热固性树脂组合物及其固化物的制作方法
1.本发明涉及低介电特性、高耐热性、高粘接性等优异且提供固化物的以热固性树脂为必需成分的热固性树脂组合物、以及由该热固性树脂组合物得到的固化物、密封材料、电路基板用材料、预浸料或层叠板。
背景技术:
2.环氧树脂、酚醛树脂等热固性树脂的粘接性、挠性、耐热性、耐化学品性、绝缘性、固化反应性优异,因此在涂料、土木粘接、浇铸成型、电气电子材料、膜材料等多方面使用。特别是在作为电气电子材料之一的印刷电路板用途中,通过对环氧树脂赋予阻燃性而得到广泛使用。
3.作为印刷电路板的用途之一的便携式设备、连接其的基站等基础设施设备随着近年来信息量的急剧增大,不断要求高功能化。特别是通过通信标准从4g变为5g,信息量进一步增加,预想需要利用高频的信号传输。因此,在印刷电路板中,为了抑制由高频引起的信号的衰减,需要介质损耗角正切更低的材料。另外,为了应对印刷电路板的细线化、高多层化,对基体树脂要求高粘接力和高耐热等特性。为了满足这些要求,使用以往的环氧树脂的基体树脂并不充分,需要功能更高的热固性树脂。
4.关于印刷电路板的基体树脂中使用的环氧树脂的低介电常数化,作为原料环氧树脂,有如下例示:对双酚a等二元酚类进行缩水甘油基化而得的化合物、对三(缩水甘油氧基苯基)烷烃类、氨基苯酚等进行缩水甘油基化而得的化合物等、对苯酚酚醛清漆等酚醛清漆类进行缩水甘油基化而得的化合物(专利文献1)。
5.在专利文献2、3中公开了为了与环氧树脂相比进一步改善耐热性、机械特性而使用含酰亚胺基的酚醛树脂的方法,通过含有酰亚胺基而改善了耐热性。另外,作为适合于改善与基材的粘接性的基体树脂的树脂,例示有对含酰亚胺基的酚醛树脂进行环氧化而得的化合物(专利文献4)。另外,在专利文献5中例示了通过使用马来酰亚胺化合物、环氧树脂和特定结构的酚固化剂而可提供改善了基板的耐热性、阻燃性的组合物,在专利文献6和专利文献7中例示了通过使用具有特定的结构的马来酰亚胺化合物而可提供粘接力、介电特性优异的组合物。
6.但是,任一文献中公开的环氧树脂都不充分满足基于近年来的高功能化的介电特性的要求,不同时满足各物性。
7.现有技术文献
8.专利文献
9.专利文献1:日本特开平5-43655号公报
10.专利文献2:日本特开平7-33858号公报
11.专利文献3:日本特开平7-10970号公报
12.专利文献4:日本特开2010-235823号公报
13.专利文献5:国际公开第2011/126070号
14.专利文献6:国际公开第2016/208667号
15.专利文献7:国际公开第2020/054526号
技术实现要素:
16.因此,本发明要解决的课题在于提供一种具有同时满足低介电性、高耐热性、高粘接性的优异的性能并在层叠、成型、粘接等用途中有用的树脂组合物及其固化物。
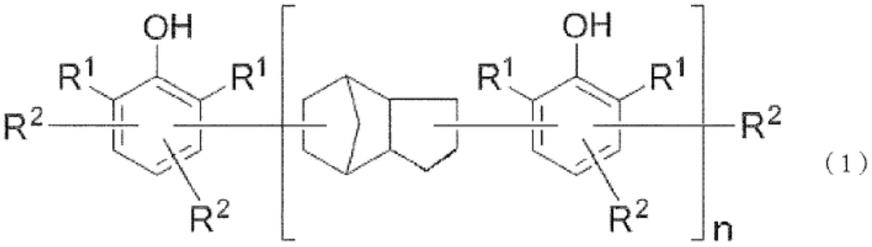
17.为了解决上述课题,本发明人进行了深入研究,结果发现含有由下式(1)表示的芳香族多羟基化合物和马来酰亚胺化合物的热固性树脂组合物同时满足前所未有的低介电特性、高玻璃化转变温度(tg)和良好的粘接强度,完成了本发明。
18.即,本发明是一种热固性树脂组合物,其特征在于,含有由下述通式(1)表示的芳香族多羟基化合物和马来酰亚胺化合物。
[0019][0020]
这里,
[0021]
r1独立地表示碳原子数1~8的烃基,
[0022]
r2独立地表示氢原子、二环戊烯基,一个以上为二环戊烯基。
[0023]
n表示重复数,其平均值为1~5的数。
[0024]
上述热固性树脂组合物优选进一步含有环氧树脂。
[0025]
另外,本发明是将上述树脂组合物固化而成的固化物,是以使用上述树脂组合物为特征的电路基板用材料、密封材料、预浸料或层叠板。
[0026]
对于本发明的树脂组合物,其固化物维持良好的粘接力,并且可以得到玻璃化转变温度高的固化物。另外,介电特性也优异,在要求低介电常数、低介质损耗角正切的层叠板和电子电路基板中发挥良好的特性。
附图说明
[0027]
图1是合成例1中得到的芳香族多羟基化合物的gpc图。
[0028]
图2是合成例1中得到的芳香族多羟基化合物的ir图。
[0029]
图3是合成例4中得到的环氧树脂的gpc图。
具体实施方式
[0030]
以下,对本发明进行详细说明。
[0031]
本发明中使用的芳香族多羟基化合物(以下也称为酚醛树脂)由上述通式(1)表示。
[0032]
在通式(1)中,r1独立地表示碳原子数1~8的烃基,优选碳原子数1~8的烷基、碳
原子数6~8的芳基、碳原子数7~8的芳烷基或烯丙基。作为碳原子数1~8的烷基,可以是直链状、支链状、环状中的任一种,例如,可以举出甲基、乙基、丙基、异丙基、正丁基、叔丁基、己基、环己基、甲基环己基等,但不限定于它们。作为碳原子数6~8的芳基,可以举出苯基、甲苯基、二甲苯基、乙基苯基等,但不限定于它们。作为碳原子数7~8的芳烷基,可以举出苄基、α-甲基苄基等,但不限定于它们。在这些取代基中,从获得的容易性和制成固化物时的反应性的观点出发,优选苯基、碳原子数1~3的烷基,特别优选甲基。
[0033]
r2独立地表示氢原子、二环戊烯基,一个以上为二环戊烯基。优选一分子中的r2每一个苯酚环平均具有0.1~1个二环戊烯基。
[0034]
二环戊烯基是来自二环戊二烯的基团,由下式(1a)或式(1b)表示。
[0035][0036]
n为重复数,表示0或1以上的数,其平均值(数均)为1~5,优选为1.1~3,更优选为1.5~2.5,进一步优选为1.6~2。作为基于gpc所得的含量,优选为n=0体处于10面积%以下的范围,n=1体处于50~70面积%的范围,n=2体以上处于20~40面积%的范围。
[0037]
酚醛树脂的分子量优选为重均分子量(mw)为400~1000、数均分子量(mn)为350~800的范围。
[0038]
对于酚醛树脂,羟基当量优选为230以上,更优选为240以上,软化点优选为120℃以下,更优选为110℃以下。
[0039]
上述酚醛树脂例如可以通过使由下述通式(2)表示的2,6-二取代酚类与二环戊二烯在三氟化硼
·
醚催化剂等路易斯酸存在下反应而得到。
[0040][0041]
这里,
[0042]
r1与上述通式(1)中的定义为相同含义。
[0043]
作为上述2,6-二取代酚类,可以举出2,6-二甲基苯酚、2,6-二乙基苯酚、2,6-二丙基苯酚、2,6-二异丙基苯酚、2,6-二(正丁基)苯酚、2,6-二(叔丁基)苯酚、2,6-二己基苯酚、2,6-二环己基苯酚、2,6-二苯基苯酚、2,6-二甲苯基苯酚、2,6-二苄基苯酚、2,6-双(α-甲基苄基)苯酚、2-乙基-6-甲基苯酚、2-烯丙基-6-甲基苯酚、2-甲苯基-6-苯基苯酚等,但是从获得的容易性和制成固化物时的反应性的观点出发,优选2,6-二苯基苯酚、2,6-二甲基苯酚,特别优选2,6-二甲基苯酚。
[0044]
上述反应中使用的催化剂为路易斯酸,具体而言为三氟化硼、三氟化硼
·
酚络合物、三氟化硼
·
醚络合物、氯化铝、氯化锡、氯化锌、氯化铁等,其中,从操作的容易性出发,优选三氟化硼
·
醚络合物。在三氟化硼
·
醚络合物的情况下,相对于二环戊二烯100质量份,催化剂的使用量为0.001~20质量份,优选为0.5~10质量份。
[0045]
作为用于在2,6-二取代类中导入上述二环戊烯基的反应方法,是使二环戊二烯
以规定的比率与2,6-二取代酚类反应的方法,可以连续添加或分阶段添加(分批逐次添加两次以上)二环戊二烯,间歇地进行反应。对于比率,相对于2,6-二取代酚类1摩尔,二环戊二烯为0.25~2倍摩尔。
[0046]
对于连续添加二环戊二烯使其反应时的比率,相对于2,6-二取代酚类,二环戊二烯为0.25~1倍摩尔,优选为0.28~1倍摩尔,更优选为0.3~0.5倍摩尔。在分批逐次添加二环戊二烯使其反应的情况下,整体优选为0.8~2倍摩尔,更优选为0.9~1.7倍摩尔。应予说明,各阶段中的二环戊二烯的使用比率优选为0.28~1倍摩尔。
[0047]
作为确认在由上述通式(1)表示的酚醛树脂中导入二环戊烯基的方法,可以使用质谱法和ft-ir测定。
[0048]
在使用质谱法的情况下,可以使用电喷雾质谱法(esi-ms)、场解吸法(fd-ms)等。通过对利用gpc等将核体数不同的成分分离而得的样品实施质谱法,可以确认导入了二环戊烯基。
[0049]
在使用ft-ir测定法的情况下,将溶解于thf等有机溶剂的样品涂布于krs-5池上,利用ft-ir测定使有机溶剂干燥而得到的带样品薄膜的池时,来自苯酚核中的c-o伸缩振动的峰出现在1210cm
-1
附近,仅在导入了二环戊烯基的情况下,来自二环戊二烯骨架的烯烃部位的c-h伸缩振动的峰出现在3040cm
-1
附近。以直线连接目标峰的起点和终点作为基线,将从峰的顶点到基线的长度设为峰高时,可以通过3040cm
-1
附近的峰(a
3040
)与1210cm
-1
附近的峰(a
1210
)的比率(a
3040
/a
1210
),来定量二环戊烯基的导入量。可以确认其比率越大物性值越好,用于满足目标物性的优选比率(a
3040
/a
1210
)为0.05以上,更优选为0.10以上,特别是0.10~0.30。
[0050]
本反应优选将2,6-二取代酚类和催化剂装入反应器中并用1~10小时滴加二环戊二烯的方式。
[0051]
反应温度优选为50~200℃,更优选为100~180℃,进一步优选为120~160℃。反应时间优选为1~10小时,更优选为3~10小时,进一步优选为4~8小时。
[0052]
反应结束后,加入氢氧化钠、氢氧化钾、氢氧化钙等碱而使催化剂失活。然后,加入甲苯、二甲苯等芳香族烃类、甲基乙基酮、甲基异丁基酮等酮类等溶剂进行溶解,水洗,然后在减压下回收溶剂,由此可以得到目标酚醛树脂。应予说明,优选使二环戊二烯尽可能全部反应,使2,6-二取代酚类的一部分未反应,优选为10%以下未反应,将其减压回收。
[0053]
反应时,根据需要可以使用苯、甲苯、二甲苯等芳香族烃类、氯苯、二氯苯等卤代烃类、乙二醇二甲醚、二乙二醇二甲醚等醚类等溶剂。
[0054]
通过使用这样的芳香族多羟基化合物,可以得到本发明的热固性树脂组合物。
[0055]
本发明的热固性树脂组合物中含有的双马来酰亚胺化合物没有特别限定,例如可以举出n-苯基马来酰亚胺、n-羟基苯基马来酰亚胺、4,4’-二苯甲烷双马来酰亚胺、聚苯甲烷马来酰亚胺、间苯撑双马来酰亚胺、对苯撑双马来酰亚胺、2,2’-[4-(4-马来酰亚胺苯氧基)苯基]丙烷、3,3’-二甲基-5,5’-二乙基-4,4’-二苯甲烷双马来酰亚胺、双(3,5-二甲基-4-马来酰亚胺苯基)甲烷、双-(3-乙基-5-甲基-4-马来酰亚胺苯基)甲烷、双(3,5-二乙基-4-马来酰亚胺苯基)甲烷、4-甲基-1,3-亚苯基双马来酰亚胺、4,4’-二苯基醚双马来酰亚胺、4,4’-二苯砜双马来酰亚胺、1,3-双(3-马来酰亚胺苯氧基)苯、1,3-双(4-马来酰亚胺苯氧基)苯、n,n’-亚乙基二马来酰亚胺、n,n’-六亚甲基
二马来酰亚胺、由下述通式(3)表示的马来酰亚胺化合物等、这些马来酰亚胺化合物的预聚物、或马来酰亚胺化合物与胺化合物的预聚物等。
[0056][0057]
这里,
[0058]
x为式(3a)、(3b)、(3c)中的任一个,
[0059]
r3独立地表示碳原子数1~5的烷基或芳香族基团。
[0060]
r4独立地表示氢原子或甲基。
[0061]
a表示0~4,优选为0或1。
[0062]
b表示0~3,优选为0或1。
[0063]
n为重复数,平均值为1~10,优选为1~5。
[0064]
本发明的热固性树脂组合物将马来酰亚胺化合物和酚醛树脂作为必需成分。酚醛树脂相对于树脂混合物中的马来酰亚胺化合物100质量份的含量优选为5~150质量份,更优选为10~130质量份,进一步优选为20~50质量份。作为用于得到本发明的热固性树脂组合物的酚醛树脂,除本发明的芳香族多羟基化合物以外,根据需要也可以并用一种或两种以上的各种酚醛树脂。优选酚醛树脂中至少30质量%为由上述通式(1)表示的芳香族多羟基化合物,更优选含有50质量%以上。在比其少的情况下,有介电特性恶化的风险。
[0065]
作为可以在本发明的热固性树脂组合物中使用的酚醛树脂系固化剂,具体例可以举出双酚a、双酚f、双酚c、双酚k、双酚z、双酚s、四甲基双酚a、四甲基双酚f、四甲基双酚s、四甲基双酚z、二羟基二苯基硫醚、4,4’-硫代双(3-甲基-6-叔丁基苯酚)等双酚类、儿茶酚、间苯二酚、甲基间苯二酚、对苯二酚、单甲基对苯二酚、二甲基对苯二酚、三甲基对苯
二酚、单叔丁基对苯二酚、二叔丁基对苯二酚等二羟基苯类、二羟基萘、二羟基甲基萘、二羟基甲基萘、三羟基萘等羟基萘类、lc-950pm60(shin-at&c公司制)等含磷酚固化剂、shonol brg-555(爱克工业株式会社制)等苯酚酚醛清漆树脂、dc-5(日铁化学&材料株式会社制)等甲酚酚醛清漆树脂、芳香族改性苯酚酚醛清漆树脂、双酚a酚醛清漆树脂、resitop tpm-100(群荣化学工业株式会社制)等三羟基苯甲烷型酚醛清漆树脂、萘酚酚醛清漆树脂等酚类、萘酚类和/或双酚类与醛类的缩合物、sn-160、sn-395、sn-485(日铁化学&材料株式会社制)等酚类、萘酚类和/或双酚类与二甲苯二醇的缩合物、酚类和/或萘酚类与异丙烯基苯乙酮的缩合物、酚类、萘酚类和/或双酚类与二环戊二烯的反应产物、酚类、萘酚类和/或双酚类与联苯系交联剂的缩合物等被称为所谓的酚醛清漆酚醛树脂的酚化合物等。从获得容易性的观点出发,优选苯酚酚醛清漆树脂、二环戊二烯型酚醛树脂、三羟基苯甲烷型酚醛清漆树脂、芳香族改性苯酚酚醛清漆树脂等。
[0066]
在酚醛清漆酚醛树脂的情况下,作为酚类,可以举出苯酚、甲酚、二甲酚、丁基苯酚、戊基苯酚、壬基苯酚、丁基甲基苯酚、三甲基苯酚、苯基苯酚等,作为萘酚类,可以举出1-萘酚、2-萘酚等,此外可以举出上述双酚类。作为醛类,可以例示甲醛、乙醛、丙醛、丁醛、戊醛、己醛、苯甲醛、氯醛、溴醛、乙二醛、丙二醛、丁二醛、戊二醛、己二醛、庚二醛、癸二醛、丙烯醛、巴豆醛、水杨醛、邻苯二甲醛、羟基苯甲醛等。作为联苯系交联剂,可以举出双(羟甲基)联苯、双(甲氧基甲基)联苯、双(乙氧基甲基)联苯、双(氯甲基)联苯等。
[0067]
本发明的热固性树脂组合物除马来酰亚胺化合物和酚醛树脂以外,还可以含有环氧树脂。环氧树脂的含量在热固性树脂组合物中优选为10~80质量%,更优选为20~70质量%。另外,相对于马来酰亚胺化合物100质量份,环氧树脂的含量优选为10~300质量份,更优选为20~280质量份。
[0068]
作为环氧树脂,分子中具有2个以上环氧基的通常的环氧树脂都可以使用。若举例,则可以举出双酚a型环氧树脂、双酚f型环氧树脂、四甲基双酚f型环氧树脂、联苯型环氧树脂、双酚芴型环氧树脂、双酚s型环氧树脂、双硫醚型环氧树脂、双萘基芴型环氧树脂、氢醌型环氧树脂、间苯二酚型环氧树脂、萘二醇型环氧树脂、苯酚酚醛清漆型环氧树脂、苯乙烯化苯酚酚醛清漆型环氧树脂、甲酚酚醛清漆型环氧树脂、烷基酚醛清漆型环氧树脂、双酚酚醛清漆型环氧树脂、萘酚酚醛清漆型环氧树脂、联苯芳烷基苯酚型环氧树脂、β-萘酚芳烷基型环氧树脂、二萘酚芳烷基型环氧树脂、α-萘酚芳烷基型环氧树脂、萘二醇芳烷基型环氧树脂、三苯甲烷型环氧树脂、二环戊二烯型环氧树脂、对由结构式(1)表示的芳香族多羟基化合物进行环氧化而得的二环戊二烯型环氧树脂、亚烷基二醇型环氧树脂、脂肪族环状环氧树脂、二氨基二苯甲烷四缩水甘油胺、氨基苯酚型环氧树脂、含磷环氧树脂、氨基甲酸酯改性环氧树脂、含唑烷酮环的环氧树脂,但是不限定于它们。另外,这些环氧树脂可以单独使用,也可以并用两种以上。从获得容易性的观点出发,进一步优选使用萘二醇型环氧树脂、苯酚酚醛清漆型环氧树脂、芳香族改性苯酚酚醛清漆型环氧树脂、甲酚酚醛清漆型环氧树脂、α-萘酚芳烷基型环氧树脂、二环戊二烯型环氧树脂、含磷环氧树脂、含唑烷酮环的环氧树脂。
[0069]
进而,本发明的树脂组合物中可以根据需要配合固化促进剂。如果使用固化促进剂,则能够与酰亚胺基进行交联反应的化合物、以及含羟基的酰亚胺化合物中包含的羟基与酰亚胺基发生加成反应而交联,因此固化物显示出良好的物性。
[0070]
如果举出固化促进剂的例子,则有胺类、咪唑类、有机膦类、路易斯酸等,具体而言,有1,8-二氮杂双环(5,4,0)十一烯-7、三乙二胺、苄基二甲胺、三乙醇胺、二甲基氨基乙醇、三(二甲基氨基甲基)苯酚等叔胺、2-甲基咪唑、2-苯基咪唑、2-乙基-4-甲基咪唑、2-苯基-4-甲基咪唑、2-十七烷基咪唑等咪唑类、三丁基膦、甲基二苯基膦、三苯基膦、二苯基膦、苯基膦等有机膦类、有机膦类与醌化合物的加成反应产物、四苯基
·
四苯基硼酸盐、四苯基
·
乙基三苯基硼酸盐、四丁基
·
四丁基硼酸盐等四取代
·
四取代硼酸盐、2-乙基-4-甲基咪唑
·
四苯基硼酸盐、n-甲基吗啉
·
四苯基硼酸盐等四苯硼盐等。作为添加量,相对于树脂组合物100质量份为0.2~5质量份的范围。
[0071]
本发明的树脂组合物可以根据需要含有填充材料、硅烷偶联剂、抗氧化剂、脱模剂、消泡剂、乳化剂、触变性赋予剂、平滑剂、阻燃剂、颜料等其他添加剂等。
[0072]
作为填充材料,具体而言,可以举出熔融二氧化硅、结晶二氧化硅、氧化铝、氮化硅、氢氧化铝、勃姆石、氢氧化镁、滑石、云母、碳酸钙、硅酸钙、氢氧化钙、碳酸镁、碳酸钡、硫酸钡、氮化硼、碳、碳纤维、玻璃纤维、氧化铝纤维、二氧化硅氧化铝纤维、碳化硅纤维、聚酯纤维、纤维素纤维、芳香族聚酰胺纤维、陶瓷纤维、微粒橡胶、热塑性弹性体、颜料等。作为使用填充材料的理由,可以举出耐冲击性的提高效果。另外,在使用氢氧化铝、勃姆石、氢氧化镁等金属氢氧化物的情况下,具有作为阻燃助剂起作用而阻燃性提高的效果。
[0073]
在将树脂组合物制成板状基板等的情况下,从其尺寸稳定性、弯曲强度等方面考虑,可以举出纤维状填充材料作为优选的填充材料。更优选可以举出使用将玻璃纤维编织成网眼状的纤维状基材的填充材料的玻璃纤维基板。
[0074]
相对于树脂组合物(固体成分)100质量份,填充材料的配合量优选为1~150质量份,更优选为10~70质量份。如果配合量多,则有固化物变脆而可能得不到充分的机械物性。另外,如果配合量少,则有可能无法发挥固化物的耐冲击性的提高等填充材料的配合效果。
[0075]
相对于树脂组合物(固体成分)100质量份,其他添加剂的配合量优选为0.01~20质量份的范围。
[0076]
通过对本发明的树脂组合物进行加热固化,可以得到固化物。作为用于得到固化物的方法,可以优选使用浇铸成型、压缩成型,传递成型等、通过制成树脂片材、带树脂的铜箔、预浸料等形态进行层叠并加热加压固化而制成层叠板等方法。此时的温度通常为150~300℃的范围,固化时间通常为10分钟~5小时左右。
[0077]
本发明的树脂组合物可以通过将上述各成分均匀混合来得到。树脂组合物可以用与以往已知的方法同样的方法容易地制成固化物。作为固化物,可以举出层叠物、浇铸成型物、成型物、粘接层、绝缘层、膜等成型固化物。
[0078]
作为使用树脂组合物的用途,可以举出印刷电路板材料、柔性电路板用树脂组合物、增层基板用层间绝缘材料等电路基板用绝缘材料、半导体密封材料、导电糊料、导电膜、增层用粘接膜、树脂浇铸成型材料、粘接剂等。这些各种用途中,在印刷电路板材料、电路基板用绝缘材料、增层用粘接膜用途中,可以作为将电容器等无源部件、ic芯片等有源部件埋入基板内的所谓电子部件内置用基板用的绝缘材料使用。其中,从高阻燃性、高耐热性、低介电特性和溶剂溶解性等特性出发,优选用于印刷布线板材料、柔性电路板用树脂组合物、
增层基板用层间绝缘材料等电路基板(层叠板)用材料及半导体密封材料。
[0079]
作为使用本发明的树脂组合物得到的密封材料,有胶带状半导体芯片用、灌注型液封用、底部填充用、半导体的层间绝缘膜用等,可以优选用于这些用途。为了将树脂组合物制备为半导体密封材料用,可以举出如下方法:将根据需要配合于树脂组合物的无机填充材料、偶联剂、脱模剂等添加剂预备混合后,使用挤出机、捏合机、辊等充分熔融混合直至均匀。此时,作为无机填充材料,通常使用二氧化硅,优选在树脂组合物中配合70~95质量%的无机填充材料。
[0080]
在将这样得到的树脂组合物用作半导体封装的情况下,可以举出如下方法:对树脂组合物进行浇铸成型或使用传递成型机、注塑成型机等进行成型,进一步在180~250℃下加热固化0.5~5小时,由此得到成型物。在用作胶带状密封材料的情况下,可以举出如下方法:对其进行加热而制作半固化片材,制成密封材料胶带,然后将该密封材料胶带置于半导体芯片上,加热至100~150℃使其软化进行成型,在180~250℃下使其完全固化。另外,在用作灌注型液态密封材料的情况下,可以将得到的树脂组合物根据需要溶解于溶剂中,然后涂布于半导体芯片或电子部件上,直接使其固化。
[0081]
本发明的树脂组合物可以溶解于有机溶剂中而制备成清漆状态。作为可以使用的有机溶剂,可以举出甲醇、乙醇等醇系溶剂、丙酮、甲基乙基酮、甲基异丁基酮、环己酮等酮系溶剂、四氢呋喃等醚系溶剂、二甲基甲酰胺、二甲基乙酰胺、n-甲基吡咯烷酮等含氮原子的溶剂、二甲基亚砜等含硫原子的溶剂等,可以混合使用1种或2种以上。只要是工业上可得到的有机溶剂,就没有特别限定,但是从溶解性、操作性的观点出发,优选甲基乙基酮、二甲基甲酰胺。
[0082]
本发明的树脂组合物可以在制成溶解于有机溶剂的组合物清漆后,含浸于玻璃布、芳香族聚酰胺无纺布、液晶聚合物等聚酯无纺布等纤维状物中,然后除去溶剂,制成预浸料。另外,可以将组合物清漆涂布于铜箔、不锈钢箔、聚酰亚胺膜、聚酯膜等片状物上,然后进行干燥,由此制成粘接片材。
[0083]
在使用上述预浸料来形成层叠板的情况下,可以将一片或多片预浸料层叠,在单侧或两侧配置金属箔而构成层叠物,对该层叠物进行加压加热而使预浸料固化、一体化,得到层叠板。这里,作为金属箔,可以使用铜、铝、黄铜、镍等的单独、合金、复合的金属箔。作为对层叠物进行加热加压的条件,只要在树脂组合物固化的条件下适当调整而进行加热加压即可,但是如果加压过低,则有时在所得到的层叠板的内部残留气泡,电特性降低,因此优选在满足成型性的条件下进行加压。加热温度优选为160~250℃,更优选为170~220℃。加压压力优选为0.5~10mpa,更优选为1~5mpa。加热加压时间优选为10分钟~4小时,更优选为40分钟~3小时。进而,可以将这样得到的单层的层叠板作为内层材料而制作多层板。在这种情况下,首先利用加成法、减成法等对层叠板形成电路,对形成的电路表面实施黑化处理,得到内层材料。在该内层材料的单面或两侧的电路形成面利用预浸料或粘接片材形成绝缘层,并且在绝缘层的表面形成导体层,从而形成多层板。
[0084]
实施例
[0085]
列举实施例和比较例来对本发明进行具体说明,但是本发明不限定于它们。只要没有特别说明,则“份”表示质量份,“%”表示质量%,“ppm”表示质量ppm。另外,测定方法分别通过以下方法进行测定。
[0086]
·
羟基当量:
[0087]
依据jis k 0070标准进行测定,单位以“g/eq.”表示。应予说明,只要没有特别说明,则芳香族多羟基化合物的羟基当量是指酚性羟基当量。
[0088]
·
软化点:
[0089]
依据jis k 7234标准、环球法进行测定。具体而言,使用自动软化点装置(株式会社meitec制,asp-mg4)。
[0090]
·
铜箔剥离强度和层间粘接力:
[0091]
依据jis c 6481进行测定,层间粘接力是在第7层与第8层之间进行剥离测定。
[0092]
·
相对介电常数和介质损耗角正切:
[0093]
依据ipc-tm-650 2.5.5.9,使用材料分析仪(agilent technologies公司制),通过电容法求出频率1ghz下的相对介电常数和介质损耗角正切,由此进行评价。
[0094]
·
玻璃化转变温度(tg):
[0095]
依据jis c 6481标准进行测定。由利用动态粘弹性测定装置(株式会社日立高新技术科学制,exstar dms6100)在5℃/分钟的升温条件下进行测定时的tanδ峰顶来表示。
[0096]
·
gpc(凝胶渗透色谱法)测定:
[0097]
使用在主体(东曹株式会社制,hlc-8220gpc)中串联地具备柱(东曹株式会社制,tskgelg4000hxl、tskgelg3000hxl、tskgelg2000hxl)的装置,柱温设为40℃。另外,洗脱液使用四氢呋喃(thf),流速设为1ml/分钟,检测器使用差示折射率检测器。测定试样使用50μl的将样品0.1g溶解于10ml的thf中并利用微滤器进行过滤而得的样品。数据处理使用东曹株式会社制gpc-8020型号ii版本6.00。
[0098]
·
ir:
[0099]
使用傅立叶变换型红外分光光度计(perkin elmer precisely制,spectrum one ft-ir spectrometer 1760x),池使用krs-5,将溶解于thf的样品涂布于池上,进行干燥,然后测定波数650~4000cm《sup》-1《/sup》的吸光度。
[0100]
·
esi-ms:
[0101]
使用质谱仪(岛津制作所制,lcms-2020),使用乙腈和水作为流动相,测定溶解于乙腈的样品,由此进行质量分析。
[0102]
实施例、比较例中使用的缩写如下。
[0103]
[马来酰亚胺化合物]
[0104]
m1:苯甲烷马来酰亚胺(大和化成工业株式会社制,bmi-2300)
[0105]
m2:合成例5中得到的马来酰亚胺树脂
[0106]
[芳香族多羟基化合物]
[0107]
p1:合成例1中得到的芳香族多羟基化合物
[0108]
p2:合成例2中得到的芳香族多羟基化合物
[0109]
p3:合成例3中得到的芳香族多羟基化合物
[0110]
p4:二环戊二烯型酚醛树脂(群荣化学工业株式会社制,gdp-6140,羟基当量196,软化点130℃)
[0111]
p5:联苯芳烷基型酚醛树脂(明和化成株式会社制,meh-7851,羟基当量223)
[0112]
[环氧树脂]
[0113]
e1:合成例4中得到的环氧树脂
[0114]
e2:联苯芳烷基型环氧树脂(日本化药株式会社制,nc-3000,环氧当量274,软化点60℃)
[0115]
[固化促进剂]
[0116]
c1:2-乙基-4-甲基咪唑(四国化成工业株式会社制,curezol2e4mz)
[0117]
合成例1
[0118]
在包括具备搅拌机、温度计、氮气吹入管、滴液漏斗和冷却管的玻璃制分离烧瓶的反应装置中,装入2,6-二甲酚140份、47%bf3醚络合物9.3份(相对于最初添加的二环戊二烯为0.1倍摩尔),一边搅拌一边加热至110℃。保持在该温度下,同时用1小时滴加二环戊二烯86.6份(相对于2,6-二甲酚为0.57倍摩尔)。进一步在110℃的温度下反应3小时后,保持在该温度下,同时用1小时滴加二环戊二烯68份(相对于2,6-二甲酚为0.44倍摩尔)。再在120℃下反应2小时。加入氢氧化钙14.6份。进一步添加10%的草酸水溶液45份。然后,加热至160℃进行脱水后,在5mmhg的减压下加热至200℃将未反应的原料蒸发除去。加入甲基异丁基酮(mibk)700份溶解产物,加入80℃的温水200份进行水洗,分离除去下层的水层。然后,在5mmhg的减压下加热至160℃将mibk蒸发除去,得到274份的红褐色的芳香族多羟基化合物(p1)。是羟基当量为299、软化点97℃的树脂,吸收比(a
3040
/a
1210
)为0.17。测定基于esi-ms(负)所得到的质谱,结果确认到m-=253、375、507、629。分别将所得到的芳香族多羟基化合物(p1)的gpc示于图1,将ft-ir示于图2。基于gpc所得的mw为690,mn为510,n=0体含量为6.5面积%,n=1体含量为61.5面积%,n=2体以上的含量为32.0面积%。图1的a表示通式(1)的n=1体与通式(1)的无r2加成体的n=1体的混合物,b表示通式(1)的n=0体。图2的c表示来自二环戊二烯骨架的烯烃部位的c-h伸缩振动的峰,d表示由苯酚核的c-o伸缩振动引起的吸收。
[0119]
合成例2
[0120]
在与合成例1同样的反应装置中装入2,6-二甲酚140份、47%bf3醚络合物9.3份(相对于最初添加的二环戊二烯为0.1倍摩尔),一边搅拌一边加热至110℃。保持在该温度下,同时用1小时滴加二环戊二烯86.6份(相对于2,6-二甲酚为0.57倍摩尔)。再在110℃的温度下反应3小时后,保持在该温度下,同时用1小时滴加二环戊二烯90.6份(相对于2,6-二甲酚为0.60倍摩尔)。进一步在120℃下反应2小时。加入氢氧化钙14.6份。再添加10%的草酸水溶液45份。然后,在加热至160℃进行脱水后,在5mmhg的减压下加热至200℃将未反应的原料蒸发除去。加入740份的mibk溶解产物,加入80℃的温水200份进行水洗,分离除去下层的水层。然后,在5mmhg的减压下加热至160℃将mibk蒸发除去,得到310份的红褐色的芳香族多羟基化合物(p2)。是羟基当量为341、软化点104℃的树脂,吸收比(a
3040
/a
1210
)为0.27。测定基于esi-ms(负)所得到的质谱,结果确认到m-=253、375、507、629。基于gpc所得的mw为830,mn为530,n=0体含量为5.9面积%,n=1体含量为60.1面积%,n=2体以上的含量为34.0面积%。
[0121]
合成例3
[0122]
在与合成例1同样的反应装置中装入2,6-二甲酚140份、47%bf3醚络合物9.3份(相对于最初添加的二环戊二烯为0.1倍摩尔),一边搅拌一边加热至110℃。保持在该温度下,同时用1小时滴加二环戊二烯86.6份(相对于2,6-二甲酚为0.57倍摩尔)。进一步在110
℃的温度下反应3小时后,保持在该温度下,同时用1小时滴加二环戊二烯34.0份(相对于2,6-二甲酚为0.22倍摩尔)。再在120℃下反应2小时。加入氢氧化钙14.6份。进一步添加10%的草酸水溶液45份。然后,加热至160℃进行脱水后,在5mmhg的减压下加热至200℃将未反应的原料蒸发除去。加入608份的mibk溶解产物,加入80℃的温水200份进行水洗,分离除去下层的水层。然后,在5mmhg的减压下加热至160℃将mibk蒸发除去,得到253份的红褐色的芳香族多羟基化合物(p3)。是羟基当量为243、软化点92℃的树脂,吸收比(a
3040
/a
1210
)为0.11。测定基于esi-ms(负)所得到的质谱,结果确认到m-=253、375、507、629。基于gpc所得的mw为460,mn为380,n=0体含量为5.6面积%,n=1体含量为66.4面积%,n=2体以上的含量为28.0面积%。
[0123]
合成例4
[0124]
在与合成例1同样的反应装置中加入合成例1中得到的芳香族多羟基化合物(p1)100份、表氯醇155份和二乙二醇二甲醚46份并加热至65℃。在125mmhg的减压下,保持在63~67℃的温度,同时用4小时滴加49%氢氧化钠水溶液30.9份。在此期间,使表氯醇与水共沸,将流出来的水依次除去至体系外。反应结束后,在5mmhg、180℃的条件下回收表氯醇,加入277份的mibk溶解产物。然后,加入80份的水溶解副产的食盐,静置而将下层的食盐水分离除去。利用磷酸水溶液进行中和后,对树脂溶液进行水洗、过滤,直至水洗液成为中性。在5mmhg的减压下加热至180℃,蒸馏除去mibk,得到113份的红褐色透明的2,6-二甲酚
·
二环戊二烯型环氧树脂。是环氧当量为358、总氯含量520ppm、软化点80℃的树脂。基于gpc所得的mw为870,mn为570。将得到的环氧树脂(e1)的gpc示于图3。
[0125]
合成例5
[0126]
在安装有温度计、冷却管、迪安-斯达克(dean-stark)共沸蒸馏分离器、搅拌机的烧瓶中,装入苯胺100份和甲苯50份,在室温下用1小时滴加35%盐酸39.2份。滴加结束后,进行加热,将共沸的水和甲苯冷却、分液,然后仅将作为有机层的甲苯送回体系内进行脱水。接着,将4,4’-双(氯甲基)联苯33.6份保持在60~70℃,同时用1小时进行添加,进一步在该温度下进行2小时反应。反应结束后,一边进行升温一边将甲苯蒸馏除去,使体系内为195~200℃,在该温度下进行15小时反应。然后,一边冷却一边将30%氢氧化钠水溶液86份以在体系内不激烈回流的方式缓慢滴加,将在80℃以下进行升温时蒸馏除去的甲苯送回体系内,在70℃~80℃下进行静置。将分离的下层的水层除去,重复进行反应液的水洗直至清洗液成为中性。接着,利用旋转蒸发器在加热减压下(200℃、0.6kpa)从油层蒸馏除去过量的苯胺和甲苯,由此得到47份的芳香族胺树脂。
[0127]
接下来,在上述烧瓶中装入马来酸酐75份和甲苯150份,进行加热,将共沸的水和甲苯冷却、分液后,仅将作为有机层的甲苯送回体系内进行脱水。接下来,一边将体系内保持为80~85℃,一边用1小时滴加使上述芳香族胺树脂100份溶解于n-甲基-2-吡咯烷酮100份中而得的树脂溶液。滴加结束后,在相同温度下进行2小时反应,加入对甲苯磺酸1.5份,将在回流条件下共沸的缩合水和甲苯冷却、分液,然后仅将作为有机层的甲苯送回体系内进行脱水,同时进行20小时反应。反应结束后,追加100份的甲苯,重复进行水洗将对甲苯磺酸和过量的马来酸酐除去,进行加热而通过共沸将水从体系内除去。接着,将反应溶液浓缩,得到133份的马来酰亚胺树脂。
[0128]
实施例1
[0129]
配合100份的马来酰亚胺m1、40份的合成例1中得到的树脂、1.5份的2e4mz,溶解于甲基乙基酮(mek)中而得到树脂浓度50%的树脂组合物清漆。
[0130]
将得到的树脂组合物清漆含浸于玻璃布(日东纺织株式会社制,wea7628xs13,0.18mm厚)中。将含浸的玻璃布在150℃的热风循环烘箱中干燥10分钟,得到预浸料。将得到的预浸料8片与铜箔(三井金属矿业株式会社制,3ec-iii,厚度35μm)上下重叠,在130℃
×
15分钟+220℃
×
120分钟的温度条件下进行2mpa的真空压制,得到1.6mm厚的层叠板。将层叠板的铜箔剥离强度、tg的测定结果示于表1。
[0131]
另外,将得到的预浸料拆开,制成通过100目的筛的粉状的预浸料粉末。将得到的预浸料粉末放入氟树脂制的模具中,在130℃
×
15分钟+220℃
×
120分钟的温度条件下进行2mpa的真空压制,得到50mm见方
×
2mm厚的固化树脂试验片。将试验片的相对介电常数和介质损耗角正切的测定结果示于表1。
[0132]
实施例2~7、比较例1~5
[0133]
以表1的配方的配合量(份)配合,使用与实施例1同样的装置,通过同样的操作,得到树脂组合物清漆,进而得到层叠板和固化树脂试验片。
[0134]
进行与实施例1同样的试验,将其结果示于表1。
[0135][0136]
工业上的可利用性
[0137]
本发明的树脂组合物的介电性、耐热性、粘接性优异,可以用于层叠、成型、粘接等各种用途,特别是有效用作高速通信设备的电子材料。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1