苯并吡喃酮化合物的制造方法及用于其的新型的中间体与流程
1.本发明涉及一种由下述化学式1表示的2-(2,5-二氟苯基)-3-羧基-7-氯-(4h)-4-苯并吡喃酮的制造方法及用于其的新型的中间体。
2.[化学式1]
[0003][0004]
背景技术:
[0005]
tnf为一种细胞因子,正常的tnf对炎症反应起着重要的作用,但过量产生的tnf会刺激巨噬细胞,诱发细胞过度的炎症反应,导致类风湿性关节炎、炎症性肠疾病、银屑病、强直性脊柱炎等各种tnf相关疾病。作为用于治疗tnf相关疾病的tnf抑制剂,有依那西普、阿达木单抗、英夫利西单抗等。这些是生物医药,具有价格高、需反复注射、会产生耐性、难以保管的缺点。
[0006]
在韩国登记专利第10-1934651号中,作为直接结合于tnf而抑制tnf活性的新型的化合物,公开有具有下述化学式1的结构的化合物即2-(2,5-二氟苯基)-3-羧基-7-氯-(4h)-4-苯并吡喃酮(以下,称为“化学式1的化合物”),其具有优异的tnf的细胞毒性抑制效果及tnf的细胞结合抑制效果,抑制由tnf引起的细胞信号传导来对脓毒症、类风湿性关节炎、炎症性肠疾病、脓毒症-诱导急性肾损伤等显示出治疗效果,显示出可与市售中的阿达木单抗及依那西普匹敌的效果。
[0007]
[化学式1]
[0008][0009]
在韩国登记专利第10-1934651号中公开有如下所示的制造方法,当r3、r4及r6为氢、r5为cl时,根据该制造方法,能够经过共3个阶段制造出所述化学式1的化合物。
[0010][0011]
但是,为了合成上述公开的制造方法的起始物质,需要几个阶段的追加工序,具体而言,能够由4-氯-2-羟基苯甲酸经过如下所示的4个阶段的追加工序来合成。
[0012][0013]
即,为了将4-氯-2-羟基苯甲酸作为起始物质制造出所述化学式1的化合物,需要经过共7个阶段的工序。
[0014]
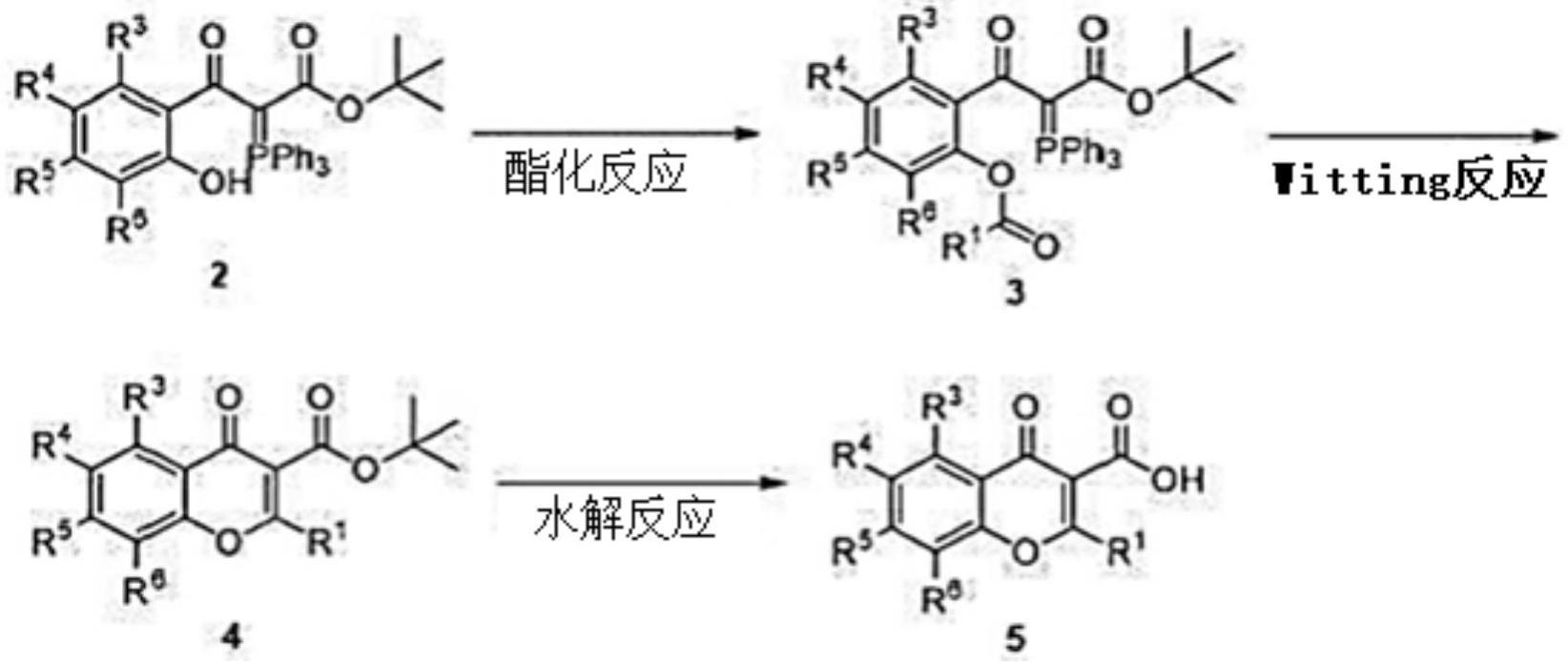
在所述制造方法中,为了合成所述化学式1的化合物,首先,对4-氯-2-羟基苯甲酸导入正膦(phosphorane)基,接着,导入2,5-二氟苯甲酰氧基之后,通过wittig反应合成4-苯并吡喃酮化合物,根据该制造方法,将导入正膦基的反应和wittig反应分为独立的步骤,因此反应步骤增加,为了首先导入正膦基,需要进一步伴有羟基的上保护反应及脱保护反应,因此反应步骤增加。并且,所述制造方法的收率小于15%,非常低,因此存在不适合于大量合成的问题点。
[0015]
因此,为了在商业上生产所述化学式1的化合物,需要更高效且经济的制造方法。
[0016]
为此,本发明人等开发出能够以高收率大量合成,更高效且经济的新型的制造方法来完成了本发明。
[0017]
现有技术文献
[0018]
专利文献
[0019]
专利文献1:韩国登记专利第10-1934651号
技术实现要素:
[0020]
发明要解决的技术课题
[0021]
本发明提供一种能够高效且经济地合成下述化学式1的化合物且能够以高收率大量制造的下述化学式1的化合物的新型的制造方法。
[0022]
[化学式1]
[0023][0024]
并且,本发明提供一种作为用于化学式1的化合物的制造方法的新型的中间体的下述化学式2及3的化合物。
[0025]
[化学式2]
[0026][0027]
[化学式3]
[0028][0029]
并且,本发明提供一种化学式2及3的化合物的制造方法。
[0030]
用于解决技术课题的手段
[0031]
本发明提供一种新型的下述化学式2及3的化合物。
[0032]
[化学式2]
[0033][0034]
[化学式3]
[0035][0036]
本发明提供一种所述化学式2的化合物的制造方法。
[0037]
在一实施方式中,能够使4-氯-2-羟基苯甲酸与2,5-二氟苯甲酰氯进行反应而制造所述化学式2的化合物。所述反应可以在-20至0
°
下,在选自由四氢呋喃(thf)、醚、甲基叔丁基醚(mtbe)、二氯甲烷(dcm)、乙腈(acn)、乙醚及二恶英组成的组中的一种以上的反应溶剂中,在选自由吡啶、三乙胺(tea)、二异丙基乙胺(dipea)、二甲氨基吡啶(dmap)、氢氧化钾、碳酸钾、乙酸钠、二甲基吡啶、碳酸钠、碳酸氢钠、碳酸氢钾及碳酸铯组成的组中的一种以上的碱的存在下进行,但并不限于此。
[0038]
本发明提供一种所述化学式3的化合物的制造方法。
[0039]
在一实施方式中,能够使所述化学式2的化合物与草酰氯进行反应而制造所述化学式3的化合物。所述反应可以在常温下,在选自由二氯甲烷(dcm)及二甲基甲酰胺(dmf)组成的组中的一种以上的反应溶剂中进行,但并不限于此。
[0040]
本发明提供一种下述化学式1的化合物的制造方法。
[0041]
[化学式1]
[0042][0043]
所述化学式1的化合物能够通过包括以下步骤的制造方法制造:使所述化学式3的化合物与下述化学式4的化合物进行反应而获得下述化学式5的化合物的步骤;及对下述化学式5的化合物进行酸处理而获得所述化学式1的化合物的步骤。
[0044]
[化学式4]
[0045][0046]
[化学式5]
[0047]
[0048]
并且,将使所述化学式3的化合物与所述化学式4的化合物进行反应之后获得的反应混合物调节为ph7以下,能够减少杂质的生成。获得所述化学式5的化合物的步骤可以在选自由甲苯、四氢呋喃(thf)及二氯甲烷(dcm)组成的组中的一种以上的反应溶剂中,在选自由n,o-双(三甲基甲硅烷基)乙酰胺、三乙胺(tea)及二异丙基乙胺(dipea)组成的组中的一种以上的碱的存在下进行,但并不限于此。获得所述化学式1的化合物的步骤可以在选自由乙酸乙酯(ea)、甲醇、异丙醇(i-proh)及甲基叔丁基醚(mtbe)组成的组中的一种以上的反应溶剂中进行,所述酸可以为盐酸或三氟乙酸,但并不限于此。
[0049]
在另一实施方式中,所述化学式1的化合物能够通过包括以下步骤的制造方法制造:使所述化学式2的化合物与草酰氯进行反应而获得所述化学式3的化合物的步骤;使所述化学式3的化合物与所述化学式4的化合物进行反应而获得所述化学式5的化合物的步骤;及对所述化学式5的化合物进行酸处理而获得所述化学式1的化合物的步骤。
[0050]
获得所述化学式3的化合物的步骤可以在选自由二氯甲烷(dcm)及二甲基甲酰胺(dmf)组成的组中的一种以上的反应溶剂中进行,但并不限于此。
[0051]
在此,通过使所述化学式2的化合物与草酰氯的反应之后制造出的所述化学式3的化合物与甲醇进行反应来确认是否制造出下述化学式6的化合物,由此能够确认所述化学式3的化合物的制造与否及收率。能够通过以下步骤来进行:通过确认下述化学式6的化合物的获得与否来确认难以分离的所述化学式3的化合物的获得与否之后,使所述化学式3的化合物与所述化学式4的化合物进行反应而获得所述化学式5的化合物。
[0052]
[化学式6]
[0053][0054]
在又一实施方式中,所述化学式1的化合物能够通过包括以下步骤的制造方法制造:使4-氯-2-羟基苯甲酸与2,5-二氟苯甲酰氯进行反应而获得所述化学式2的化合物的步骤;使所述化学式2的化合物与草酰氯进行反应而获得所述化学式3的化合物的步骤;使所述化学式3的化合物与所述化学式4的化合物进行反应而获得所述化学式5的化合物的步骤;及对所述化学式5的化合物进行酸处理而获得所述化学式1的化合物的步骤。
[0055]
获得所述化学式2的化合物的步骤可以在-20至0℃下,在选自由四氢呋喃(thf)、醚、甲基叔丁基醚(mtbe)、二氯甲烷(dcm)、乙腈(acn)、乙醚及二恶英组成的组中的一种以上的反应溶剂中,在选自由吡啶、三乙胺(tea)、二异丙基乙胺(dipea)、二甲氨基吡啶(dmap)、氢氧化钾、碳酸钾、乙酸钠、二甲基吡啶、碳酸钠、碳酸氢钠、碳酸氢钾及碳酸铯组成的组中的一种以上的碱的存在下进行,但并不限于此。
[0056]
在一实施方式中,所述化学式1的化合物能够利用下述反应式1制造。
[0057]
[反应式1]
[0058][0059]
本发明提供一种所述化学式1的化合物的iv型结晶的制造方法。
[0060]
在一实施方式中,能够将所述化学式1的化合物添加到甲基异丁基酮(mibk)中而制造化学式1的化合物的iv型结晶。将所述化学式1的化合物添加到所述甲基异丁基酮(mibk)中的温度可以为15至25℃。
[0061]
在一实施方式中,能够通过基于本发明的所述化学式1的化合物的制造方法制造所述化学式1的化合物之后,将所述化学式1的化合物添加到甲基异丁基酮(mibk)中,来制造化学式1的化合物的iv型结晶。
[0062]
在一实施方式中,能够包括以下步骤来制造化学式1的化合物的iv型结晶:使4-氯-2-羟基苯甲酸与2,5-二氟苯甲酰氯进行反应而获得所述化学式2的化合物的步骤;使所述化学式2的化合物与草酰氯进行反应而获得所述化学式3的化合物的步骤;使所述化学式3的化合物与所述化学式4的化合物进行反应而获得所述化学式5的化合物的步骤;对所述化学式5的化合物进行酸处理而获得所述化学式1的化合物的步骤;及将所述化学式1的化合物添加到甲基异丁基酮(mibk)中而获得iv型结晶的化学式1的化合物的步骤。
[0063]
所述化学式1的化合物的iv型结晶可以在选自由11.3、14.7、14.9、16.9、17.2、22.5及25.7组成的组中的4个以上的衍射角2θ
±
0.2
°
处显示出包含特征峰的x-射线粉末衍射(xrpd)光谱。所述iv型结晶可以在10℃/min的升温条件的差示扫描量热仪(dsc)分析时在约160℃至约170℃的范围内具有吸热峰,或者,热重(tga)曲线在160℃以下时无重量损失。
[0064]
本发明提供一种通过基于本发明的所述化学式1的化合物的制造方法制造的所述化学式1的化合物。
[0065]
本发明提供一种tnf过表达疾病的治疗或预防用组合物,其包含通过基于本发明的所述化学式1的化合物的制造方法制造的所述化学式1的化合物,该tnf过表达疾病选自由类风湿性关节炎、幼年类风湿性关节炎、炎症性肠疾病、克罗恩病、溃疡性结肠炎、银屑病、斑块状银屑病、幼年斑块状银屑病、银屑病性关节炎、多关节型幼年特发性关节炎、肠白塞病、强直性脊柱炎、中轴型脊柱炎、幼年附着点炎相关关节炎、骨关节炎、风湿性多肌痛、多发性硬化症、全身性红斑狼疮、气喘、干燥综合征、肺炎、慢性阻塞性肺病、肉样瘤病、环状肉芽肿、韦格纳肉芽肿、动脉硬化症、脉管炎、心力衰竭、心肌梗塞、肾损伤、肾炎、移植物抗宿主病、痴呆、阿尔茨海默病、帕金森病、疼痛、葡萄膜炎、白塞病、化脓性汗腺炎、毛发红糠疹、糖尿病性类脂质渐进性坏死、坏疽性脓皮病、斯维特综合征、角层下脓疱性皮肤病、硬皮
病、皮肌炎、脓毒症及脓毒性休克组成的组。
[0066]
发明效果
[0067]
在本发明的制造方法中,为了合成所述化学式1的化合物,先进行对4-氯-2-羟基苯甲酸导入2,5-二氟苯甲酰氧基的步骤之后,在使由化学式4表示的2-(三苯基亚正膦基)乙酸叔丁酯(tert-butyl 2-(triphenylphosphoranylidene)acetate)进行反应的步骤中,使正膦中间体化合物的合成及通过wittig反应的4-苯并吡喃酮化合物的合成成为一个步骤,从而与以往的制造方法相比,减少了制造步骤。
[0068]
并且,在本发明中,通过分离出作为反应中间体化合物的所述化学式3的化合物,使得能够确认中间体化合物的收率及反应的进行与否,由此提高了所述化学式1的化合物的收率。
[0069]
并且,在本发明中,通过调节使所述化学式3的化合物与所述化学式4的化合物进行反应之后获得的反应混合物的ph来减少杂质的生成,从而进一步提高了收率。
[0070]
因此,在本发明中,省略以往的制造方法中不必要的工序,降低反应速度,经济且高效率地合成所述化学式1的化合物,并且,通过中间体的分离及ph调节,使得能够以更高的收率获得所述化学式1的化合物。
[0071]
由此,能够通过本发明的所述化学式1的化合物的制造方法在商业上大量生产所述化学式1的化合物。
[0072]
具体实施方式
[0073]
以下,通过实施例对本发明进行更详细的说明,但下述实施例仅用于说明的目的,并不旨在限定本技术发明的范围。
[0074][0075]
[实施例1]4-氯-2-((2,5-二氟苯甲酰基)氧基)苯甲酸的制造
[0076]
[反应式2]
[0077][0078]
将四氢呋喃(thf)(15l)、4-氯-2-羟基苯甲酸(3.9kg)、吡啶(1.79kg)在15~30℃下放入反应器中,一边搅拌一边冷却至-20~0℃。在另外的装置中制造四氢呋喃(thf)(45l)、2,5-二氟苯甲酰氯(3.99kg)溶液之后,缓慢地滴加到所述混合物中。完成反应之后,在-10~0℃下,向反应物中添加了乙酸乙酯(ea)。添加蒸馏水结束反应之后,将反应温度上升至常温。加入乙酸乙酯(ea)(20l)之后,用水(20l)清洗后,去除了水层。用1m的hcl(9l)清洗了有机层。用水(9l)清洗有机层,直至ph成为4~5。将有机层减压浓缩之后,溶解于二甲基甲酰胺(dmf)(18l)中,加入水(72l)之后,在常温下搅拌了6小时。过滤生成的固体之后,
用水(9l)清洗两次,将固体在40~50℃下真空干燥17小时,获得了4-氯-2-((2,5-二氟苯甲酰基)氧基)苯甲酸6.4kg(纯度:99%,收率:89.5%)。
[0079]
在上述反应中,代替吡啶而使用二异丙基乙胺(dipea)、氢氧化钾(koh)、碳酸钾(k2co3)或乙酸钠(ch3coona)时,获得了73.1%、52.4%、39.0%或55.4%纯度的4-氯-2-((2,5-二氟苯甲酰基)氧基)苯甲酸。
[0080]
代替四氢呋喃(thf)而在醚、甲基叔丁基醚(mtbe)或二氯甲烷(dcm)中进行上述反应时,获得了77.1%、54.6%或47.4%纯度的4-氯-2-((2,5-二氟苯甲酰基)氧基)苯甲酸。
[0081]
当使用二异丙基乙胺(dipea)在乙腈(acn)中进行上述反应时,获得了61.1%纯度的4-氯-2-((2,5-二氟苯甲酰基)氧基)苯甲酸,当使用三乙胺(tea)在二氯甲烷(dcm)中进行上述反应时,获得了37.5%纯度的4-氯-2-((2,5-二氟苯甲酰基)氧基)苯甲酸,当使用三乙胺(tea)在甲基叔丁基醚(mtbe)中进行上述反应时,获得了58.3%纯度的4-氯-2-((2,5-二氟苯甲酰基)氧基)苯甲酸,当同时使用三乙胺(tea)及二甲氨基吡啶(dmap)在二氯甲烷(dcm)中进行上述反应时,获得了41.8%纯度的4-氯-2-((2,5-二氟苯甲酰基)氧基)苯甲酸。
[0082][0083]
[实施例2]5-氯-2-(氯羰基)苯基2,5-二氟苯甲酸酯(5-chloro-2-(chlorocarbonyl)phenyl 2,5-difluorobenzoate)的制造
[0084]
[反应式3]
[0085][0086]
将二氯甲烷(dcm)(35l)及4-氯-2-((2,5-二氟苯甲酰基)氧基)苯甲酸(6.2kg)加入反应器中之后,将二甲基甲酰胺(dmf)(10ml)加入反应器中。在常温下一边搅拌一边缓慢地滴加草酰氯(5.0kg)之后,进一步搅拌了3小时。将所述混合物减压浓缩,加入正庚烷(35l),在40℃以下搅拌了2小时。浓缩进行至正庚烷成为约50~60l之后,冷却至反应液的温度成为常温,进而,又搅拌了1~2小时。过滤生成的固体,用正庚烷(9l)清洗之后,在30~40℃下真空干燥30小时,获得了5-氯-2-(氯羰基)苯基2,5-二氟苯甲酸酯5.84kg(纯度:100%,收率:88.2%)。
[0087][0088]
[实施例3]7-氯-2-(2,5-二氟苯基)-4-氧代-4h-色烯-3-羧酸叔丁酯的制造
[0089]
[反应式4]
[0090][0091]
步骤1:
[0092]
将甲苯(36l)、2-(三苯基亚正膦基)乙酸叔丁酯(7.78kg)及n,o-双(三甲基甲硅烷基)乙酰胺(7.01kg)在常温下加入反应器中。将反应液一边搅拌一边冷却至-5~5℃之后,将5-氯-2-(氯羰基)苯基2,5-二氟苯甲酸酯(5.7kg)缓慢地添加到反应器中之后,搅拌了22小时。使用5%碳酸氢钠水溶液结束反应之后,添加二氯甲烷(dcm),并将反应液的温度上升至25~35℃。用水(10l)清洗所述反应混合物,直至水相的ph成为7以下。
[0093]
步骤2:
[0094]
对有机层在95~105℃下进行回流搅拌。然后,将所述反应混合物冷却至50~60℃,进行浓缩之后,将甲苯残留物在异丙醇(ipa)(15l)中浆液(sulyrry)化,直至残留物成为1.0%以下。将所述混合物的温度调整为17~23℃之后,为了结晶化而在17~23℃下搅拌了所述混合物,直至母液中的7-氯-2-(2,5-二氟苯基)-4-氧代-4h-色烯-3-羧酸叔丁酯的重量成为2.0wt%以下。过滤悬浮液,用异丙醇(3l)清洗固体之后,将固体在30~40℃下真空干燥了17小时,直至异丙醇(ipa)及甲苯的总含量成为1.0wt%以下。然后,进行冷却,使温度成为20~30℃,获得了7-氯-2-(2,5-二氟苯基)-4-氧代-4h-色烯-3-羧酸叔丁酯4.46kg(纯度:99.8%,收率:65.4%)。
[0095]
在上述反应中,代替n,o-双(三甲基甲硅烷基)乙酰胺而使用三乙胺(tea)或二异丙基乙胺(dipea)时,获得了76.0%或70.0%纯度的7-氯-2-(2,5-二氟苯基)-4-氧代-4h-色烯-3-羧酸叔丁酯。
[0096]
代替甲苯而在四氢呋喃(thf)或二氯甲烷(dcm)中进行上述反应时,获得了56.1%或21.7%纯度的7-氯-2-(2,5-二氟苯基)-4-氧代-4h-色烯-3-羧酸叔丁酯。
[0097][0098]
[实施例4]2-(2,5-二氟苯基)-3-羧基-7-氯-(4h)-4-苯并吡喃酮的制造
[0099]
[反应式5]
[0100][0101]
将乙酸乙酯(ea)(12.6l)及7-氯-2-(2,5-二氟苯基)-4-氧代-4h-色烯-3-羧酸叔丁酯(4.2kg)加入反应器中之后,在45~55℃下使其完全溶解之后,冷却至15~25℃。向反应液中缓慢地滴加4m的hcl/乙酸乙酯(ea)(21.0l)溶液之后,搅拌了42小时。分别用正庚烷(22.5l)及水(22.5l)清洗了生成的固体。在40~50℃下真空干燥了27小时。
[0102]
将甲基异丁基酮(mibk)8.2kg加入反应器中,并添加了2-(2,5-二氟苯基)-3-羧
基-7-氯-(4h)-4-苯并吡喃酮。将所述混合物在15~25℃下搅拌了23小时,直至2-(2,5-二氟苯基)-3-羧基-7-氯-(4h)-4-苯并吡喃酮的晶型成为iv型。过滤固体,用甲基叔丁基醚(mtbe)清洗之后,在30~40℃下真空干燥26小时,获得了2-(2,5-二氟苯基)-3-羧基-7-氯-(4h)-4-苯并吡喃酮2.75kg(纯度:99.9%,收率:75.4%)。
[0103]
代替乙酸乙酯(ea)而在甲醇、异丙醇(i-proh)或甲基叔丁基醚(mtbe)中进行上述反应时,获得了52.2%、28.3%或85.8%纯度的2-(2,5-二氟苯基)-3-羧基-7-氯-(4h)-4-苯并吡喃酮。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1