用于新抗原分析的方法和系统与流程
用于新抗原分析的方法和系统
1.相关申请的交叉引用
2.本技术要求2020年6月13日递交的美国临时申请号63/038,816的权益,其公开内容通过引用整体并入本文。
3.以电子方式提交的序列表的引用
4.随本文以电子方式提交的文件名seqlisting-neoantigen-pct.txt,大小91741字节,创建日期2021年6月13日的序列表的内容通过引用整体并入本文。
技术领域
5.本发明大体上涉及蛋白质组学分析领域,并且具体地,涉及凭借液相色谱和质谱(lc-ms)进行的蛋白质组学分析,并且更具体地,涉及用于分析小肽(诸如新抗原)的基于lc-ms的方法和系统。
背景技术:
6.能够对由携带癌症相关突变的基因编码的蛋白进行加工,并且携带突变的肽通过人类白细胞抗原(hla)分子能呈现在细胞表面上。这种肽被称为新抗原(neoantigens)。新抗原是真正个性化和癌症特异性的,这使其成为理想的抗癌治疗靶点(schumacher&schreiber,2015)。新抗原通过其t细胞受体可以被t细胞识别,这是癌症免疫疗法的基础(lauss et al.,2017;ott et al.,2017;riaz et al.,2016;sahin et al.,2017;schumacher&schreiber,2015)。为了鉴别潜在的药物靶点,近来,报道了包括全免疫肽组的深度剖析和靶向检测方法以及t细胞反应性的间接测定在内的许多技术来揭示新抗原的库(bassani-sternberg et al.,2016;danilova et al.,2018;wang et al.,2019)。我们之前报道了一种称为“mana-srm”的技术,作为从大约2亿个组织培养肿瘤细胞中检测新抗原的基础研究平台,该技术比之前公布的用于此目的的通常需要超过20至30亿个细胞的技术(bassani-sternberg et al.,2016;wang et al.,2019)灵敏10倍以上。mana-srm是用于基于组织培养的新抗原测定的理想工具,并且已能够开发靶向人类肿瘤抑制基因tp53和癌基因k-ras中最频繁突变热点的第一个现成癌症疫苗(douglass et al.,2021;hsiue et al.,2021)。然而,鉴于活检或手术切除可获得的肿瘤组织量有限,mana-srm的灵敏度和鲁棒性对于常规临床应用仍然是不可行的。在建立基于新抗原的个性化癌症治疗之前,迫切需要一种更灵敏、快速和可重复的平台来分析临床样品中的新抗原。
7.近年来,质谱平台经历了一系列突破性的改进(bekker-jensen et al.,2020;meier et al.,2018;zubarev&makarov,2013)。与此同时,蛋白质组分析正在从数小时的测定转变为短短21分钟的操作,且在覆盖范围上几乎没有妥协(bache et al.,2018)。这些改进极大地革新了基于质谱的蛋白质组学,使其更适合临床应用。
技术实现要素:
8.本技术提供了质谱法的进一步改进,其覆盖数个领域,包括样品制备、硬件配置和
自动化,以及质谱分析等。更具体地,本技术提供了一种集成方法和系统(下文中称为“valid-neo线路”),用于检测并量化来自临床样品的新抗原,而无需大量的人工样品处理。
9.在第一方面,提供了通过检测手段来表征靶标肽的方法。该方法包括以下步骤:
10.(1)引入至少一种保护性分子来与所述靶标肽混合,其中所述至少一种保护性分子中的每一种配置为具有与所述靶标肽相似的特征,且进一步配置为能够通过所述检测手段与所述靶标肽区分开;以及
11.(2)应用所述检测手段来表征所述靶标肽。
12.本文中,上述检测手段可包括质谱分析,并且因此,所述至少一种保护性分子中的每一种配置为具有能够通过质谱分析与所述靶标肽区分开的m/z值。
13.除质谱分析之外,本文涵盖的检测手段也可涉及其他手段。示例可包括:使用靶标肽序列特异性抗体,当保护性肽或化合物与靶标肽被设计成呈现不同的荧光信号时使用荧光检测方法;当靶标肽与pcr可扩增信号相关时使用基于聚合酶链反应(pcr)的检测方法;当靶标肽与具体测序信息相关时使用基于核酸序列的检测方法等。事实上,任何可以区别性检测靶标肽和至少一种保护性化合物的检测方法都可以应用于该方法,并且被认为是在本技术的范围内。
14.如在此以及本技术的其它部分所使用的,术语靶标肽的“表征”可包括靶肽的检测(即鉴定、定性等)和量化的一者或两者。
15.如本文所使用的,术语“保护性分子(guard molecule)”是指在样品制备的一个或多个步骤以及处理线路中与靶标肽共存的分子,其主要用于保护靶标肽以免于在分析中损失。
16.具体地,如果检测手段涉及质谱分析,则保护性分子可配置为具有与靶标肽相似的特征(例如,相似的疏水性和电荷状态),并且具有可通过下游分析仪器与靶标肽区分开的特征,诸如当通过质谱仪分析时,具有能够与靶标肽区分开的不同m/z值。
17.在另一检测手段中,诸如使用肽-序列特异性抗体来检测靶标肽,其中该序列特异性抗体可用于选择性检测靶标肽且不与保护性化合物相互作用。
18.无论使用何种检测手段,保护性分子与靶标肽的共存都可以确保保护性分子在样品制备中的行为与靶标肽相似,从而作为阻断剂防止靶标肽在样品制备和处理过程期间与靶标肽可能相互作用的任何表面或任何物质的非特异性结合。
19.如本文所使用的,术语“相似特性”或“相似性”由样品制备和处理过程中采用的分离和分级机制来大致定义,其通常可指其中两种分子,诸如第一分子(例如靶标分子)和第二分子(例如保护性分子)在相同ph环境下具有基本相同的疏水性和电荷状态的情况。例如,在样品制备线路仅包括反相柱的某种情形下,可用于本文公开的方法中的保护性分子可具有与靶标分子相似的疏水性。在使用尺寸排阻柱分离分析物的另一种情形下,可用于本文公开的方法中的保护性分子可具有与靶标分子基本相似的分子量,以从而能够在样品制备和处理过程中与靶标分子共存。在又一种情形下,可以使用上述两种类型的柱,因此对于保护性分子的选择,使用具有相同序列的重同位素标记的肽或仅具有一个氨基酸不同于靶标肽的肽作为它们的保护性肽是保护靶标肽免受损失的最可行的方法。
20.如本文所使用的,术语“m/z值”是基于质谱的肽分析领域的普通技术人员常规所知的,其通常是指某一分子的质荷比。众所周知的是,目前的质谱技术能够容易地分离分子
量相差约1/30000质子(即1道尔顿)的两种分子。例如,分光计可以分离分子量为100.0001的第一分子与分子量为100.0002的第二分子。因此,用具有不同分子量或不同电荷的另一氨基酸残基取代一个氨基酸残基,或者用重同位素标记一个或多个氨基酸残基通常会引起至少1道尔顿的差异,并且会使得靶标肽和保护性肽容易地彼此分离。
21.根据所述方法的某些实施方式,所述至少一种保护性分子包括保护性肽。在本文中,存在若干可能的实施方式。
22.在第一实施方式中,保护性肽可具有与靶标肽相同的氨基酸残基序列,且保护性肽中的至少一个氨基酸残基是重同位素标记的。
23.如本文所使用的,标记在某些氨基酸残基上的术语“重同位素”是指如下事实:在保护性肽中,存在至少一个氨基酸残基是重同位素标记的氨基酸残基,诸如具有c
13
和/或n
15
标记的赖氨酸残基(即,来自氨基酸的标准c
12
元素被重同位素元素c
13
替代,n
14
元素可被重同位素元素n
15
替代)。
24.在第二实施方式中,保护性肽中仅有一个氨基酸残基不同于靶标肽。本文中,与靶标肽中的相应氨基酸残基相比,保护性肽中的区别氨基酸残基可以具有相同的特征(即疏水性、相同ph下的电荷状态等),但是具有不同的分子量。例如对于一种靶标肽:kras_q61h新抗原序列ildtagheey(seq id no.496,参见表1),可以使用几种可能的保护性肽,其可包括以下非限制性示例:
25.1)ildtaghdey(seq id no.507),通过用d取代靶标肽的8号位的氨基酸残基e来获得;
26.2)ivdtagheey(seq id no.518),通过用v取代靶标肽的2号位的氨基酸残基l获得;和
27.3)ildsagheey(seq id no.529),通过用s取代靶标肽的4号位的氨基酸残基t获得。
28.凭借这些上述氨基酸残基取代(即e/d、l/v和s/t),这些保护性肽中的每一个在样品制备过程中都可以表现出与靶标肽相似的行为,但仍然通过质谱能够容易地进行区分。
29.在第三实施方式中,保护性肽中的至少两个氨基酸残基可不同于靶标肽。例如,对于上述靶标肽kras_q61h新抗原序列ildtagheey(seq id no.496),一种可能的保护性肽可以是ivdtaghdey(seq id no.540),其通过用v取代2号位的氨基酸残基l并用d取代8号位的氨基酸残基e而获得。
30.在第四实施方式中,保护性肽可具有相对于靶标肽的乱序序列(scrambled sequence)。例如,用于上述示例性靶标序列ildtagheey(seq id no.496)的可能的保护性肽可以是eyilgedtah(seq id no.541),其中它们都具有相同的氨基酸残基组成,但具有不同的序列,因此通过可行的分析方法,诸如使用序列特异性抗体或通过质谱仪能够容易地区分。
31.在其他实施方式中,上述至少一种保护性分子可包括非肽化合物。与靶标肽行为相似的任何化合物(即相同的疏水性、在相同ph下相同的电荷状态等)在样品制备中可作为阻断剂来以防止靶标肽与靶标肽可相互作用的表面的非特异性结合,并且此外,保护性化合物可通过分析性过程来区分,例如具有不同的质量和/或m/z来通过序列特异性抗体或质谱仪进行区分。
32.根据方法的某些实施方式,靶标肽是新抗原肽,并且新抗原肽的示例可包括但不限于,kras_q61h、kras_q61l、kras_q61r、idh2_r140q、tp53_y220c、tp53_r248w、tp53_r213l、kras_g12v_9mer、kras_g12v_10mer、kras_g12d_9mer或kras_g12d_10mer。这些示例将涵盖在如下示出的本技术的实施方式1中。注意的是,除了表征新抗原肽之外,该方法还可以用于表征不是新抗原的其他肽。本文对此没有限制。
33.根据该方法的一些实施方式,其中所述靶标肽为新抗原肽,所述新抗原肽来自从受试者获得的组织样品,并且在引入至少一种保护性分子以与所述靶标肽混合的步骤(1)之前,所述方法进一步包括组织样品制备步骤。该组织制备步骤可包括以下子步骤:
34.(a)提供组织样品,其中所述组织样品为冷冻组织样品;
35.(b)在至少8000psi的冲击力下研磨所述冷冻组织样品,从而获得冷冻的单细胞组织粉末;以及
36.(c)处理所述冷冻的单细胞组织粉末,然后获得经处理的组织样品。
37.本文中,受试者可以是人,但也可以是另一种哺乳动物物种,例如猴、猿、狗、小鼠、大鼠等。也可以是非哺乳动物物种。组织样品可以是手术切除肿瘤样品,或者可以是活检样品。
38.本文中,在组织样品制备步骤的子步骤(a)中,组织样品可为冷冻组织样品,其优选可为速冻在液氮中的组织样品。组织样品可以是通过活检或通过手术切除从受试者新鲜获得的,并且可以是ffpe(福尔马林固定石蜡包埋)组织样品,其在研磨子步骤(b)之前通过液氮处理。其他形式的组织样品也是可能的,并且应当被认为由本文的公开内容所涵盖。
39.根据某些实施方式,子步骤(b)中研磨冷冻组织样品的冲击力可为约10000psi。在其他实施方式中,冲击力可为约12000psi、15000psi等。
40.根据某些实施方式,处理所述冷冻的单细胞组织粉末,然后获得经处理的组织样品的子步骤(c)包括:裂解、超声处理和离心,其中所述经处理的组织样品可来自离心后的上清液。
41.在组织样品制备步骤的子步骤(c)中,通过组织裂解来处理冷冻的单细胞组织粉末,以裂解经研磨的组织样品中的细胞,随后进行超声处理以使细胞中的基因组dna片段化。离心后,上清液成为含有待表征的新抗原肽的经处理的组织样品。组织样品制备步骤的更多细节可见于本技术的实施方式1。
42.根据某些实施方式,在处理所述冷冻的单细胞组织粉末,然后获得经处理的组织样品的子步骤(c)之后,所述方法可进一步包括:
43.对从离心后的沉淀物中获得的基因组dna进行分析。
44.根据一些实施方式,在组织样品制备步骤之后且在引入至少一种保护性分子以与靶标肽混合的步骤(1)之前,该方法进一步包括人类白细胞抗原(hla)分子富集步骤,该富集步骤包括:
45.使所述经处理的组织样品通过hla富集柱,其中所述hla富集柱包括其上固定有抗-hla抗体的基质。
46.根据一些实施方式,在hla分子富集步骤之后,该方法进一步包括洗脱步骤,该洗脱步骤包括:
47.将具有低ph的洗脱缓冲液施加于所述hla富集柱,从而获得含有所述新抗原肽的
洗脱物。
48.在本文中,所述洗脱缓冲液包含所述至少一种保护性分子。
49.根据一些实施方式,在所述洗脱步骤之后且在应用质谱分析以表征靶标肽的步骤(2)之前,所述方法进一步包括清洁步骤(clean-up step),该清洁步骤包括:
50.(a)使所述洗脱物通过捕集柱至少一次,从而将所述新抗原肽捕集在所述捕集柱中,其中所述捕集柱包括能够与所述新抗原肽结合但对杂质具有较低或没有结合亲和力的基质;以及
51.(b)洗脱所述捕集柱,从而获得经清洁的(cleaned)洗脱物。
52.根据一些实施方式,在所述清洁步骤之后且在应用质谱分析以表征靶标肽的步骤(2)之前,所述方法进一步包括纯化步骤,该纯化步骤包括:
53.使所述经清洁的洗脱物通过尺寸排阻柱(sec柱,或sec)以收集含新抗原肽的级分。
54.本文中,任选地,在通过该经清洁的洗脱物之前,可向该经清洁的洗脱物加入肽梯(peptide ladder),诸如下文所述的neo-sec梯,其意在界定收集新抗原的边界。具体地,可以监测特征色谱峰以指示收集的起点(例如,在neo-sec梯的示例中代表2000da的峰)和终点(例如,在neo-sec梯的示例中代表800da的峰)。
55.对于上文所示方法的组织制备步骤、hla分子富集步骤、洗脱步骤、清洁步骤、纯化步骤和质谱分析步骤,可在本技术的实施方式1中找到更多细节。
56.根据所述方法的某些实施方式,hla分子富集步骤、洗脱步骤、清洁步骤和纯化步骤中的至少两个连续步骤可操作性地连接,从而基本上实现自动化处理而很少或没有人工干预。
57.根据某些实施方式,hla分子富集步骤、洗脱步骤、清洁步骤和纯化步骤中的基本上所有步骤可操作地连接,并且基本上是自动化的。
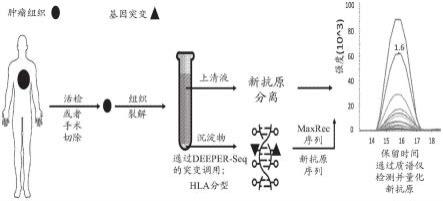
58.根据优选实施方式,使含新抗原肽的样品通过质谱仪的质谱分析步骤,即如上所述的步骤(2)可以进一步与上游纯化步骤可操作地连接以实现自动化。换句话说,整个样品处理过程,包括hla分子富集步骤、洗脱步骤、清洁步骤、纯化步骤和质谱分析步骤能够在很少或没有人工干预下实现自动化。
59.应注意的是,这些步骤中的每一个可不限于本文公开的步骤,并可通过本领域普通技术人员已知的替代方式来实现,然而通过应用如本文公开的这些步骤,可实现样品制备、hla分子富集、洗脱、清洁、纯化和质谱分析的自动化。被称为“valid-neo”线路的集成系统和方法基本上需要很少或不需要人工干预,从而在不同诊断中心和医院中能够确保高灵敏度、可再现性和可移植性。
60.在第二方面,进一步提供了一种能够实施上述方法的系统。该系统包括以下部件:
61.(1)组织样品研磨装置,配置为在组织样品制备步骤中向冷冻组织样品施加至少8000psi的冲击力,以从而获得冷冻的单细胞组织粉末;
62.(2)hla富集柱,包括其上固定有抗-hla抗体的基质,并且配置为允许在组织样品制备步骤中获得的经处理的组织样品的通过,从而在hla分子富集步骤中富集hla分子;
63.(3)捕集柱,包括能够与新抗原肽结合但对杂质具有较低或无结合亲和力的基质,并且配置为在清洁步骤中将新抗原肽捕集于其中;
64.(4)尺寸排阻柱(sec),配置为在纯化步骤中纯化新抗原肽;以及
65.(5)质谱仪,配置为实施质谱分析步骤。
66.在本技术的实施方式1中,更详细地描述了组织样品研磨装置(称为“uniceller”)的一个实施方式。
67.根据该系统的某些实施方式,hla富集柱、捕集柱、尺寸排阻柱(sec)和质谱仪的至少两个相邻部件依次且可操作地彼此连接,从而允许自动化。
68.根据某些实施方式,hla富集柱、捕集柱、尺寸排阻柱(sec)和质谱仪中的所有部件依次且可操作地彼此连接,从而允许自动化。
附图说明
69.图1示出了根据本技术某些实施方式的valid-neo线路的一般过程;
70.图2a和图2b一起示出了根据本技术某些实施方式的valid-neo线路中新抗原分离和纯化的流程;
71.图3a至图3f一起示出了总结患者的癌症类型、其肿瘤的所选遗传突变特征,以及侧翼有癌症驱动基因上最高流行突变位点的可能新抗原序列的表;
72.图4示出了uniceller和其他三种传统手段之间的蛋白质提取效率的比较结果;
73.图5示出了valid-neo线路分析的再现性评估结果;以及
74.图6示出了通过sec柱纯化新抗原的hplc色谱图。
具体实施方式
75.为了进一步描述上文提供的新抗原分析方法和系统,以下提供了一个具体实施方式(即实施方式1)。
76.实施方式1
77.图1示出了valid-neo线路分析新抗原肽的一般过程。具体地,首先通过活检或手术切除从受试者获取肿瘤组织样品,然后通过组织裂解、超声处理和离心处理来处理该肿瘤组织样品,以获得hla分子(在上清液中)和基因组dna(在沉淀物中)。通过deeper-seq线路(wang et al.,2017)对来自沉淀物的肿瘤dna进行基因组测序,以揭示患者的特异性突变。进行突变调用(mutation calling),并建立新抗原和新抗原肽的潜在序列(称为“maxrec序列”)。然后通过多步过程从组织裂解物的上清液中分离和纯化新抗原,如图2a和图2b所示和下文所述,随后通过质谱直接检测并量化新抗原。
78.图2a和图2b示出了根据本技术某些实施方式的valid-neo线路中新抗原分离和纯化的多步骤过程的流程。如这两幅图所示,valid-neo系统中主要有5个步骤来获得新抗原,步骤1)通过重复上样抗体柱来富集hla;步骤2)洗脱hla分子并分离新抗原,然后上样捕集柱;步骤3)清洁新抗原并去除hla分子;步骤4)洗脱新抗原并上样sec柱;步骤5)通过sec柱纯化新抗原。
79.在valid-neo线路中新抗原分离和纯化的多步骤过程的该实施方式中,注意的是,系统中还包括一系列的阀(参见图中的“阀1至阀5”)、一系列的泵(参见泵1至泵5)、一系列的dad检测器(参见“检测器1至检测器3”)和级分收集器。图2a和图2b中还示出了系统中每个部件的示意性配置和连接。
80.如所示出的,每个阀包括总共6个端口(#1-6),每个端口可操作且可控地连接至其他装置,诸如“抗体柱”(即hla富集柱)、“捕集柱”、“sec柱”、泵、dad检测器和级分收集器的入口或出口。每个泵配置为提供驱动流体在线路中沿预定方向(如图中箭头所示)流动的驱动力,并且每个端口配置为基于其接收的控制信号以受控方式打开或关闭。每个dad检测器配置为检测其接收的流体的特定参数。处理器(未示出)与上述部件中的每一个通信地连接,并且配置为基于从dad检测器传输的检测信号,以程序化方式控制上述部件中的每一个的协调工作。例如,处理器可以控制阀的每个端口的打开/关闭状态,并且可以控制每个泵的启动或停止以及流速。从而,该系统的每个部件的协调工作可以实现自动化样品处理,使得经处理的组织样品(即,含hla/新抗原的样品,或图1中的“上清液”)在进入质谱仪进行表征之前,以自动化和受控的方式流过富集柱、捕集柱和sec柱。
81.材料和方法
82.肿瘤样品
83.从bioivt获得总共10名患者的肿瘤样品。该研究由complete omics inc.和bioivt的人体研究机构审查委员会批准,并符合健康保险可便携性和责任法案。患者的癌症类型及其肿瘤的所选遗传突变特征列于图3a至图3f所示的表中。在该表中,粗体和下划线的氨基酸残基代表新抗原肽序列中感兴趣的突变。
84.valid-neo的构建
85.valid-neo是由新抗原检测所必需的五个步骤组成的集成系统,包括:1)富集hla分子,2)从抗体柱洗脱新抗原,3)清洁新抗原,4)从捕集柱洗脱新抗原,5)通过sec柱纯化新抗原。该集成系统由一系列串联的hplc系统、一台质谱仪和一套优化的缓冲液(包括maxrec系统)组成。
86.从组织样品提取hla分子
87.从bioivt(bioivt,ny)获得人肿瘤新鲜冷冻组织。将50mg冷冻组织包裹在铝箔中,使得组织块被至少四层铝箔覆盖。将包裹的组织块在液氮中速冻。使用uniceller(complete omics inc,md)来从组织块产生单细胞水平粉末,并且该过程可以重复5次,直到组织块被完全研磨成冷冻的单细胞粉末,其中uniceller是涉及用于对冷冻组织包施加强冲击的内部搭建装置(in-house built device)。将1ml nl缓冲液(complete omics inc,md)加入到组织粉末中,并将组织悬浮液转移到蛋白质lo-bind管中,随后通过bioruptor 300进行五轮超声处理(能量水平4.5,任务步骤30秒,延迟步骤59秒)。在冰上孵育组织裂解物1小时,在此期间,每10分钟对悬浮液上下移液20次,并且每10分钟进行一次额外的超声处理循环。在4℃下离心组织裂解物30分钟,并将澄清的上清液转移到新的蛋白lo-bind管中。用4倍体积的nc缓冲液(complete omics inc.,md)稀释含有hla分子的上清液,之后准备用于hla分子分离。
88.通过抗体柱在线富集hla分子
89.通过基于dmp(庚二酸二甲酯)的交联反应,将抗-hla抗体(克隆w6/32)固定在蛋白a琼脂糖珠(thermofisher scientific,ma)上。然后将50ml珠装入hla富集柱,并用1l nc缓冲液(complete omics inc,md)冲洗。将hla-新抗原悬浮液通过0.22μm过滤器来过滤,用4倍体积的nc缓冲液稀释,并直接注入到hla富集柱上。将流出液收集到样品环中,并重新注入该色谱柱。该注入重复4次,使悬浮液通过抗体柱总共5次。在重复上样期间,hla分子从流
动相中耗尽并被色谱柱捕获,而hla-悬浮液被泵推入系统的nc缓冲液逐渐稀释。重复上样确保了hla分子与色谱柱的有效结合,并且流动相对样品的后续稀释有助于提高清洗效率和减少非特异性结合。然后用nc缓冲液以1ml/min的速度冲洗抗体柱20分钟,以除去未结合的蛋白质和杂质(包括盐和洗涤剂)。
90.新抗原肽的在线洗脱和抗体柱的再生
91.用递增梯度(在5分钟内从0到100%)的ne缓冲液(complete omics inc,md)通过色谱柱,随后用100% ne缓冲液以1ml/min持续冲洗2分钟来进行新抗原肽的洗脱。然后通过运行递增梯度(在5分钟内从0到100%)的nn缓冲液(complete omics inc,md),并用nc缓冲液以1ml/min冲洗1小时来中和抗体柱。随后使洗脱的hla分子和新抗原肽经受进一步纯化。
92.新抗原肽的在线分离和纯化
93.注入含新抗原肽的hla洗脱物,使其通过捕集柱总共5次,随后用10ml 0.1%甲酸洗涤。使用流动相溶剂a:在水中的0.1%甲酸和流动相溶剂b:在乙腈中的0.1%甲酸来通过三次乙腈梯度循环以从捕集柱中洗脱经清洁的肽。梯度从0%溶剂b开始,在30秒内增加到60%溶剂b,然后在30秒内降低到0%溶剂b,并且该1分钟梯度步骤以1ml/min的跟随速率重复三次,随后用100%溶剂b以2ml/min的高速冲洗1分钟。在初始梯度变化发生后30秒,收集后续物,并在冲洗步骤结束后1分钟停止该收集。收集了总共4.5ml的新抗原肽悬浮液,其中估计含有30%乙腈和0.1%甲酸。将收集的新抗原悬浮液直接上样到填充有孔径为1.7μm颗粒的sec柱(waters,ma)上。在分析之前,将neo-sec梯(complete omics,md)加入到系统,以界定收集新抗原的边界。监测特征色谱峰以指示收集的起点(代表2000da的峰)和终点(代表800da的峰)。收集含有分离的新抗原肽的流出液,并在质谱分析前进行冷冻干燥。
94.质谱方法的开发
95.合成在患者癌症基因组中侧翼有基因突变的重同位素标记的新抗原肽。用两步法对检测参数进行优化。步骤1)用理论碰撞能量以及低于和高于理论值5ev的两个额外碰撞能量(对于每次跃迁,总共三个碰撞能量值)检测每种肽的所有可能的离子(从第一个到最后一个)。选择最高丰度的跃迁来用于下一轮优化。步骤2)对从在先步骤中选择的高丰度跃迁(对于肽靶标的每个电荷状态,》20次跃迁)进行进一步优化,其中对于每次跃迁,测试9个碰撞能量值,包括理论碰撞能量值以及低于和高于理论值的步长为2ev的4个阶梯值。经过两轮优化后,手动调整检测参数,以避免从参考人类肿瘤样品制备的valid-neo基质中共同检测到的杂质产生假阳性信号,并且选择平均8至10个跃迁作为每个靶标的特征跃迁。在每批分析之前和之后,使用制造商的调谐混合物,然后使用myprot-srm调谐升压器(tuning booster)(complete omics,md)对安捷伦6495c三重四极杆质谱仪进行调谐。在每次测定之前,为了确保质谱仪在整个研究中的稳定和一致的性能,对myprot-srm性能标准品(complete omics,md)进行分析,其中该myprot-srm性能标准品是跨越宽范围质量(m/z 100-1400)和大范围疏水性的标准肽的混合物。每次运行前记录系统性能得分。
96.预调整系统以确保最高灵敏度
97.为了实现测定的最高灵敏度,开发了一种策略,以通过用与被检测肽“相似”的肽对系统进行预调整和共处理来确保样品损失最小。用于确保测定回收率最大的肽称为maxrec肽。创建maxrec预测算法,以基于期望从线路中检测到的靶标肽的序列、疏水性和可
检测性(在质谱仪中检测到的信号强度)来生成maxrec肽序列。本研究中使用的maxrec肽序列示出在表1中,其中粗体和下划线字体的氨基酸残基代表感兴趣的突变(即靶标突变),斜体字体的氨基酸残基代表maxrec肽中使用的改变的残基。所有maxrec肽都是以高纯度(》99.9%)来合成。在每次测定之前,将含有浓度为100飞摩尔/μl的maxrec肽的缓冲体系注入valid-neo线路。通过线路的maxrec肽的浓度远远高于从临床样品中的靶标肽中可能观察到的浓度。在注入临床样品前,用nc缓冲液冲洗valid-neo线路30分钟,以耗尽过量的未结合maxrec肽。
98.表1.本研究中使用的maxrec肽
[0099][0100]
数据存储
[0101]
本文中报道的数据经由proteome xchange存储于peptideatlas srm实验库(passel)(标识符pass01588)。
[0102]
结果
[0103]
为了最大限度地从肿瘤组织样品回收hla分子,在不解冻样品的情况下快速将冷冻组织匀浆化成单细胞粉末至关重要。为此,开发了一种称为“uniceller”的设备,其能够对冷冻组织块施加强冲击力(~10000psi)。组织粉末通过uniceller来生产,然后被快速溶解在新抗原裂解(nl)缓冲液(材料和方法)中,接着重复移液和程序化超声处理(材料和方法)。通过该过程,表明能够从组织样品中提取几乎100%的hla分子,这代表了与使用包括杜恩斯匀浆器(dounce homogenizer)、探头超声波破碎仪和珠式破碎器(bead ruptor)在内的传统方法相比,更高的回收效率(参见图4,其显示了uniceller和其它三种传统方法之间的回收效率的比较结果。具体对于该实验,通过不同的方法处理相同量(50mg)的肿瘤组织来提取hla分子,上述不同方法包括使用杜恩斯匀浆器、探头超声波破碎仪、珠式破碎器和uniceller。w6/32抗体用于印迹)。使用uniceller时观察到的较高产量表明,作为主要位于细胞表面的蛋白质复合物,hla分子在提取过程中可能容易受到温度变化和液体悬浮液
中的苛刻机械力的影响。此外,来自uniceller组的沉淀物中几乎没有hla分子,这表明当施加强机械冲击力以快速产生单细胞水平干组织粉末,然后立即在裂解缓冲液中进行适度但重复的hla提取时,能够实现较高的提取效率。
[0104]
处理从uniceller组织裂解物获得的沉淀物以提取基因组dna(参见图1)。使用专有的双rna探针捕获一组选定的癌症驱动基因的单链外泌区,并通过deeper-seq线路进行测序(wang et al.,2017)。只有当观察到点突变时,即两条dna链上的互补残基对来自相同的dna双链分子时,才进行突变调用,如前文所述(wang et al.,2017)。选择在高频率突变的癌症驱动基因k-ras和tp53以及稍微低频率突变的驱动基因idh2中带有热点突变的9个肿瘤样品来用于进一步评估潜在的新抗原呈递。
[0105]
基于抗体柱的亲和色谱法比常规免疫沉淀法更有效且更具成本效益,因此在valid-neo线路中被用于富集hla分子(moser&hage,2010)。为了实现高的富集效率,用相对于从典型样品(50至100mg湿组织,具有50%肿瘤质量)中富集hla分子所需的量20倍过量的抗体(50mg抗体)填充抗体缀合的柱,此外,进行重复的样品上样以确保抗体与新抗原捕获(nc)缓冲液(参见图2a和图2b,材料和方法)中hla分子之间的结合。通过从色谱柱上洗脱并在酸性新抗原洗脱(ne)缓冲液(材料和方法)中孵育,hla复合物被洗脱,并且新抗原肽与hla分子上解离(参见图2a和图2b)。然后通过新抗原中和(nn)缓冲液使hla富集柱再生。该色谱柱在对于本研究中分析的新抗原没有观察到明显的性能损失下可用于至少30次富集和洗脱过程(参见图5,示出了valid-neo线路分析的再现性评价结果。具体对于该实验,通过相同neotrue valid-ne线路对10个肿瘤样品进行处理,以评估它们的内源性新抗原呈递。对于每种新抗原,进行三次重复的neotrue valid-neo测定。对于每种新抗原的检测,在第1次重复与第2次重复之间以及第2次重复与第3次重复之间不同抗原,存在9次neotrue valid-neo测定。
[0106]
通过填充有c18小孔球形二氧化硅颗粒(直径)的捕集柱从新抗原肽中分离hla分子和其他大蛋白。新抗原(分子量约1.5kda)明显小于hla分子(分子量约41kda),并将进入孔中,因此被主要位于孔内的c18基质有效保留。大多数hla分子和其他大蛋白不能被柱有效保留。然后用0.1%甲酸清洗结合到捕集柱上的新抗原,以除去hla分子和其他杂质(参见图2a和图2b)。然后将新抗原洗脱到由30%乙腈和0.1%甲酸组成的悬浮液中。向悬浮液中加入neo-sec梯(complete omics inc,md),该neo-sec梯包含具有2000da和800da特征分子量的两组肽(材料和方法),并使悬浮液通过尺寸排阻柱(下文简称为sec或sec柱)进行分级(参见图2a和图2b)。在洗脱过程中,通过二极管阵列检测器(即dad)持续测量280nm波长处的吸光度。在观察到2000da的特征峰后开始收集新抗原级分,在观察到800da的特征峰前停止收集(参见图6,显示了通过sec柱纯化新抗原的hplc色谱图。具体地,新抗原样品与neo-sec梯一起上样到sec柱上。观察在2000da和800da处的两个特征峰,这标志着含新抗原级分的边界)。收集两个特征峰之间的流出液并冻干。然后,在预先优化的条件(材料和方法)对新抗原样品经历valid-neo质谱分析。
[0107]
为了进一步提高回收率和线路的灵敏度,开发了一种新抗原回收系统,称为“最大回收(maxrec)系统”。maxrec中的关键要素是一组序列与靶标肽略有不同(仅相差1或2个氨基酸)的肽,并且它们以比内源性新抗原高得多的丰度重新悬浮在maxrec系统中。maxrec肽被设计成模拟靶标肽的物理特征,以便使系统中的非特异性结合机会饱和,从而最小化由
于这种非特异性相互作用导致的靶标肽损失。尽管与靶标肽具有相似的物理特征,但maxrec肽在化学上是不同的,并且基于现代质谱仪的高分辨率能够容易地与新抗原靶标区分开来(参见图2a和图2b)。valid-neo系统中的三重四极杆质谱仪设置为完全不察觉maxrec肽,并且maxrec系统的存在不会降低平台的灵敏度,这是此类仪器的特点。对valid-neo新抗原分离系统进行预调整,并向样品中加入maxrec肽(参见上表1)。通过maxrec系统,能够实现本研究中所有新抗原肽的检测性能的显著提高,其中灵敏度平均提高了27倍(从2倍到67倍不等)(结果参见下表2)。
[0108]
表2.通过不同方法的新抗原量化
[0109][0110]
已经表明:几乎所有mhc i类相关新抗原的长度为8至12个氨基酸(sarkizova et al.,2020)。对于每个样品,可以以大规模平行的方式直接测定侧翼有癌症驱动基因上最高流行突变位点的所有潜在新抗原序列(每个错义突变位点最多50个可能的新抗原肽),而无需任何预测,从而避免了不确定性(参见图3a至图3f示出的表格)。使用valid-neo线路,对九个新鲜冷冻肿瘤样品进行分析以检测和量化每个患者的个性化新抗原,并对它们在mana-srm和当前集成valid-neo线路之间的相对性能进行比较(参见表2)。在q61位点具有kras突变的患者在每个肿瘤细胞表面上呈现平均6.6个拷贝的新抗原,在侧翼有g12位点的新抗原在每个肿瘤细胞上呈现平均32.1个拷贝。tp53_新抗原的呈递率较低,范围为每个细胞1至8个拷贝,与每个细胞呈递6.1个拷贝的新抗原的idh2_突变相似。每个测定进行三次,并充分评估管线的再现性(参见表2,图5)。
[0111]
讨论
[0112]
传统上,细胞毒性化疗是癌症的主要治疗剂,无论给定患者个体的疾病遗传背景如何(bonadonna&valagussa,1983;chan et al.,2012;savage et al.,2009;yagoda&petrylak,1993)。虽然细胞毒性化疗仍然是许多癌症的一线治疗,但是对癌症的进一步分子表征已经促进了基于小分子或抗体的药剂的开发,这些药剂可以治疗具有相同疾病遗传基础的患者亚群(sawyers,2004;scaltriti&baselga,2006;sharkey&goldenberg,2006)。随着下一代测序(ngs)的发展,明显的是,每个个体的癌症都有其自身的遗传特征,患者之间的癌症驱动基因突变有不同程度的重叠(bagnyukova et al.,2010;cancer genome atlas research et al.,2013;chin et al.,2011;vogelstein et al.,2013)。近年来,高度个性化的癌症治疗方法通过靶向患者的特异性新抗原取得了成功,为这种高度个性化治
discovery science to personalized medicine.nat med,17(3),297-303.
[0123]
9.danilova,l.,anagnostou,v.,caushi,j.x.,sidhom,j.w.,guo,h.,chan,h.y.,suri,p.,tam,a.,zhang,j.,asmar,m.e.,marrone,k.a.,naidoo,j.,brahmer,j.r.,forde,p.m.,baras,a.s.,cope,l.,velculescu,v.e.,pardoll,d.m.,housseau,f.,&smith,k.n.(2018,aug).the mutation-associated neoantigen functional expansion of specific t cells(manafest)assay:a sensitive platform for monitoring antitumor immunity.cancer immunol res,6(8),888-899.
[0124]
10.douglass,j.,hsiue,e.h.,mog,b.j.,hwang,m.s.,dinapoli,s.r.,pearlman,a.h.,miller,m.s.,wright,k.m.,azurmendi,p.a.,wang,q.,paul,s.,schaefer,a.,skora,a.d.,molin,m.d.,konig,m.f.,liu,q.,watson,e.,li,y.,murphy,m.b.,pardoll,d.m.,bettegowda,c.,papadopoulos,n.,gabelli,s.b.,kinzler,k.w.,vogelstein,b.,&zhou,s.(2021,mar 1).bispecific antibodies targeting mutant ras neoantigens.sci immunol,6(57).
[0125]
11.hsiue,e.h.,wright,k.m.,douglass,j.,hwang,m.s.,mog,b.j.,pearlman,a.h.,paul,s.,dinapoli,s.r.,konig,m.f.,wang,q.,schaefer,a.,miller,m.s.,skora,a.d.,azurmendi,p.a.,murphy,m.b.,liu,q.,watson,e.,li,y.,pardoll,d.m.,bettegowda,c.,papadopoulos,n.,kinzler,k.w.,vogelstein,b.,gabelli,s.b.,&zhou,s.(2021,mar 1).targeting a neoantigen derived from a common tp53_mutation.science.
[0126]
12.jurtz,v.,paul,s.,andreatta,m.,marcatili,p.,peters,b.,&nielsen,m.(2017,nov 1).netmhcpan-4.0:improved peptide-mhc class i interaction predictions integrating eluted ligand and peptide binding affinity data.j immunol,199(9),3360-3368.
[0127]
13.lauss,m.,donia,m.,harbst,k.,andersen,r.,mitra,s.,rosengren,f.,salim,m.,vallon-christersson,j.,torngren,t.,kvist,a.,ringner,m.,svane,i.m.,&jonsson,g.(2017,nov 23).mutational and putative neoantigen load predict clinical benefit of adoptive t cell therapy in melanoma.nat commun,8(1),1738.
[0128]
14.lu,y.c.,yao,x.,crystal,j.s.,li,y.f.,el-gamil,m.,gross,c.,davis,l.,dudley,m.e.,yang,j.c.,samuels,y.,rosenberg,s.a.,&robbins,p.f.(2014,jul 1).efficient identification of mutated cancer antigens recognized by t cells associated with durable tumor regressions.clin cancer res,20(13),3401-3410.
[0129]
15.meier,f.,brunner,a.d.,koch,s.,koch,h.,lubeck,m.,krause,m.,goedecke,n.,decker,j.,kosinski,t.,park,m.a.,bache,n.,hoerning,o.,cox,j.,rather,0.,&mann,m.(2018,dec).online parallel accumulation-serial fragmentation(pasef)with a novel trapped ion mobility mass spectrometer.mol cell proteomics,17(12),2534-2545.
[0130]
16.moser,a.c.,&hage,d.s.(2010,apr).immunoaffinity chromatography:an introduction to applications and recent developments.bioanalysis,2(4),769-790.
[0131]
17.ott,p.a.,hu,z.,keskin,d.b shukla,s.a.,sun,j.,bozym,d.j.,zhang,w.,luoma,a.,giobbie-hurder,a.,peter,l.,chen,c.,olive,o.,carter,t.a.,li,s.,lieb,d.j.,eisenhaure,t.,gjini,e.,stevens,j.,lane,w.j.,javeri,i.,nellaiappan,k.,salazar,a.m.,daley,h.,seaman,m.,buchbinder,e.i.,yoon,c.h.,harden,m.,lennon,n.,gabriel,s.,rodig,s.j.,barouch,d.h.,aster,j.c.,getz,g.,wucherpfennig,k.,neuberg,d.,ritz,j.,lander,e.s.,fritsch,e.f.,hacohen,n.,&wu,c.j.(2017,jul13).an immunogenic personal neoantigen vaccine for patients with melanoma.nature,547(7662),217-221.
[0132]
18.riaz,n.,morris,l.,havel,j.j.,makarov,v.,desrichard,a.,&chan,t.a.(2016,aug).the role of neoantigens in response to immune checkpoint blockade.int immunol,28(8),411-419.
[0133]
19.sahin,u.,derhovanessian,e.,miller,m.,kloke,b.p.,simon,p.,lower,m.,bukur,v.,tadmor,a.d.,luxemburger,u.,schrors,b.,omokoko,t.,vormehr,m.,albrecht,c.,paruzynski,a.,kuhn,a.n.,buck,j.,heesch,s.,schreeb,k.h.,muller,f.,ortseifer,i.,vogler,i.,godehardt,e.,attig,s.,rae,r.,breitkreuz,a.,tolliver,c.,suchan,m.,martic,g.,hohberger,a.,sorn,p.,diekmann,j.,ciesla,j.,waksmann,o.,bruck,a.k.,witt,m.,zillgen,m.,rothermel,a.,kasemann,b.,langer,d.,bolte,s.,diken,m.,kreiter,s.,nemecek,r.,gebhardt,c.,grabbe,s.,holler,c.,utikal,j.,huber,c.,loquai,c.,&tureci,o.(2017,jul 13).personalized rna mutanome vaccines mobilize poly-specific therapeutic immunity against cancer.nature,547(7662),222-226.
[0134]
20.sarkizova,s.,klaeger,s.,le,p.m.,li,l.w.,oliveira,g.,keshishian,h.,hartigan,c.r.,zhang,w.,braun,d.a.,ligon,k.l.,bachireddy,p.,zervantonakis,i.k.,rosenbluth,j.m.,ouspenskaia,t.,law,t.,justesen,s.,stevens,j.,lane,w.j.,eisenhaure,t.,lan zhang,g.,clauser,k.r.,hacohen,n.,carr,s.a.,wu,c.j.,&keskin,d.b.(2020,feb).a large peptidome dataset improves hla class iepitope prediction across most of the human population.nat biotechnol,38(2),199-209.
[0135]
21.savage,p.,stebbing,j.,bower,m.,&crook,t.(2009,jan).why does cytotoxic chemotherapy cure only some cancers?nat clin pract oncol,6(1),43-52.
[0136]
22.sawyers,c.(2004,nov 18).targeted cancer therapy.nature,432(7015),294-297.
[0137]
23.scaltriti,m.,&baselga,j.(2006,sep 15).the epidermal growth factor receptor pathway:a model for targeted therapy.clin cancer res,12(18),5268-5272.
[0138]
24.schumacher,t.n.,&schreiber,r.d.(2015,apr 3).neoantigens in cancer immunotherapy.science,348(6230),69-74.
[0139]
25.sharkey,r.m.,&goldenberg,d.m.(2006,jul-aug).targeted therapy of cancer:new prospects for antibodies and immunoconjugates.ca cancer j clin,56
(4),226-243.
[0140]
26.vogelstein,b.,papadopoulos,n.,velculescu,v.e.,zhou,s.,diaz,l.a.,jr.,&kinzler,k.w.(2013,mar 29).cancer genome landscapes.science,339(6127),1546-1558.
[0141]
27.wang,q.,douglass,j.,hwang,m.s.,hsiue,e.h.,mog,b.j.,zhang,m.,papadopoulos,n.,kinzler,k.w.,zhou,s.,&vogelstein,b.(2019,nov).direct detection and quantification of neoantigens.cancer immunol res,7(11),1748-1754.
[0142]
28.wang,q.,wang,x.,tang,p.s.,o’leary g,m.,&zhang,m.(2017,jun 13).targeted sequencing of both dna strands barcoded and captured individually by rna probes to identify genome-wide ultra-rare mutations.sci rep,7(1),3356.
[0143]
29.yagoda,a.,&petrylak,d.(1993,feb 1).cytotoxic chemotherapy for advanced hormone-resistant prostate cancer.cancer,71(3 suppl),1098-1109.
[0144]
30.zubarev,r.a.,&makarov,a.(2013,jun 4).orbitrap mass spectrometry.anal chem,85(11),5288-5296.
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1