[0082]
流速:1.0ml/min
[0083]
检测:uv 200-274nm,pda
[0084]
测定波长:225nm。
[0085]
hplc的测定波长优选为210~230nm的范围。其原因在于:虽于其他波长亦可进行测定,但有时会由于其吸收波长的差异而与实际含量相差甚远。又,亦可通过nmr算出其比率。于使用nmr的情形时,可使用
13
c-nmr根据于1-2ppm出现的亚异丙基的峰强度比来算出比率。
[0086]
本发明的一个实施方案的顺丁烯二酰亚胺树脂于溶剂中的溶解性及与其他树脂的溶解性较高,可作为顺丁烯二酰亚胺溶液(以下亦简称为清漆)或可硬化性树脂组合物处理。又,本发明的一个实施方案的顺丁烯二酰亚胺树脂的硬化物的电特性、耐热性、耐候性、吸湿性、及阻燃性优异。
[0087]
作为可使用的溶剂,例如可列举:丙酮、甲基乙基酮、环己酮等酮类;苯、甲苯、二甲苯、四甲基苯、环己烷等烃类;乙二醇二甲醚、乙二醇二乙醚、二丙二醇二甲醚、二丙二醇二乙醚、三乙二醇二甲醚、三乙二醇二乙醚等二醇醚类;乙酸乙酯、乙酸丁酯、甲基溶纤剂乙酸酯、乙基溶纤剂乙酸酯、丁基溶纤剂乙酸酯、卡必醇乙酸酯、丙二醇单甲醚乙酸酯、戊二酸二烷基酯、琥珀酸二烷基酯、己二酸二烷基酯等酯类;γ-丁内酯等环状酯类;石油醚、石油脑、氢化石油脑、溶剂石油脑等石油系溶剂等,这些之中,优选为酮类、烃类、酯类,进而优选为烃类,尤其优选为芳族烃类。这些可单独地使用,亦可并用两种以上。溶剂于所获得的清漆中去除溶剂的固形物成分浓度通常为10~80重量%,优选为以成为20~70重量%的范围使用。
[0088]
再者,通过使顺丁烯二酰亚胺树脂溶解于溶剂而可制作顺丁烯二酰亚胺溶液,但
优选为将合成时使用的溶剂直接供作顺丁烯二酰亚胺溶液。其原因在于:工业上的废弃物及能源消耗减少。
[0089]
继而,对本实施方案的顺丁烯二酰亚胺树脂的制造方法进行说明,但并不限定于本制造方法。
[0090]
[芳族胺树脂的制造方法]
[0091]
本实施方案的顺丁烯二酰亚胺树脂可使用由下述式(5)表示的芳族胺树脂作为前驱物。
[0092][0093]
(式(5)中,n为重复数,其平均值为1<n<5)
[0094]
由上述式(5)表示的芳族胺树脂的制造方法并无特别限定。例如,于专利文献4中,揭示有以下内容:使苯胺与间二异丙烯苯或间二(α-羟基异丙基)苯于酸性催化剂的存在下以180~250℃进行反应,由此获得上述式(5)中的n=1体作为主要成分。于该n=1体中,包含三种异构物,即由下述式(6)表示的1-(邻氨基异丙苯基)-3-(对氨基异丙苯基)苯、由下述式(7)表示的1,3-双(对氨基异丙苯基)苯、及由下述式(8)表示的1,3-双(邻氨基异丙苯基)苯。
[0095][0096]
于专利文献4中,进而将作为副成分而包含的n=2~5体通过晶析而精制,获得纯度为98%的1,3-双(对氨基异丙苯基)苯。又,于专利文献5中,使1,3-双(对氨基异丙苯基)苯进行顺丁烯二酰亚胺化,合成n,n'-(1,3-亚苯基二(2,2-亚丙基)二对亚苯基)双顺丁烯二酰亚胺,从而获得结晶产物,但为了使其溶解于溶剂,需要进行加热,若加热后于室温放置,则于数小时会析出结晶。因此,于对树脂组合物进行调整的情形时亦存在结晶析出的可能性,n,n'-(1,3-亚苯基二(2,2-亚丙基)二对亚苯基)双顺丁烯二酰亚胺的浓度越高,结晶化的可能性越高。为了制作印刷配线板及复合材,将玻璃布或碳纤维含浸于清漆而使树脂附着,但若结晶析出,则无法进行含浸作业,另一方面,若为了保持溶解状态而升高温度,则组合物的反应会加快,清漆的可使用时间变短。
[0097]
于本发明的一个实施方案的制造方法中,通过使用具有分子量分布且含有非对称胺的胺树脂来合成顺丁烯二酰亚胺树脂,可提高溶剂溶解性以及其硬化物中的介电特性。
[0098]
于由上述式(5)表示的芳族胺树脂中,由上述式(6)表示的芳族胺化合物以hplc面
积百分率计,优选为30面积%以上且未达60面积%,进而优选为30面积%以上且未达55面积%,尤其优选为35面积%以上且未达50面积%。
[0099]
于合成由上述式(5)表示的芳族胺树脂时,所使用的酸性催化剂可列举:盐酸、磷酸、硫酸、甲酸、氯化锌、氯化铁、氯化铝、对甲苯磺酸、甲磺酸等酸性催化剂等。于本实施方案中,优选为盐酸、对甲苯磺酸、甲磺酸等质子酸。这些可单独使用,亦可并用两种以上。催化剂的使用量相对于所使用的苯胺,通常为1~30重量%,优选为1~17重量%,进而优选为1~12重量%,尤其优选为1~7重量%,若催化剂的使用量过多,则目标的非对称结构的化合物较少,优先形成具有对称结构的化合物。若催化剂的使用量过少,则不仅减慢反应进行,且亦存在反应无法完成的情况,故不佳。
[0100]
关于反应,根据需要,可使用甲苯、二甲苯等有机溶剂进行,亦可于无溶剂下进行。例如,将酸性催化剂添加至苯胺与溶剂的混合溶液后,于催化剂包含水的情形时,优选为通过共沸将水自体系内去除。其后,添加二异丙烯苯或二(α-羟基异丙基)苯后,将溶剂自体系内去除,并同时升温,于140~190℃,优选为160~190℃进行5~50小时,优选为5~30小时的反应。于反应温度过高的情形时,非对称结构会于生成后再结合,优先形成对称结构,因此无法发挥目标的溶剂溶解性及电特性。由于在使用二(α-羟基异丙基)苯时会产生副产物水,因此于升温时水与溶剂共沸并同时自体系内去除。反应结束后,通过碱性水溶液中和酸性催化剂后,将非水溶性有机溶剂加入至油层,反复水洗直至废水变为中性为止,其后,于加热减压下去除溶剂及过量的苯胺衍生物。于使用活性白土或离子交换树脂的情形时,于反应结束后过滤反应液,去除催化剂。
[0101]
又,由于根据反应温度及催化剂的种类的不同会产生副产物二苯胺,因此视需要去除二苯胺为佳。于高温、高真空下,或者使用水蒸气蒸馏等手段,将二苯胺衍生物去除至1重量%以下,优选为0.5重量%以下,更优选为0.2重量%以下。
[0102]
本实施方案的顺丁烯二酰亚胺树脂例如通过以下方式获得:于溶剂及催化剂的存在下,使由上述式(5)表示的芳族胺树脂与顺丁烯二酸或顺丁烯二酸酐(以下亦称为“马来酸酐”)进行加成或进行脱水缩合反应。
[0103]
[顺丁烯二酰亚胺树脂的制造方法]
[0104]
关于反应中使用的溶剂,由于需要将于反应中生成的水自体系内去除,因此使用非水溶性的溶剂。非水溶性的溶剂例如可列举:甲苯、二甲苯等芳族溶剂;环己烷、正己烷等脂族溶剂;二乙醚、二异丙醚等醚类;乙酸乙酯、乙酸丁酯等酯系溶剂;甲基异丁基酮、环戊酮等酮系溶剂等,但并不限定于这些,亦可并用2种以上。
[0105]
又,除了上述非水溶性溶剂以外,亦可并用非质子性极性溶剂。例如可列举:二甲基砜、二甲基亚砜、二甲基甲酰胺、二甲基乙酰胺、1,3-二甲基-2-咪唑啶酮、n-甲基-2-吡咯啶酮等,亦可并用2种以上。于使用非质子性极性溶剂的情形时,优选为使用沸点比并用的非水溶性溶剂的沸点高者。
[0106]
又,反应中使用的催化剂为酸性催化剂,并无特别限定,例如可列举:对甲苯磺酸、羟基对甲苯磺酸、甲磺酸、硫酸、磷酸等。酸性催化剂的使用量相对于芳族胺树脂,通常为0.1~10重量%,优选为1~5重量%。于酸催化剂较多的情形时,对位位向性变强,有对结晶性及介电特性产生不良影响的担忧。
[0107]
例如,将由上述式(5)表示的芳族胺树脂溶解于甲苯与n-甲基-2-吡咯啶酮,向其
中添加马来酸酐,生成酰胺酸,其后添加对甲苯磺酸,自体系内去除于回流条件下生成的水,并同时进行反应。
[0108]
或者,将马来酸酐溶解于甲苯,于搅拌下添加由上述式(5)表示的芳族胺树脂的n-甲基-2-吡咯啶酮溶液,生成酰胺酸,其后添加对甲苯磺酸,自体系内去除于回流条件下生成的水,并同时进行反应。
[0109]
或者,将马来酸酐溶解于甲苯,添加对甲苯磺酸,于搅拌、回流状态滴加由上述式(5)表示的芳族胺树脂的n-甲基-2-吡咯啶酮溶液,并同时将于中途共沸而来的水除至体系外,且使甲苯返回至体系内,并同时进行反应(以上为第一段反应)。
[0110]
于任一方法中,马来酸酐相对于由上述式(5)表示的芳族胺树脂的胺基,通常使用1~3倍当量,优选为使用1.2~2.0倍当量。
[0111]
为了减少未闭环的酰胺酸,于上述列出的顺丁烯二酰亚胺化反应后,将水加入反应溶液,使之分离为树脂溶液层与水层,过量的顺丁烯二酸或顺丁烯二酸酐、非质子性极性溶剂、催化剂等于水层侧溶解,因此将其分液去除,进而反复进行同样的操作,彻底去除过量的顺丁烯二酸或顺丁烯二酸酐、非质子性极性溶剂、及催化剂。亦可再次向去除了过量的顺丁烯二酸或顺丁烯二酸酐、非质子性极性溶剂、及催化剂的有机层的顺丁烯二酰亚胺树脂溶液添加催化剂,再次进行于加热回流条件下的残留酰胺酸的脱水闭环反应(以上为第二段反应)。通过进行本方法,可获得酸值较低的顺丁烯二酰亚胺树脂溶液,但于本实施方案中,即使仅进行第一段反应也无妨。
[0112]
再脱水闭环反应的时间通常为1~5小时,优选为1~3小时,亦可根据需要添加上述非质子性极性溶剂。反应结束后,进行冷却,反复水洗直至水洗水变为中性为止。其后,于加热减压下通过共沸脱水将水去除后,可将溶剂蒸馏去除,或者加入其他溶剂,从而将树脂溶液调整至所需浓度,亦可将溶剂完全蒸馏去除,制成固形树脂而取出。
[0113]
继而,对本实施方案的可硬化性树脂组合物进行说明。
[0114]
于本实施方案的可硬化性树脂组合物,可含有能够与本实施方案的顺丁烯二酰亚胺树脂进行交联反应的化合物。作为该化合物,只要为具有胺基、氰酸基、酚性羟基、醇性羟基、烯丙基、甲基烯丙基、丙烯酰基、甲基丙烯酰基、乙烯基、共轭二烯基等可与顺丁烯二酰亚胺树脂进行交联反应的官能团(或结构)的化合物,则并无特别限定。
[0115]
由于胺化合物与顺丁烯二酰亚胺化合物进行交联反应,因此可使用由上述式(5)表示的芳族胺树脂。由于顺丁烯二酰亚胺树脂亦可自聚合,因此亦可单独使用。又,亦可并用除了由上述式(5)表示的芳族胺树脂以外的胺化合物或除了由上述式(1)表示的顺丁烯二酰亚胺树脂以外的顺丁烯二酰亚胺化合物。
[0116]
本实施方案的可硬化性树脂组合物中的由上述式(1)表示的顺丁烯二酰亚胺树脂的含量优选为10重量%以上,更优选为15重量%以上,进而优选为20重量%。于上述范围的情形时,关于硬化物的物性,呈现以下趋势:机械强度变高,剥离强度亦变高,进而耐热性亦变高。
[0117]
作为可掺合至本实施方案的可硬化性树脂组合物的胺化合物,可使用先前公知的胺化合物。作为胺化合物的具体例子,可列举:二亚乙基三胺、三亚乙基四胺、四亚乙基五胺、间苯二甲胺、三甲基六亚甲基二胺、2-甲基戊二胺、二乙基氨基丙胺、异佛酮二胺、1,3-双氨基甲基环己烷、双(4-氨基环己基)甲烷、双(4-氨基-3-甲基环己基)甲烷、降莰烯二胺、
1,2-二氨基环己烷、二氨基二苯甲烷、间苯二胺、二氨基二苯砜、二氰二胺、聚氧丙烯二胺、聚氧丙烯三胺、n-氨基乙基哌嗪、苯胺-福马林树脂等,但并不限定于这些。这些可单独地使用,亦可并用两种以上。
[0118]
又,专利文献3的权利要求书所记载的芳族胺树脂的低吸湿性、阻燃性、及介电特性优异,故尤佳。
[0119]
作为可掺合至本实施方案的可硬化性树脂组合物的顺丁烯二酰亚胺化合物,可使用先前公知的顺丁烯二酰亚胺化合物。作为顺丁烯二酰亚胺化合物的具体例子,可列举:4,4'-二苯基甲烷双顺丁烯二酰亚胺、聚苯基甲烷顺丁烯二酰亚胺、间亚苯基双顺丁烯二酰亚胺、2,2'-双[4-(4-顺丁烯二酰亚胺苯氧基)苯基]丙烷、3,3'-二甲基-5,5'-二乙基-4,4'-二苯基甲烷双顺丁烯二酰亚胺、4-甲基-1,3-亚苯基双顺丁烯二酰亚胺、4,4'-二苯醚双顺丁烯二酰亚胺、4,4'-二苯砜双顺丁烯二酰亚胺、1,3-双(3-顺丁烯二酰亚胺苯氧基)苯、1,3-双(4-顺丁烯二酰亚胺苯氧基)苯等,但并不限定于这些。这些可单独地使用,亦可并用两种以上。顺丁烯二酰亚胺化合物的掺合量以重量比计,优选为于本实施方案的顺丁烯二酰亚胺树脂的5倍以下的范围,更优选为于2倍以下的范围。
[0120]
又,专利文献3的权利要求所记载的顺丁烯二酰亚胺树脂的低吸湿性、阻燃性、及介电特性优异,故尤佳。
[0121]
作为可掺合至本实施方案的可硬化性树脂组合物的氰酸酯化合物,可使用先前公知的氰酸酯化合物。作为氰酸酯化合物的具体例子,可列举通过使酚类与各种醛的缩聚物、酚类与各种二烯化合物的聚合物、酚类与酮类的缩聚物、及双酚类与各种醛的缩聚物等与卤化氰进行反应而获得的氰酸酯化合物,但并不限定于这些。这些可单独地使用,亦可使用两种以上。
[0122]
作为上述酚类,可列举:苯酚、烷基取代苯酚、芳族取代苯酚、萘酚、烷基取代萘酚、二羟基苯、烷基取代二羟基苯、二羟基萘等。
[0123]
作为上述各种醛,可列举:甲醛、乙醛、烷基醛、苯甲醛、烷基取代苯甲醛、羟基苯甲醛、萘甲醛、戊二醛、苯二甲醛、巴豆醛、桂皮醛等。
[0124]
作为上述各种二烯化合物,可列举:二环戊二烯、萜烯类、乙烯基环己烯、降莰二烯、乙烯基降莰烯、四氢茚、二乙烯苯、二乙烯基联苯、二异丙烯基联苯、丁二烯、异戊二烯等。
[0125]
作为上述酮类,可列举:丙酮、甲基乙基酮、甲基异丁基酮、苯乙酮、二苯基酮等。
[0126]
又,合成方法于日本特开2005-264154号公报记载的氰酸酯化合物的低吸湿性、阻燃性、及介电特性优异,因此作为氰酸酯化合物尤佳。
[0127]
于本实施方案的可硬化性树脂组合物,可进而掺合环氧树脂。作为可掺合的环氧树脂,可使用先前公知的任一环氧树脂。作为环氧树脂的具体例子,可列举:使酚类与各种醛的缩聚物、酚类与各种二烯化合物的聚合物、酚类与酮类的缩聚物、双酚类与各种醛的缩聚物、及醇类等进行缩水甘油化而获得的环氧丙基醚系环氧树脂;以4-乙烯基-1-环己烯二环氧化物、3,4-环氧环己基甲基-3,4'-环氧环己烷羧酸酯等为代表的脂环式环氧树脂;以四环氧丙基二氨基二苯基甲烷(tgddm)、三环氧丙基对氨基苯酚等为代表的缩水甘油胺系环氧树脂;环氧丙酯系环氧树脂等,但并不限定于这些。这些可单独地使用,亦可使用两种以上。
[0128]
又,以通过使酚类与双卤化甲基芳烷基衍生物或芳烷基醇衍生物进行缩合反应而获得的苯酚芳烷基树脂为原料,与表氯醇进行脱氯化氢反应,由此获得环氧树脂,由于由此获得的环氧树脂的低吸湿性、阻燃性、及介电特性优异,因此作为环氧树脂尤佳。
[0129]
于掺合环氧树脂的情形时,掺合量并无特别限定,以重量比计优选为顺丁烯二酰亚胺树脂的0.1~10倍,更优选为顺丁烯二酰亚胺树脂的0.2~4倍的范围。若环氧树脂的掺合量为顺丁烯二酰亚胺树脂的0.1倍以下,则有硬化物变脆的担忧,若为10倍以上,则有介电特性降低的担忧。
[0130]
于本实施方案的可硬化性树脂组合物,可进而掺合具有酚树脂的化合物。
[0131]
作为可掺合的酚树脂,可使用先前公知的任一酚树脂。作为酚树脂的具体例子,可列举:双酚类(双酚a、双酚f、双酚s、联苯酚、双酚ad等)、酚类(苯酚、烷基取代苯酚、芳族取代苯酚、萘酚、烷基取代萘酚、二羟基苯、烷基取代二羟基苯、二羟基萘等)与各种醛(甲醛、乙醛、烷基醛、苯甲醛、烷基取代苯甲醛、羟基苯甲醛、萘甲醛、戊二醛、苯二甲醛、巴豆醛、桂皮醛等)的缩聚物;酚类与各种二烯化合物(二环戊二烯、萜烯类、乙烯基环己烯、降莰二烯、乙烯基降莰烯、四氢茚、二乙烯苯、二乙烯基联苯、二异丙烯基联苯、丁二烯、异戊二烯等)的聚合物;酚类与酮类(丙酮、甲基乙基酮、甲基异丁基酮、苯乙酮、二苯基酮等)的缩聚物;酚类与芳族二甲醇类(苯二甲醇、α,α,α',α'-苯二甲醇、联苯二甲醇、α,α,α',α'-联苯二甲醇等)的缩聚物;酚类与芳族二氯甲基类(α,α'-二氯二甲苯、双氯甲基联苯等)的缩聚物;双酚类与各种醛的缩聚物;及它们的改性物,但并不限定于这些。这些可单独地使用,亦可使用两种以上。
[0132]
又,通过使酚类与上述双卤化甲基芳烷基衍生物或芳烷基醇衍生物进行缩合反应而获得的苯酚芳烷基树脂由于低吸湿性、阻燃性、及介电特性优异,故而作为酚树脂尤佳。
[0133]
又,于上述酚树脂具有烯丙基或甲基烯丙基的情形时,对于顺丁烯二酰亚胺基的反应性比对于羟基的反应性良好,因此硬化速度变快,并且交联点增多,因此强度及耐热性提高,故较佳。
[0134]
又,亦可掺合上述酚树脂的羟基经烯丙化而成的烯丙醚体及经甲基烯丙化而成的甲基烯丙醚体,由于羟基被醚化,因此吸水性降低。
[0135]
于本实施方案的可硬化性树脂组合物,可进而掺合具有酸酐基的化合物。
[0136]
作为具有可掺合的酸酐基的化合物,可使用先前公知的任一者。作为具有酸酐基的化合物的具体例子,可列举:1,2,3,4-丁烷四羧酸二酐、1,2,3,4-环丁烷四羧酸二酐、1,2,3,4-环戊烷四羧酸二酐、1,2,4,5-环己烷四羧酸二酐、焦蜜石酸二酐、5-(2,5-二侧氧四氢呋喃基)-3-甲基-3-环己烯-1,2-二羧酸酐、4-(2,5-二侧氧四氢呋喃-3-基)-1,2,3,4-四氢萘-1,2-二羧酸酐等。
[0137]
具有酸酐基的化合物可单独使用或混合2种以上使用。又,酸酐基与胺反应,结果成为酰胺酸,若进而以200℃~300℃进行加热,则通过脱水反应成为酰亚胺结构,从而成为耐热性非常优异的材料。
[0138]
于本实施方案的可硬化性树脂组合物,可视需要掺合硬化用的催化剂(硬化促进剂)。例如可列举:2-甲基咪唑、2-乙基咪唑、2-苯基咪唑、2-乙基-4-甲基咪唑、2-十一基咪唑、1-氰基乙基-2-乙基-4-甲基咪唑等咪唑类;三乙胺、三亚乙二胺、2-(二甲基氨基甲基)苯酚、1,8-二氮双环(5,4,0)十一-7-烯、三(二甲基氨基甲基)苯酚、苄基二甲基胺等胺类;
三苯基膦、三丁基膦、三辛基膦等膦类;辛酸锡、辛酸锌、二顺丁烯二酸二丁基锡、环烷酸锌、环烷酸钴、油酸锡等有机金属盐;氯化锌、氯化铝、氯化锡等金属氯化物;二叔丁基过氧化物、二异丙苯基过氧化物等有机过氧化物;偶氮双异丁腈、偶氮双二甲基戊腈等偶氮化合物;盐酸、硫酸、磷酸等无机酸;三氟化硼等路易斯酸;碳酸钠、氯化锂等盐类等。于本实施方案中,尤其优选为掺合上述胺类、膦类、以及有机过氧化物。硬化用催化剂的掺合量相对于合计为100重量份的可硬化性树脂组合物,优选为10重量份以下,更优选为5重量份以下的范围。
[0139]
于本实施方案中,尤其是就介电特性的方面而言,优选为掺合聚丁二烯及其改性物、苯乙烯丁二烯共聚物等可硬化性的聚合物,进而优选为掺合经取代或未经取代的聚苯醚、聚苯乙烯、氟树脂,尤其优选为于该分子中具有1,2-乙烯基、丙烯酰基、甲基丙烯酰基、烯丙基、甲基烯丙基、乙烯苯、茚等结构作为官能团者。就介电特性、膜特性等方面而言,尤其优选为具有1,2-乙烯基的苯乙烯丁二烯共聚物、或者与具有丙烯酰基或甲基丙烯酰基的聚苯醚等的组合。
[0140]
进而,于本实施方案的可硬化性树脂组合物,可视需要掺合公知的添加剂。作为可使用的添加剂的具体例子,可列举:环氧树脂用硬化剂、丙烯腈共聚物的改性物、聚乙烯、聚酰亚胺、顺丁烯二酰亚胺系化合物、顺丁烯二酰亚胺树脂、氰酸酯系化合物、氰酸酯树脂、聚硅氧凝胶、聚硅氧油、以及二氧化硅、氧化铝、碳酸钙、石英粉、铝粉、石墨、滑石、粘土、氧化铁、氧化钛、氮化铝、石绵、云母、玻璃粉末等无机填充材、如硅烷偶合剂的填充材的表面处理剂、脱模剂、碳黑、酞青蓝、酞青绿等着色剂。所述添加剂的掺合量相对于100重量份的可硬化性树脂组合物,优选为1000重量份以下,更优选为700重量份以下的范围。
[0141]
本实施方案的可硬化性树脂组合物的制备方法并无特别限定,可仅将各成分均匀混合,或者可将各成分进行预聚化。例如,于存在催化剂或不存在催化剂的条件下、存在溶剂或不存在溶剂的条件下,对顺丁烯二酰亚胺树脂与氰酸酯化合物进行加热,由此进行预聚化。同样地,可添加本实施方案的顺丁烯二酰亚胺树脂,并根据需要添加环氧树脂、胺化合物、顺丁烯二酰亚胺系化合物、氰酸酯化合物、酚树脂、酸酐化合物、及其他添加剂来进行预聚化。关于各成分的混合或预聚化,于不存在溶剂的条件下例如使用挤出机、捏合机、辊等,于存在溶剂的条件下使用具有搅拌装置的反应釜等。
[0142]
通过使本实施方案的可硬化性树脂组合物加热熔融而低粘度化,并使其含浸于玻璃纤维、碳纤维、聚酯纤维、聚酰胺纤维、氧化铝纤维等增强纤维,可获得预浸体。又,通过使上述清漆含浸于如上所述的纤维并进行加热干燥,亦可获得预浸体。作为使用的纤维,尤其是于高频用途中,优选为使用不仅使用了e玻璃,且使用了ne玻璃等低介电玻璃、石英玻璃等的玻璃纤维。所述纤维物的掺合量相对于树脂总量100体积%,优选为10体积%~70体积%,尤其优选为20体积%~65体积%。又,亦可与填料等进行组合来处理,于此情形时,纤维及填料的总计相对于树脂总量100体积%,优选为80体积%以下。若超过80体积%,则不仅片材变脆,且于随后的硬化时无法表现出流动性,从而难以作为基板进行处理。
[0143]
将上述预浸体裁剪成所需的形状,根据需要与铜箔等积层后,通过加压成形法、高压釜成形法、或片缠绕成形法等对积层物施加压力,并同时使可硬化性树脂组合物加热硬化,由此可获得电气电子用积层板(印刷配线板)、或碳纤维增强材料。
[0144]
[实施例]
[0145]
以下,通过实施例及比较例对本发明进行具体说明,但本发明并不限定于以下例子。再者,本文中“份”及“%”分别表示“重量份”及“重量%”。软化点及熔融粘度通过下述方法进行测定。
[0146]
软化点:通过以jis k-7234为依据的方法进行测定。
[0147]
酸值:通过以jis k-0070:1992为依据的方法进行测定。
[0148]
·
gpc(凝胶渗透层析法)分析
[0149]
gpc:dgu-20a3r、lc-20ad、sil-20aht、rid-20a、spd-20a、cto-20a、cbm-20a(均由岛津制作所制造)
[0150]
管柱:shodex kf-603、kf-602
×
2、kf-601
×2[0151]
连结洗脱液:四氢呋喃
[0152]
流速:0.5ml/min
[0153]
管柱温度:40℃
[0154]
检测:ri(示差折射率检测器)。
[0155]
·
顺丁烯二酰亚胺当量
[0156]
通过gpc的面积%算出。
[0157]
·
hplc(高效液相层析法)分析
[0158]
岛津制作所股份有限公司制造的输液单元lc-20ad
[0159]
岛津制作所股份有限公司制造的光二极体阵列检测器spd-m20a
[0160]
岛津制作所股份有限公司制造的管柱烘箱cto-20a
[0161]
管柱:intersil ods-2.5μm,4.6
×
250mm 40℃
[0162]
流动相a:乙腈(an)
[0163]
流动相b:水(w)
[0164]
时间程式:
[0165]
0-28min.an/w=30%/70%
→
100%/0%
[0166]
28-40min.an/w=100%/0%
[0167]
流速:1.0ml/min
[0168]
检测:uv 200-274nm,pda
[0169]
测定波长:225nm。
[0170]
[合成例1]
[0171]
向安装有温度计、冷却管、迪安斯达克共沸蒸馏分离器、及搅拌机的烧瓶添加373份的苯胺、150份的甲苯、194份的间二(α-羟基异丙基)苯、及75份的活性白土(日本活性白土制造的e),蒸馏去除水及甲苯,并同时耗费6小时将体系内升温至170℃,于此温度反应20小时。其后,冷却至室温,加入600份的甲苯,并通过过滤而去除活性白土。继而,用旋转蒸发器于加热减压下将过量的苯胺与甲苯自油层蒸馏去除,由此获得320份的由上述式(5)表示的芳族胺树脂(sa-1)。芳族胺树脂(sa-1)的胺当量为180.5g/eq,软化点为45℃。通过gpc分析(ri),n=1体为78.8面积%,平均的n为1.22。将通过hplc分析得出的n=1体中的异构物的比率示于表1。又,其邻位位向与对位位向的面积比(o/p)为28%。
[0172]
邻位位向与对位位向的通过hplc分析得出的面积比(o/p)=(由上述式(7)表示的芳族胺化合物的面积%)
×
2+(由上述式(6)表示的芳族胺化合物的面积%)/(由上述式(8)
表示的芳族胺化合物的面积%)
×
2+(由上述式(6)表示的芳族胺化合物的面积%)。
[0173]
[合成例2]
[0174]
向安装有温度计、冷却管、迪安斯达克共沸蒸馏分离器、及搅拌机的烧瓶添加804份的苯胺、432份的甲苯、及450份的35%盐酸,升温并同时将水与甲苯蒸馏去除,使体系内为170℃,于该温度耗费1.5小时滴加158份的1,3-二异丙烯苯,于此温度反应30小时。其后,冷却并同时以体系内不会剧烈回流的方式缓慢滴加87份的30%氢氧化钠水溶液,于80℃以下加入432份的甲苯,并于70℃~80℃静置。去除分离的下层的水层,反复水洗反应液,直至洗净液变为中性为止。继而,用旋转蒸发器于加热减压下将过量的苯胺与甲苯自油层蒸馏去除后,加入864份的甲苯进行加热溶解,其后,加入864份的环己烷,进行晶析、过滤、干燥,由此获得303份的由上述式(5)表示的芳族胺树脂(sa-2)。通过gpc分析(ri),n=1体为98%以上,平均的n为1。将通过hplc分析得出的n=1体中的异构物的比率示于表1。又,其邻位位向与对位位向的面积比(o/p)为0%。
[0175]
[合成例3]
[0176]
向安装有温度计、冷却管、迪安斯达克共沸蒸馏分离器、及搅拌机的烧瓶添加373份的苯胺、250份的甲苯、及10份的35%盐酸,升温并同时将水与甲苯蒸馏去除,使体系内为170℃,于该温度耗费1.5小时滴加194份的1,3-二异丙烯苯,于此温度反应45小时。其后,冷却并同时以体系内不会剧烈回流的方式缓慢滴加17.4份的30%氢氧化钠水溶液,于80℃以下加入300份的甲苯,并于70℃~80℃静置。去除分离的下层的水层,反复水洗反应液,直至洗净液变为中性为止。继而,用旋转蒸发器于加热减压下将过量的苯胺与甲苯自油层蒸馏去除,由此获得306份的由上述式(5)表示的芳族胺树脂(a-1)。芳族胺树脂(a-1)的胺当量为187.9g/eq,软化点为59℃。通过gpc分析(ri),n=1体为62.5面积%,平均的n为1.45。将通过hplc分析得出的n=1体中的异构物的比率示于表1。又,其邻位位向与对位位向的面积比(o/p)为176%。
[0177]
[合成例4~12]
[0178]
于合成例3中改变使用催化剂、催化剂量、苯胺量、反应温度、反应时间,合成胺树脂a-2至a-10。将结果示于表1。
[0179]
[合成例13]
[0180]
向安装有温度计、冷却管、迪安斯达克共沸蒸馏分离器、及搅拌机的烧瓶添加192份的苯胺、123份的甲苯、及194份的1,3-二异丙烯苯,搅拌并同时添加21份的35%盐酸。其后,升温并同时将水与甲苯蒸馏去除,使体系内为160℃,反应24小时。其后,冷却并同时以体系内不会剧烈回流的方式缓慢滴加30份的30%氢氧化钠水溶液,于80℃以下加入20份的甲苯,于70℃~80℃静置。去除分离的下层的水层,反复水洗反应液,直至洗净液变为中性为止。继而,用旋转蒸发器于加热减压下将过量的苯胺与甲苯自油层蒸馏去除后,随后用旋转蒸发器于加热减压下将过量的苯胺与甲苯自油层蒸馏去除,由此获得158份的由上述式(5)表示的芳族胺树脂(a-11)。芳族胺树脂(a-13)的胺当量为186g/eq,软化点为58.8℃。通过gpc分析(ri),n=1体为64.9面积%,将通过hplc分析得出的n=1体中的异构物的比率示于表1。
[0181]
[表1]
[0182][0183]
hcl:35%盐酸水溶液(纯正化学制造)。
[0184]
[比较例1]
[0185]
向安装有温度计、冷却管、迪安斯达克共沸蒸馏分离器、及搅拌机的烧瓶添加147份的顺丁烯二酸酐、300份的甲苯、及4份的甲磺酸,并设为加热回流状态。继而,保持回流状态,并同时耗费3小时滴加以下树脂溶液,即197份的芳族胺树脂(sa-1)溶解于95份的n-甲基-2-吡咯啶酮及100份的甲苯而制成的树脂溶液。于此期间,将于回流条件共沸而来的缩合水与甲苯于迪安斯达克共沸蒸馏分离器内进行冷却、分液后,作为有机层的甲苯返回至体系内,水向体系外排出。结束滴加树脂溶液后,保持回流状态,进行脱水操作,并同时反应6小时。反应结束后,反复水洗四次,去除甲磺酸及过量的顺丁烯二酸酐,通过于70℃以下的加热减压下甲苯与水共沸,自体系内去除水。继而,加入2份的甲磺酸,于加热回流状态反应2小时。反应结束后,反复水洗四次,直至水洗水变为中性为止,其后,通过于70℃以下的加热减压下甲苯与水共沸,自体系内去除水,于加热减压下将甲苯完全蒸馏去除,由此获得由上述式(1)表示的顺丁烯二酰亚胺树脂(sm-1)。所获得的顺丁烯二酰亚胺树脂(sm-1)的软化点为100℃,酸值为9mg koh/g。通过gpc分析(ri),n=1体为73%,平均的n为1.37。将通过hplc分析得出的n=1体中的异构物的比率示于表2。又,其邻位位向与对位位向的面积比(o/p)为30%。
[0186]
[比较例2]
[0187]
向安装有温度计、冷却管、迪安斯达克共沸蒸馏分离器、及搅拌机的烧瓶添加147份的顺丁烯二酸酐、300份的甲苯、及3.3份的甲磺酸,并设为加热回流状态。继而,保持回流状态,并同时耗费3小时滴加以下树脂溶液,即172份的1,3-双(对氨基异丙苯基)苯(sa-2)溶解于66份的n-甲基-2-吡咯啶酮及100份的甲苯而制成的树脂溶液。于此期间,将于回流条件下共沸而来的缩合水与甲苯于迪安斯达克共沸蒸馏分离器内进行冷却、分液后,作为
有机层的甲苯返回至体系内,水向体系外排出。结束滴加树脂溶液后,保持回流状态,进行脱水操作,并同时反应2小时。反应结束后,反复水洗四次,去除甲磺酸及过量的顺丁烯二酸酐,通过于70℃以下的加热减压下甲苯与水共沸,自体系内去除水。继而,加入1.7份的甲磺酸,于加热回流状态反应2小时。反应结束后,反复水洗三次,直至水洗水变为中性为止,其后,通过于70℃以下的加热减压下甲苯与水共沸,自体系内去除水,继而,于加热减压下将甲苯完全蒸馏去除,由此获得237份的顺丁烯二酰亚胺树脂(sm-2)。所获得的顺丁烯二酰亚胺树脂(sm-2)的软化点为91℃,酸值为3mg koh/g。通过gpc分析(r1),n=1体为95%,平均的n为1.02。将n=1体中的异构物的比率示于表2。又,其邻位位向与对位位向的面积比(o/p)为0%。
[0188]
[实施例1]
[0189]
向安装有温度计、冷却管、迪安斯达克共沸蒸馏分离器、及搅拌机的烧瓶添加147份的顺丁烯二酸酐、300份的甲苯、及4份的甲磺酸,并设为加热回流状态。继而,保持回流状态,并同时耗费3小时滴加以下树脂溶液,即197份的芳族胺树脂(a-1)溶解于95份的n-甲基-2-吡咯啶酮及100份的甲苯而制成的树脂溶液。于此期间,将于回流条件下共沸而来的缩合水与甲苯于迪安斯达克共沸蒸馏分离器内进行冷却、分液后,作为有机层的甲苯返回至体系内,水向体系外排出。结束滴加树脂溶液后,保持回流状态,进行脱水操作,并同时反应6小时。反应结束后,反复水洗四次,去除甲磺酸及过量的顺丁烯二酸酐,通过于70℃以下的加热减压下甲苯与水共沸,自体系内去除水。继而,加入2份的甲磺酸,于加热回流状态反应2小时。反应结束后,反复水洗四次,直至水洗水变为中性为止,其后,通过于70℃以下的加热减压下甲苯与水共沸,自体系内去除水,继而,关于甲苯,于加热减压下蒸馏去除溶剂,直至树脂浓度达到大约70-80%为止,其后,追加甲苯,将树脂浓度调整至60%。由此,获得含有顺丁烯二酰亚胺树脂(m-1)的顺丁烯二酰亚胺溶液(v1)。通过gpc分析(ri),n=1体为57%,平均的n为1.80。将通过hplc分析得出的n=1体中的异构物的比率示于表2。分取50份所获得的树脂溶液,于加热减压下,完全蒸馏去除溶剂,由此获得由上述式(1)表示的顺丁烯二酰亚胺树脂(m-1)。所获得的顺丁烯二酰亚胺树脂(m-1)的软化点为115℃,酸值为7.5mg koh/g。又,其邻位位向与对位位向的面积比(o/p)为163%。
[0190]
[实施例2]
[0191]
向安装有温度计、冷却管、迪安斯达克共沸蒸馏分离器、及搅拌机的烧瓶添加147份的顺丁烯二酸酐、300份的甲苯、及4份的甲磺酸,并设为加热回流状态。继而,保持回流状态,并同时耗费3小时滴加以下树脂溶液,即197份的芳族胺树脂(a-2)溶解于95份的n-甲基-2-吡咯啶酮及100份的甲苯而制成的树脂溶液。于此期间,将于回流条件下共沸而来的缩合水与甲苯于迪安斯达克共沸蒸馏分离器内进行冷却、分液后,作为有机层的甲苯返回至体系内,水向体系外排出。结束滴加树脂溶液后,保持回流状态,进行脱水操作,并同时反应6小时。反应结束后,反复水洗四次,去除甲磺酸及过量的顺丁烯二酸酐,通过于70℃以下的加热减压下甲苯与水共沸,自体系内去除水。继而,加入2份的甲磺酸,于加热回流状态反应2小时。反应结束后,反复水洗四次,直至水洗水变为中性为止,其后,通过于70℃以下的加热减压下甲苯与水共沸,自体系内去除水,继而,将甲苯于加热减压下完全蒸馏去除,由此获得由上述式(1)表示的顺丁烯二酰亚胺树脂(m-2)。所获得的顺丁烯二酰亚胺树脂(m-2)的软化点为111℃,酸值为8mg koh/g。通过gpc分析(ri),n=1体为56%,平均的n为1.78。
将通过hplc分析得出的n=1体中的异构物的比率示于表2。又,其邻位位向与对位位向的面积比(o/p)为120%。
[0192]
[实施例3~5]
[0193]
对于合成例6、8、9中所获得的胺树脂,与实施例1同样地进行合成。将结果示于表2。
[0194]
[实施例14]
[0195]
向安装有温度计、冷却管、迪安斯达克共沸蒸馏分离器、及搅拌机的烧瓶添加147份的顺丁烯二酸酐、300份的甲苯、及8份的甲磺酸,并设为加热回流状态。继而,保持回流状态,并同时耗费3小时滴加以下树脂溶液,即190份的芳族胺树脂(a-11)溶解于95份的n-甲基-2-吡咯啶酮及100份的甲苯而制成的树脂溶液。于此期间,将于回流条件下共沸而来的缩合水与甲苯于迪安斯达克共沸蒸馏分离器内进行冷却、分后,作为有机层的甲苯返回至体系内,水向体系外排出。结束滴加树脂溶液后,保持回流状态,进行脱水操作,并同时反应6小时。
[0196]
反应结束后,反复水洗四次,去除甲磺酸及过量的顺丁烯二酸酐,通过于70℃以下的加热减压下甲苯与水共沸,自体系内去除水。继而,加入2份的甲磺酸,于加热回流状态反应2小时。反应结束后,反复水洗四次,直至水洗水变为中性为止,其后,通过于70℃以下的加热减压下甲苯与水共沸,自体系内去除水,继而,关于甲苯,于加热减压下将溶剂蒸馏去除,直至树脂浓度达到大约70-80%为止,其后,追加甲苯,将树脂浓度制备为60%。由此,获得含有顺丁烯二酰亚胺树脂(m-11)的顺丁烯二酰亚胺溶液(v-11)。通过gpc分析(ri),n=1体为48.1%,平均的n为1.85。将通过hplc分析得出的n=1体中的异构物的比率示于表2。又,其邻位位向与对位位向的面积比(o/p)为127%。
[0197]
分取50份的所获得的树脂溶液,于加热减压下,完全蒸馏去除溶剂,由此获得由上述式(1)表示的顺丁烯二酰亚胺树脂(m-11)。所获得的顺丁烯二酰亚胺树脂(m-11)的软化点为126℃,酸值为9.5mg koh/g。
[0198]
[表2]
[0199][0200][0201]
[比较例3、4及实施例6~10]
[0202]
将比较例1、2、实施例1~5中所获得的顺丁烯二酰亚胺树脂以树脂部分为60%的
方式溶解于甲苯,获得顺丁烯二酰亚胺树脂溶液vsm-1、2、vm-1~5。将所获得的顺丁烯二酰亚胺树脂溶液于-10℃保管三个月后,观察有无结晶析出。将观察到结晶析出者设为
×
,未观察到结晶析出者设为
○
,并示于表3。
[0203]
[表3]
[0204][0205]
根据表3,确认到本实施方案的顺丁烯二酰亚胺树脂的溶液稳定性优异。
[0206]
[实施例11、12、比较例5、6]
[0207]
对于通过实施例1、3所获得的顺丁烯二酰亚胺树脂m-1、m-3、及通过比较例1、2所获得的sm-1、sm-2各50重量份,掺合0.75重量份的催化剂(dcp,二异丙苯基过氧化物,化药诺力昂公司制造),并于250℃硬化2小时。自所获得的硬化物切出2.5mm
×
50mm
×
0.25mm的板,使用空腔共振机(aet公司制造的adms01oc1)对在1ghz及10ghz的频率的介电常数及介电损耗正切进行测定,将所测定的结果示于表4。
[0208]
[表4]
[0209] 比较例5比较例6实施例11实施例12顺丁烯二酰亚胺sm-1sm-2m-1m-3介电常数(1ghz)2.712.722.69-介电损耗正切(1ghz)0.00170.0020.0016-介电常数(10ghz)2.662.682.622.59介电损耗正切(10ghz)0.00190.00260.00160.0016
[0210]
※
表中的
“‑”
表示未测定。
[0211]
根据表4,确认到本实施方案的顺丁烯二酰亚胺树脂不仅于1ghz,且于10ghz亦具有优异的介电特性。
[0212]
[实施例13、比较例7]
[0213]
对于由实施例4所获得的顺丁烯二酰亚胺树脂m-4、及由比较例1所获得的sm-1各50重量份,掺合1.5重量份的催化剂(2e4mz:2-乙基-4-甲基咪唑(东京化成工业公司制造)),并于250℃硬化2小时。自所获得的硬化物切出50mm
×
50mm
×
0.25mm的板,使用平衡型圆板共振器法(共振器:使用fatec制造的平衡型圆板共振器),对在25℃、150℃各频率的介电损耗正切进行测定,将所测定的结果示于表5。
[0214]
[表5]
[0215][0216]
根据表5,确认到本实施方案的顺丁烯二酰亚胺树脂即使于高频区域亦具有优异的介电特性。又,确认到本实施方案的顺丁烯二酰亚胺树脂因温度导致的差异亦小,亦具有较高的环境耐性。
[0217]
[实施例15、比较例8、9]
[0218]
使用由实施例14所获得的顺丁烯二酰亚胺树脂(m-11)、由比较例1所获得的顺丁烯二酰亚胺树脂(sm-1)、及由比较例2所获得的顺丁烯二酰亚胺树脂(sm-2),以表6记载的比例(重量份)掺合各种环氧树脂、硬化剂、及硬化促进剂,通过混合辊进行混炼并平板化后,通过转移成形制备树脂成形体,于200℃硬化2小时。针对以下项目测定由此所获得的硬化物的物性,将所测定的结果示于表6。又,针对各硬化物,通过动态粘弹性装置(dma)测定弹性模量根据温度的变化。将结果示于图1。
[0219]
·
tma tg:通过热机械特性装置测定玻璃化转变温度。
[0220]
·
dma弹性模量:通过动态粘弹性试验机(dma)测定。
[0221]
·
td5(5%热重量减少温度):针对粉碎所获得的硬化物而得到的粉状物,使用通过100网目的筛网且无法通过200网目的筛网的样本,通过tg-dta测定热分解温度。其是以10mg的样本量、10℃/min的升温速度、及200ml/hr的空气量测定的重量减少了5%的温度。
[0222]
·
弯曲模量:以jis k-6911为根据测定。
[0223]
·
介电常数及介电损耗正切:使用空腔共振机(aet公司制造)于室温测定。
[0224]
[表6]
[0225]
[0226]
e1:nc-3000-l(日本化药制造,环氧当量为271g/eq)
[0227]
p1:
カヤハード
gph-65(日本化药制造,羟基当量200g/eq)
[0228]
c1:2e4mz:2-乙基-4-甲基咪唑(东京化成工业公司制造)。
[0229]
根据表6的结果,确认到实施例15的硬化物于自室温至低温的范围具有较高的弹性模量,有助于基板的刚性,并且于回流焊时的温度范围亦维持较高的弹性模量。又,由于可确认到耐热性(tg)亦较高,且于热时的稳定性(耐热分解td5)方面亦优异,因此可谓通过热产生的耐候性优异。进而,于介电特性方面,确认到可抑制通过掺合环氧树脂产生的介电特性劣化的影响,介电常数与介电损耗正切皆优异。
[0230]
[实施例16、比较例10]
[0231]
对于由实施例14所获得的顺丁烯二酰亚胺树脂(m-11)及由比较例1所获得的顺丁烯二酰亚胺树脂(sm-1)各50重量份,以0.75重量份掺合催化剂(dcp,二异丙苯基过氧化物,化药诺力昂公司制造),进行175℃的转移成形,其后,于250℃硬化2小时。自所获得的硬化物切出2.5mm
×
50mm
×
0.25mm的板,于干燥后及于室温浸渍于水24小时后,使用空腔共振机(aet公司制造),于10ghz的频率对其介电常数及介电损耗正切进行测定,将所测定的结果示于表7。
[0232]
[表7]
[0233][0234]
根据表7的结果,确认到实施例16的硬化物于吸水试验后的介电特性降低较少,吸湿性较低,因此耐候性优异。
[0235]
本技术基于2020年3月11日申请的日本专利申请2020-41840号,其内容作为参考而引入于此。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1