用于制备1,2,4,6-四-O-乙酰基-3-叠氮基-3-脱氧-D-吡喃半乳糖苷的大规模方法与流程
用于制备1,2,4,6-四-o-乙酰基-3-叠氮基-3-脱氧-d-吡喃半乳糖苷的大规模方法
技术领域
1.本发明涉及制备1,2,4,6-四-o-乙酰基-3-叠氮基-3-脱氧-α/β-d-吡喃半乳糖苷,特别是制备1,2,4,6-四-o-乙酰基-3-叠氮基-3-脱氧-β-d-吡喃半乳糖苷的方法,所述方法可以放大规模。所述方法的参数稳定,并且所述方法适用于gmp生产。
背景技术:
2.特发性肺纤维化(ipf)代表了巨大的全球健康负担。它是一种病因未知的慢性病症,其中反复急性肺损伤引起进行性纤维化,导致肺结构破坏,使肺功能恶化,结果是呼吸衰竭和死亡。虽然特发性肺纤维化(ipf)是肺纤维化的原型和最常见原因,但许多呼吸系统疾病可能进展为肺纤维化,并且这通常意味着预后较差。从诊断到死亡的中位时间是2.5年,并且ipf的发病率和患病率持续上升。它仍然是少数没有有效疗法的呼吸系统病症之一,并且没有可靠的生物标志物来预测疾病进展。导致肺纤维化的机制尚不清楚,但主要围绕在由于尚且未知的原因造成的重复的上皮细胞损伤导致的伤口愈合异常。ipf的特征在于含有成纤维细胞/肌成纤维细胞的成纤维细胞病灶,其显示出对成纤维细胞因子诸如转化生长因子-β1(tgf-β1)的激活反应增加。治疗特发性肺纤维化的药物尚未满足巨大的需求。化合物3,3'-二脱氧-3,3
’‑
双-[4-(3-氟苯基)-1h-1,2,3-三唑-1-基]-1,1'-硫烷二基-二-β-d-吡喃半乳糖苷目前处于治疗ipf的临床ii期,并且制备所述化合物的方法在wo 2014/067986中有描述。式x的化合物是制备上述化合物的非常重要的中间体,并且因此需要可以放大规模的制造所述化合物的方法。
技术实现要素:
[0003]
本发明涉及用于制备1,2,4,6-四-o-乙酰基-3-叠氮基-3-脱氧-β-d-吡喃半乳糖苷的新方法,
[0004][0005]
所述方法可以扩大规模到大规模和/或工业规模,诸如30kg或更多,例如80kg或更多。例如,从2kg至80kg,诸如从4kg至80kg或从10kg至100kg。所述方法也可以用于较小规模,诸如从200g至2kg。
[0006]
在第一方面,本发明涉及用于制备具有式(x)的化合物的方法,诸如适用于大规模合成的方法,
[0007][0008]
其中所述方法包括使式ix的化合物
[0009][0010]
与乙酰化剂在碱的存在下在合适的溶剂中并且在合适的温度下反应足够的反应时间以制备式x的化合物。
[0011]
在实施方案中,将式x的化合物纯化并且分离为固体。优选地,将1,2,4,6-四-o-乙酰基-3-叠氮基-3-脱氧-β-d-吡喃半乳糖苷分离为白色固体,诸如结晶的或无定形的。
[0012]
在进一步的实施方案中,所述乙酰化剂选自以下的一种或多种:乙酸酐;酰氯;在活化剂诸如羰基二咪唑或二烷基碳二亚胺的存在下的乙酸;在脱水条件下的酸催化剂;或使用酰基酯的酯交换剂。
[0013]
在再进一步的实施方案中,所述碱选自以下的一种或多种:叔胺,诸如三乙胺或二异丙基乙胺;或芳族胺碱,诸如吡啶或咪唑,任选地在催化更强的碱诸如二甲氨基吡啶的存在下。
[0014]
在进一步的实施方案中,所述合适的溶剂选自一种或多种环状或非环状醚类溶剂,诸如1,4-二噁烷、2-甲基四氢呋喃或叔丁基甲基醚;酯溶剂,诸如乙酸乙酯或乙酸异丙酯;芳族溶剂,诸如甲苯。
[0015]
在再进一步的实施方案中,所述合适的温度是从-5℃至40℃。典型地从-5℃至35℃。
[0016]
在进一步的实施方案中,任选地向合适的溶剂中添加乙酰化剂在至少30分钟诸如至少3小时的时间段内进行。
[0017]
在再进一步的实施方案中,反应时间是从1至24小时。
[0018]
在进一步的实施方案中,所述方法包括用于制备具有式ix的化合物的前序步骤,其中所述前序步骤包括使式viii的化合物
[0019][0020]
与酸(诸如酸性阳离子交换树脂或对甲苯磺酸)在合适的溶剂中在合适的温度下反应足够的反应时间,以制备式ix的化合物。
[0021]
在实施方案中,所述酸选自一种或多种酸性阳离子交换树脂诸如amberlite ir-120h、稀盐酸或对甲苯磺酸。
[0022]
在进一步的实施方案中,所述合适的溶剂是有机溶剂和水的混合物,诸如1,4-二噁烷、2-甲基四氢呋喃、四氢呋喃或乙腈和水。
[0023]
在再进一步的实施方案中,所述合适的温度是从25℃至70℃。
[0024]
在进一步的实施方案中,反应时间是1至24小时。
[0025]
在再进一步的实施方案中,所述方法包括用于制备具有式viii的化合物的前序步骤,其中所述前序步骤包括使式vii的化合物
[0026][0027]
与合适的叠氮化物在合适的溶剂中在合适的温度下反应足够的反应时间,以制备式viii的化合物。
[0028]
在实施方案中,所述叠氮化物选自叠氮化物盐,诸如叠氮化钠或叠氮化钾。
[0029]
在进一步的实施方案中,所述合适的溶剂选自一种或多种偶极非质子溶剂,诸如二甲基甲酰胺、二甲亚砜或乙腈;或双相体系,诸如叔丁基甲基醚或类似的水不混溶性溶剂,诸如2-甲基四氢呋喃/水与相转移催化剂,诸如四丁基溴化铵。
[0030]
在再进一步的实施方案中,所述合适的温度是从0℃至30℃。
[0031]
在进一步的实施方案中,反应时间是30min至22小时。
[0032]
在再进一步的实施方案中,所述方法包括用于制备具有式vii的化合物的前序步骤,其中所述前序步骤包括使式vi的化合物
[0033][0034]
与合适的碱在合适的溶剂中反应,然后与三氟甲基化剂(trifluoromethylating agent)在合适的溶剂中在合适的温度下反应足够的反应时间,以制备式vii的化合物。
[0035]
在实施方案中,所述合适的碱是吡啶或受阻脂族叔胺,诸如二异丙基乙胺。
[0036]
在进一步的实施方案中,所述三氟甲磺酸化剂(triflating agent)选自一种或多种三氟甲磺酸酐或等效的三氟甲磺酸化剂,诸如n-苯基-双(三氟甲磺酰亚胺)。
[0037]
在再进一步的实施方案中,所述合适的溶剂独立地选自非质子溶剂,诸如叔丁基甲基醚、甲苯或四氢呋喃。
[0038]
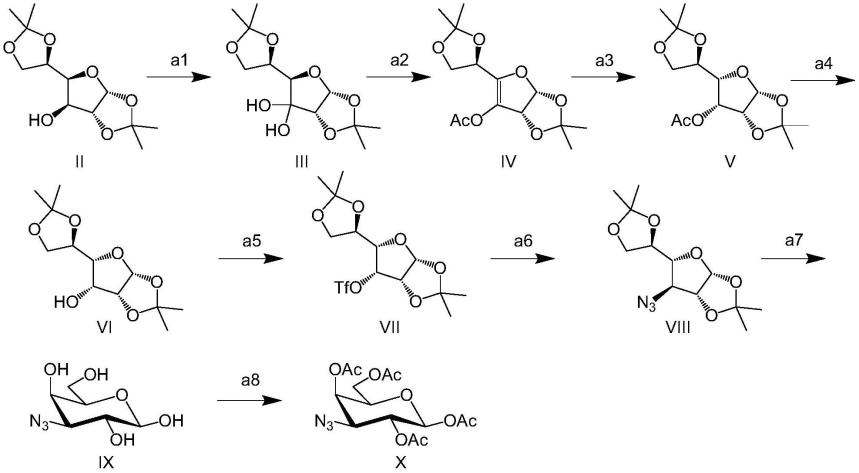
在进一步的实施方案中,所述合适的温度是从0℃至30℃。23.根据权利要求19-22中任一项所述的方法,其中所述合适的温度是从-5℃至30℃。
[0039]
在再进一步的实施方案中,反应时间是至少1小时。
具体实施方式
[0040]
式(x)的化合物具有化学名称(iupac)1,2,4,6-四-o-乙酰基-3-叠氮基-3-脱氧-β-d-吡喃半乳糖苷。式x的化合物是β端基异构体,然而,α端基异构体和β端基异构体的混合物已被lowary,t.l.和hindsgaul,o.(1994)recognition of synthetic o-methyl,epimeric,and aminoanalogues of the acceptor alpha-l-fucp-(1,2-beta-d-galp-or by the blood group a and bgene-specified glycosyltransferases.carbohydr.res.251:33-67公开。α端基异构体和β端基异构体可以通过各种方法诸如通过结晶来分离。然而,对于本发明方法,优选的目的是制备β端基异构体。
[0041]
在本文的实验部分中描述了所述方法的进一步实施方案,并且每个单独的方法以及每种起始材料构成可以形成实施方案的一部分的实施方案。
[0042]
上述实施方案应被视为是指本文描述的任一方面(诸如
‘
治疗方法’、
‘
药物组合物’、
‘
用作药剂的化合物’或
‘
用于方法中的化合物’)以及本文描述的任一个实施方案,除非指定实施方案涉及本发明的某一个或多个方面。
[0043]
本文引用的所有参考文献,包括出版物、专利申请和专利,均通过引用特此并入,其程度就好像单独且具体地指示将每个参考文献通过引用并入并且以其整体在本文中阐述一样。
[0044]
本文中使用的所有标题和子标题只是为了方便而使用,并且不应被解释为以任何方式限制本发明。
[0045]
本发明涵盖上述要素的所有可能变化的任何组合,除非本文另有指示或与上下文明显矛盾。
[0046]
如在描述本发明的上下文中使用的术语“一(a)”和“一(an)”和“所述(the)”以及类似的指称应被解释为涵盖单数和复数两者,除非本文另有指示或与上下文明显矛盾。
[0047]
本文中的值范围的列举仅旨在用作单独提及引用落入范围内的每个单独值的速记方法,除非在本文中另有指示,并且将每个单独的值并入说明书中,就好像它在本文中被单独列举一样。除非另有说明,否则本文提供的所有确切值都代表相应的近似值(例如,关于特定因素或测量值提供的所有确切示例性值可以被认为也提供相应的近似测量值,在适当时被“约”修饰)。
[0048]
除非本文另有指示或与上下文明显矛盾,否则本文所述的所有方法都可以以任何合适的顺序执行。
[0049]
除非另有指示,否则本文提供的任何和所有例子或示例性语言(例如,“诸如”)的使用仅旨在更好地阐明本发明并且不对本发明的范围构成限制。说明书中的语言都不应被解释为表明任何要素对于本发明的实践都是必不可少的,除非同样明确说明。
[0050]
本文中专利文件的引用和并入仅为方便起见,并且不反映对此类专利文件的有效性、可专利性和/或可执行性的任何看法。
[0051]
本文关于一个要素或多个要素使用的术语诸如“包含”、“具有”、“包括”或“含有”对本发明的任何方面或实施方案的描述旨在为本发明的类似方面或实施方案提供支持,所述类似方面或实施方案“由该特定的一个要素或多个要素组成”、“基本上由该特定的一个要素或多个要素组成”或“基本上包含该特定的一个要素或多个要素”,除非另有说明或与上下文明显矛盾(例如,本文描述为包含特定要素的组合物应理解为还描述了由该要素组
成的组合物,除非另有说明或与上下文明显矛盾)。
[0052]
在适用法律允许的最大范围内,本发明包括本文提出的方面或权利要求中列举的主题的所有修改和等效方案。
[0053]
本发明通过以下实施例进一步进行说明,然而,所述实施例不应被解释为限制保护范围。在前述描述和以下实施例中公开的特征可以,既单独地又以其任何组合,对于以其多样的形式实现本发明而言是重要的。
[0054]
实验
[0055][0056]
制造具有式x的化合物的当前方法涉及如下文详细描述的几个方法步骤。
[0057]
通用程序
[0058]
在25℃下在400mhz bruker avance av400波谱仪上记录核磁共振(nmr)波谱。使用残留溶剂作为内标,以ppm(δ)报告化学位移。峰多重性表示如下:s,单峰;d、双重峰;t,三重峰;q,四重峰;m,多重峰;br s,宽单峰。
[0059]
使用以下缩写:
[0060]
ac:乙酰基
[0061]
aq.:水溶液
[0062]
dcm:二氯甲烷
[0063]
dmf:n,n-二甲基甲酰胺
[0064]
rt:室温
[0065]
sat.:饱和
[0066]
tbme:叔丁基甲基醚
[0067]
tempo:(2,2,6,6-四甲基哌啶-1-基)氧基
[0068]
1,2:5,6-二-o-异亚丙基-α-d-呋喃糖(glucofuran)-3-酮糖水合物,iii
[0069]
[0070]
向在20℃下的溶解在乙酸乙酯(17.33l)和水(7.00l)的混合物中的1,2:5,6-二-o-异亚丙基-α-d-呋喃葡萄糖(glucofuranose)ii(3.5kg,13.45mol)中添加nabr(1.08kg,10.5mol)、naoac(1.66kg,20.18mol)和tempo(21g,0.13mol)。经30分钟将反应混合物从20℃冷却至12℃,并且添加次氯酸钠溶液(14%,12.9l,29.59mol)。将混合物在18℃下搅拌30分钟,然后添加2m na2s2o3水溶液直到观察到阴性的淀粉-碘化物测试。分离各相,并且将有机相用饱和nacl水溶液(3.5l)洗涤,经mgso4干燥,并且使用旋转蒸发器在真空中浓缩以得到3.72kg呈油性固体的1,2:5,6-二-o-异亚丙基-α-d-呋喃糖-3-酮糖水合物。1hnmr(400mhz,cdcl3)δ5.86(d,j=3.8hz,1h),4.43(q,j=6.3hz,1h),4.26(d,j=3.8hz,1h),4.15(dd,j=8.7,6.3hz,1h),4.09(dd,j=8.7,6.1hz,1h),3.97(br s,1h),3.91(d,j=6.5hz,1h),3.62(br s,1h),1.58(s,3h),1.50(s,3h),1.38(s,3h),1.36(s,3h)。
[0071]
可替代程序
[0072]
在27℃下,向溶解在乙酸乙酯混合物(504l)中的1,2:5,6-二-o-异亚丙基-α-d-呋喃葡萄糖ii(112kg,430mol)中添加脱矿质水(168l)、nabr(35.0kg,340mol)、naoac(53.0kg,646mol)和另外的脱矿质水(56l)。将反应混合物冷却至0℃,然后添加tempo(1.0kg,6.45mol)和另外的乙酸乙酯(56l)。向单独的容器中添加次氯酸钠(9%-12%w./w,1000l,约1649mol),并且在2.5℃下用浓硫酸:水的1:1混合物(约67l)将ph调节至11.5至12.5。将次氯酸钠溶液过滤,并且在7℃下经7.5小时添加到反应混合物中。将混合物在3℃下搅拌1小时,然后在3℃下添加20%w/w亚硫酸钠水溶液(135kg)。将混合物温热至29℃,并且分离各相。检查有机相是否不存在次氯酸钠。向水相中添加固体nacl(56kg),并且然后将其用乙酸乙酯(225l)萃取。向水相中添加固体nacl(28kg),并且然后将其用乙酸乙酯(224l)萃取。在30℃下,将合并的有机相用9%w/w氯化钠水溶液(112kg)洗涤,分离各层,并且将水层用乙酸乙酯(112l)反萃取。将合并的有机层用mgso4(56kg)干燥,过滤,将滤饼用乙酸乙酯(56l)洗涤,并且将合并的滤液在至多45℃下在真空中浓缩至392l。添加三次另外的乙酸乙酯(225l、225l和112l),进行蒸馏循环以将水控制在≤0.3%w/w并且确定溶液的测定。将1,2:5,6-二-o-异亚丙基-α-d-呋喃糖-3-酮糖水合物iii作为乙酸乙酯溶液进行至方法的下一步骤,假定119kg,100%产率。
[0073]
3-o-乙酰基-1,2:5,6-二-o-异亚丙基-α-d-赤型-己-3-烯呋喃糖(enofuranose),iv
[0074][0075]
向在20℃下的1,2:5,6-二-o-异亚丙基-α-d-呋喃糖-3-酮糖水合物iii(3.5kg,12.7mol)的吡啶(7.17l,88.7mol)溶液中,分批加入乙酸酐(3.60l,38.1mol)。将反应混合物加热至60℃并且搅拌16小时。将混合物冷却至20℃,并且加入tbme(14l)。在20℃+/-5℃下,将混合物按顺序用2m hcl水溶液(2x10.5l)、水(7l)、5%nahco3水溶液(10.5l)和饱和nacl水溶液(7l)洗涤。将有机相经mgso4干燥,并且使用旋转蒸发器在真空中浓缩以得到2.78kg呈灰白色固体的3-o-乙酰基-1,2:5,6-二-o-异亚丙基-α-d-赤型-己-3-烯呋喃糖。1h nmr(400mhz,cdcl3)δ5.95(d,j=5.5hz,1h),5.31(d,j=5.5hz,1h),4.62(t,j=6.4hz,
1h),4.03-3.94(m,2h),2.12(s,3h),1.45(s,3h),1.39(s,3h),1.36(s,3h),1.30(s,3h)。
[0076]
可替代程序
[0077]
在32℃下,向1,2:5,6-二-o-异亚丙基-α-d-呋喃糖-3-酮糖水合物iii(119kg,430mol)的乙酸乙酯溶液(从上一步骤进行而来的约301l)中,添加吡啶(85.1kg,1075mol)、二甲基氨基吡啶(dmap)(11.2kg,91.7mol)、乙酸乙酯(112l)和乙酸酐(88kg,862mol)。将反应加热至60℃持续4小时,然后冷却至27℃并且添加甲醇(11.2l)。将混合物在27℃下搅拌45分钟,冷却至12℃,添加乙酸乙酯(675l),然后在15℃下将有机溶液用2m hcl水溶液(3x336l)洗涤。在15℃下,将有机层用水(340l)洗涤,用5%nahco3水溶液洗涤三次(560l,然后2x340l),然后用5%nacl水溶液(225l)洗涤。将有机层分离并且在至多45℃下在真空中浓缩至约112l,然后添加正庚烷(225l),并且继续蒸馏。添加另外的庚烷(170l),并且在至多45℃下在真空中继续蒸馏至约112l。将残余物冷却至25℃,并且添加tbme(900l)。将溶液冷却至10℃,并且用1m hcl水溶液(3x294l)、水(340l)、5%w/w nahco3水溶液(3x560l)、然后5%w/w nacl水溶液(225l)洗涤。将有机层分离并且用amberlite ir-96树脂(4.5kg)处理,同时在至多40℃下在真空中蒸馏至约112l。将甲醇(900l)添加到残余物中,并且在30℃下将混合物用活性炭(11.2kg)处理。将混合物经助滤剂过滤,并且将3-o-乙酰基-1,2:5,6-二-o-异亚丙基-α-d-赤型-己-3-烯呋喃糖iv(93.6kg,72%th)作为在甲醇中的溶液进行至下一阶段。
[0078]
3-o-乙酰基-1,2:5,6-二-o-异亚丙基-α-d-呋喃葡萄糖,v
[0079][0080]
在20℃下,向含有10%pd/c(50%湿,142g,0.067mol)的容器中加入3-o-乙酰基-1,2:5,6-二-o-异亚丙基-α-d-赤型-己-3-烯呋喃糖iv(1.0kg,3.33mol)在甲醇(7.5l)中的溶液。使用夹套容器逐滴添加三乙基硅烷(1.60l,9.99mol)在甲醇(5.25l)中的溶液,维持温度低于35℃。将混合物在20℃下搅拌10分钟,然后通过短垫过滤,用甲醇(1.5l)洗涤以得到3-o-乙酰基-1,2:5,6-二-o-异亚丙基-α-d-呋喃葡萄糖的甲醇溶液,将其直接用于生产1,2:5,6-二-o-异亚丙基-α-d-呋喃葡萄糖vi。
[0081]
1,2:5,6-二-o-异亚丙基-α-d-呋喃葡萄糖,vi
[0082][0083]
向在室温下的3-o-乙酰基-1,2:5,6-二-o-异亚丙基-α-d-呋喃葡萄糖v(3.33mol)的甲醇溶液中,加入25wt%甲醇钠的甲醇溶液至ph 11。将反应混合物在室温下搅拌1小时,然后添加固体二氧化碳直到ph≤7.5。使用旋转蒸发器将混合物在40℃下在真空中浓缩,并且将粗残余物悬浮在乙酸乙酯(3.9l)中并且加热至60℃。允许混合物冷却至≤40℃,然后过滤,用乙酸乙酯(1.5l)洗涤。将混合物在40℃下在真空中浓缩至2.5l的体积,并且加热至60℃。添加庚烷(4.65l),并且将混合物从60℃冷却至5℃并且搅拌30分钟。通过过滤分离产
物,用庚烷(3x0.5l)洗涤。将滤饼在40℃下在真空下干燥以得到0.60kg(47%,从ii经4个步骤)呈灰白色固体的1,2:5,6-二-o-异亚丙基-α-d-呋喃葡萄糖。1h nmr(400mhz,cdcl3)δ5.78(d,j=4.1hz,1h),4.66(dd,j=6.3,4.1hz,1h),4.52-4.43(m,1h),4.26-4.19(m,2h),3.90(dd,j=8.6,5.7hz,1h),3.72(dd,j=8.6,7.2hz,1h),2.64(d,j=6.4hz,1h),1.63(s,3h),1.45(s,3h),1.43(s,3h),1.38(s,3h)。
[0084]
可替代程序
[0085][0086]
向3-o-乙酰基-1,2:5,6-二-o-异亚丙基-α-d-赤型-己-3-烯呋喃糖iv(79.9kg,266mol)在甲醇(约874l)中的溶液中,加入amberlite ira-96树脂(11.2kg),所述树脂已用水和甲醇预洗涤过。将混合物在30℃下搅拌20分钟,并且确认ph≥6.0。将混合物过滤,将滤床用甲醇(28l)洗涤并且加入10%pd/c(2.8kg)。将反应置于5至6巴的氢气下在33℃下1h,然后过滤,将滤饼用甲醇(225l)洗涤。将滤液在至多45℃下在真空中蒸馏至约560l。向残余物中加入30%甲醇钠的甲醇溶液(4.5l)以将ph调节至≥11.0,并且将混合物在28℃下搅拌2.5小时。以5.6kg的份数添加固体二氧化碳以将ph调节至≤7.5,并且将反应混合物在至多45℃下在真空中蒸馏至约224l。添加乙酸乙酯(3x340l),并且在至多45℃下在真空中蒸馏至约224l。将乙酸乙酯(225l)添加到残余物中,并且确认甲醇水平≤2.0%。在44℃下将溶液用活性炭(28kg)处理20分钟,然后通过助滤剂过滤,并且将过滤器用乙酸乙酯(225l)洗涤。将合并的滤液在至多45℃下在真空中蒸馏至约120l,并且在45℃下添加庚烷(790l)。将混合物冷却至-2℃,在该温度下保持1小时,然后过滤,并且在0℃下将滤饼用庚烷洗涤。将1,2:5,6-二-o-异亚丙基-α-d-呋喃葡萄糖在真空中在至多40℃下干燥以递送50.9kg,45%,从ii经四个步骤。
[0087]
1,2:5,6-二-o-异亚丙基-3-o-三氟甲磺酸酯-α-d-呋喃葡萄糖,vii
[0088][0089]
向在室温下的1,2:5,6-二-o-异亚丙基-α-d-呋喃葡萄糖vi(0.66kg,2.54mol)在tbme(3.3l)中的溶液中,添加吡啶(493ml,6.10mol)。将悬浮液冷却至0℃。将三氟甲磺酸酐(513ml,3.05mol)逐滴添加到混合物中,维持温度低于15℃。将反应混合物在10℃下搅拌1小时,然后依次用2m hcl水溶液(1.32l)、5%nahco3溶液(2x1.32l)和10%nacl水溶液(1.32l)洗涤。在20℃+/-5℃下将dmf(4.95l)添加到有机相中,并且通过使用旋转蒸发器在40℃下在真空中浓缩除去tbme以得到1,2:5,6-二-o-异亚丙基-3-o-三氟甲磺酸酯-α-d-呋喃葡萄糖的dmf溶液,将其直接用于生产3-叠氮基-3-脱氧-1,2:5,6-二-o-异亚丙基-α-d-呋喃半乳糖viii。
[0090]
3-叠氮基-3-脱氧-1,2:5,6-二-o-异亚丙基-α-d-呋喃半乳糖,viii
[0091][0092]
将1,2:5,6-二-o-异亚丙基-3-o-三氟甲磺酸酯-α-d-呋喃葡萄糖vii(2.54mol)在dmf(4.95l)中的溶液冷却至0℃,并且分批添加nan3(330g,5.08mol)。将混合物在5℃下搅拌30分钟,然后温热至20℃并且搅拌16小时。逐滴添加水(3.6l),维持温度在20℃
±
5℃范围内,并且将混合物用tbme(2x1.92l)萃取。将合并的萃取物按顺序用5%nahco3水溶液(3.6l)和10%nacl水溶液(3.6l)洗涤。将有机相经mgso4干燥,并且在40℃下在真空中浓缩以得到655g(91%,从vi经两个步骤)3-叠氮基-3-脱氧-1,2:5,6-二-o-异亚丙基-α-d-呋喃半乳糖。1h nmr(400mhz,cdcl3)δ5.70(d,j=4.0hz,1h),4.50(dd,j=3.9,1.8hz,1h),4.25(td,j=6.7,5.7hz,1h),3.98(dd,j=8.4,6.7hz,1h),3.85(dd,j 5.6,1.8hz,1h),3.78(dd,j=8.4,6.7hz,1h),3.73(t,j=5.6hz,1h),1.48(s,3h),1.36(s,3h),1.29(s,3h),1.29(s,3h)。
[0093]
可替代程序
[0094][0095]
向1,2:5,6-二-o-异亚丙基-α-d-呋喃葡萄糖vi(45kg,173mol)在tbme(225l)中的溶液中添加吡啶(41.0kg,519mol),并且将溶液冷却至1℃。将三氟甲磺酸酐(73.1kg,259mol)缓慢添加到悬浮液中,维持0℃,然后添加tbme(45l)。将反应混合物温热至15℃,搅拌1.5小时,然后冷却至5℃,并且在≤10℃下添加水(225l)。将混合物温热至18℃,并且分离各层。将水相用tbme(180l)萃取,将合并的有机层用5%nahco3水溶液(135l)并且然后用5%nacl水溶液(135l)洗涤。通过在至多35℃下在真空中的蒸馏进行溶剂交换为dmf,并且将所得溶液(约270l)冷却至20℃。用三乙胺将ph调节至≥8.0,并且将溶液冷却至1℃,然后分5份经1小时添加叠氮化钠(22.5kg,346mol)。将温度升高至18℃,并且将混合物搅拌3小时。将反应冷却至5℃,并且在10℃下添加水(450l)。将混合物用tbme(2x180l)萃取,并且将合并的有机萃取物用5%nahco3水溶液(135l)洗涤,然后用水(135l)洗涤。分离有机层,并且通过在至多50℃下在真空中的蒸馏进行溶剂交换为1,4-二噁烷。将3-叠氮基-3-脱氧-1,2:5,6-二-o-异亚丙基-α-d-呋喃半乳糖viii(43kg,88%,从vi经两个步骤)进行到呈在1,4-二噁烷中的溶液(约90l总体积)的1,2,4,6-四-o-乙酰基-3-叠氮基-3-脱氧-β-d-吡喃半乳糖x的制造。
[0096]
可替代方法
[0097]
向1,2:5,6-二-o-异亚丙基-α-d-呋喃葡萄糖vi(200g,0.77mol)在tbme(540ml)中的溶液中添加吡啶(112ml,1.84mol),并且将溶液冷却至5℃。将三氟甲磺酸酐(262g,0.93mol)经2小时添加到悬浮液中,维持5℃,然后添加tbme(72ml)。将反应混合物温热至10
℃并且搅拌1小时,然后通过在5℃下添加亚硫酸氢钠溶液[由预混合的50%naoh水溶液(22.0g)、水(138ml)和95%硫酸(56g)制成]淬灭。添加水(140ml),并且将混合物温热至20℃。分离各层,并且在20℃下将有机相用水(140ml)、5%nahco3水溶液(140ml)、然后18%nacl水溶液(128ml)洗涤。分离各层,并且将三乙胺盐酸盐(80g,0.58mol)和四丁基溴化铵(tbab)(24.0g,0.16mol)添加到有机相中。在20℃下添加溶解在水(200ml)中的叠氮化钠(74g,1.14mol),然后将混合物加热至50℃。将反应保持20小时,然后冷却至20℃,并且添加水(400ml)。分离各相,并且将有机相用水(200ml)洗涤,然后用18%氯化钠水溶液(120ml)洗涤。通过在至多60℃下在真空中蒸馏进行溶剂交换为2-甲基四氢呋喃(2-methf)。将3-叠氮基-3-脱氧-1,2:5,6-二-o-异亚丙基-α-d-呋喃半乳糖viii(206g,94%,从vi经两个步骤)进行到呈在2-methf中的溶液(约840ml总体积)的1,2,4,6-四-o-乙酰基-3-叠氮基-3-脱氧-β-d-吡喃半乳糖x的制造。
[0098]
3-叠氮基-3-脱氧-d-吡喃半乳糖,ix
[0099][0100]
将溶解在1,4-二噁烷(2.62l)中的3-叠氮基-3-脱氧-1,2:5,6-二-o-异亚丙基-α-d-呋喃半乳糖viii(655g,2.30mol)、水(0.65l)和强酸性阳离子交换树脂(262g)的混合物在60℃下搅拌18小时。将混合物冷却至20℃并且过滤,将树脂用4:1 1,4-二噁烷:水(1.96l)洗涤。向在室温下的滤液溶液中加入强酸性阳离子/弱碱性阴离子混合床树脂(262g),并且将混合物在室温下搅拌1小时。将混合物过滤,将树脂用4:1 1,4-二噁烷:水(1.96l)洗涤。使用旋转蒸发器将滤液在40℃下在真空中浓缩,并且用乙腈(2x0.66l)共蒸发以得到468g(99%)呈白色固体的3-叠氮基-3-脱氧-d-吡喃半乳糖。1h nmr(400mhz,cdcl3)δ5.16(d,j=3.8hz,1h),4.03(t,j=6.2hz,1h),4.00-3.94(m,2h),3.70-3.66(m,2h),3.60(dd,j=10.7,3.0hz,1h)。
[0101]
1,2,4,6-四-o-乙酰基-3-叠氮基-3-脱氧-β-d-吡喃半乳糖,x
[0102][0103]
向3-叠氮基-3-脱氧-d-吡喃半乳糖ix(770g,3.75mol)在乙酸乙酯(2.31l)和三乙胺(6.27l,45.0mol)中的悬浮液中逐滴添加乙酸酐(3.54l,37.5mol)在乙酸乙酯(1.54l)中的溶液,维持温度≤35℃。将反应混合物在30℃下搅拌18小时,然后冷却至0℃。将混合物依次用2m hcl水溶液(5.0l)、水(1.54l)、5%nahco3水溶液(1.54l)和10%nacl水溶液(1.54l)洗涤,维持温度≤25℃。将有机相在真空中浓缩,并且在室温下将残余物悬浮在乙酸乙酯(2.0l)中。将悬浮液温热至60℃直至材料完全溶解。将混合物冷却至20℃并且加入庚烷(2.89l)。将混合物冷却至0℃,搅拌1小时,然后过滤,将固体用4:1庚烷:乙酸乙酯(0.77l)洗涤。将固体在50℃下在真空下干燥以得到882g(63%)呈白色固体的1,2,4,6-四-o-乙酰基-3-叠氮基-3-脱氧-β-d-吡喃半乳糖。1h nmr(400mhz,cdcl3)δ5.70(d,j=8.3hz,1h),5.48(dd,j=3.4,0.9hz,1h),5.30(dd,j=10.6,8.3hz,1h),4.23-3.99(m,3h),3.69
叠氮基-3-脱氧-β-d-吡喃半乳糖(167g,62%,从viii经2个步骤)。
[0109]
可替代程序
[0110][0111]
在25℃下,向1,2:5,6-二-o-异亚丙基-α-d-呋喃葡萄糖vi(35kg,134mol)在tbme(130l)中的溶液中添加吡啶(31.9kg,403mol)。将悬浮液冷却至0℃,并且在≤20℃下将三氟甲磺酸酐(61.6kg,218mol)经1.5小时添加到混合物中。将反应混合物在≤20℃下搅拌15分钟,然后温热至25℃并且搅拌6小时。将混合物冷却至15℃,然后用1.5m hcl水溶液将ph调节至3.0。将混合物过滤,将滤饼用tbme(70kg)洗涤,并且分离各相。在25℃下,将有机相用5%nahco3水溶液(73kg)和10%nacl水溶液(77kg)洗涤。分离各层,并且将dmf(35l)添加到有机相中。通过在至多35℃下在真空中蒸馏除去tbme,并且将另外的dmf(133kg)添加到残余物中。继续蒸馏以得到1,2:5,6-二-o-异亚丙基-3-o-三氟甲磺酸酯-α-d-呋喃葡萄糖(vii)在dmf(约250l)中的溶液。添加三乙胺(0.25kg),并且将溶液冷却至5℃,然后经1小时分批添加nan3(17.5kg,269mol)。将混合物在5℃下搅拌30分钟,温热至25℃并且搅拌1小时。将反应冷却至10℃并且缓慢添加水(263kg),维持温度在10℃。在20℃下,将混合物用tbme萃取两次(130kg,然后106kg)。将合并的有机萃取物按顺序用5%nahco3水溶液(126kg)和10%nacl水溶液(2x116kg)洗涤。将有机相在至多50℃下在真空中浓缩至约100l。添加1,4-二噁烷(74kg)并且继续蒸馏两次至约100l。将溶液冷却至25℃,然后添加预洗涤的amberlite ir-120树脂(14kg)和水(35kg)。将混合物加热至65℃并且在65℃下搅拌24小时。将混合物冷却至25℃,过滤,并且将滤饼用4:1w/w 1,4-二噁烷:水(105kg)洗涤。将预洗涤的amberlite ira-96树脂(15.8kg)加入合并的滤液中,并且将混合物在25℃下搅拌3小时。加入三乙胺(2.5kg)以将ph调节至6.5至7.5,并且将混合物过滤,将树脂用4:1 1,4-二噁烷:水(105kg)洗涤。通过在至多65℃下在真空中浓缩将合并的滤液干燥至约120l,然后添加和蒸馏1,4-二噁烷(3x84kg)。将残余溶液冷却至25℃,并且添加三乙胺(151kg,1495mol)。在25℃下,将乙酸酐(126kg,1234mol)经4小时添加到溶液中。将反应混合物在30℃下搅拌24小时,冷却至20℃,然后添加水(228kg)和2-甲基四氢呋喃(14.7kg)。在5℃下,将反应混合物的ph用1.5m hcl水溶液(105kg)调节至1至2。将混合物在5℃下搅拌2小时,然后过滤,并且将滤饼用水(2x175kg)洗涤。将滤饼用磷酸氢二钾水溶液(20%w/w,175kg)在25℃下浆化30分钟,然后过滤,并且将固体用水(174kg)洗涤。将固体用正庚烷(47.6kg)洗涤,然后在35℃下干燥。在25℃下,将粗材料进一步在水(255kg)中浆化2小时两次,然后在35℃下干燥以得到呈白色固体的1,2,4,6-四-o-乙酰基-3-叠氮基-3-脱氧-β-d-吡喃半乳糖(24.4kg,49%,从vi经4个步骤)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1