生物活性的苯酚盐离子络合物的制作方法
1.本发明总体上涉及新的活性剂的离子型金属、磷鎓和铵苯酚盐及其用途。
2.背景
3.自然界中存在各种各样的包含苯酚基团的生物活性材料,并且在人类饮食中发挥不可或缺的作用。含苯酚的材料还存在于若干种重要的合成药物和天然药物中,例如阿扑吗啡、埃斯特纶(estron)、雌二醇、丙泊酚、邻苯基苯酚、左旋多巴(l-dopa)、多柔比星、大麻素活性分子诸如四氢大麻酚(thc)和大麻二酚(cbd)以及包含酪氨酸的肽分子或蛋白质分子。另外的实例是沙波立沙钠(salcaprozate sodium)(snac),一种用于肽和维生素的口服吸收促进剂;天然的多酚和修饰的多酚诸如姜黄素、黄酮、黄烷醇、花青素、没食子酸、咖啡酸、百里酚、水杨酸、羟基二苯乙烯类、selariscinin等等。
4.众所周知,酚类物质,诸如邻儿茶酚或对儿茶酚,由于氧化或与醛类和亲核体的反应而在水溶液中具有低的化学稳定性。还已知的是,这些酚类药物常常具有低的溶解度和生物利用度。因此,口服递送受到限制,导致生产用于口服施用的前药替代品。在一些情况下,对于减少或增强透过皮肤的渗透,诸如可以通过苯酚盐形成实现的防晒分子存在需求。
5.国际公布号wo2016/127111教导了cbd和与三个七氟烷基配体络合的镧系金属的加合物,其中cbd的oh未转化为苯酚盐阴离子。
6.一般说明
7.为了克服与含苯酚的活性材料相关的许多困难,本文公开的技术的发明人已经将这些活性材料转换为呈盐的形式的前药实体(pro-drug entity),这赋予它们:
[0008]-改进的化学稳定性;
[0009]-对氧化的降低的敏感性;以及
[0010]-改进的物理性质,诸如改进的水溶性、结晶的容易性、纯度以及其他。
[0011]
通过将化合物转换为它们相应的盐,本发明人已经允许形成多药物媒介物,该多药物媒介物包含在单一剂型内的一种或更多种、相同或不同的药物实体。换句话说,单一阳离子可以与两种或更多种、相同或不同的药物实体缔合,所述药物实体可以作为单元被施用或使用。
[0012]
因此,在最一般的意义上,本发明提供了一种分离的材料或至少一种含苯酚的活性材料的苯酚盐形式,其中所述分离的材料包含一种或更多种苯酚盐物质和呈金属盐、磷鎓或铵的形式的抗衡离子(阳离子),并且其中所述活性材料不是苯酚(c6h6o)、甲基苯酚、溴苯酚、二溴苯酚、三溴苯酚、五氯苯酚、双酚a、四溴双酚a、间苯二酚、氢醌、氢醌或萘酚。一种或更多种苯酚盐物质以使得能够实现增加的稳定性、在水性介质中有效的溶解、更有效的递送和改进的生物利用度的形式呈现。当本发明的材料产物中的每一种材料产物解离成含苯酚的化合物和金属阳离子时,本发明的材料一定不是金属加合物也不是金属配位化合物,如本领域中已知的。
[0013]
本发明的材料被视为分离的材料。术语“分离的“意指材料既不是外源的材料(accidental material),也不是保留在生产该材料的汤或介质中并且未被分离的中间材
料。因此,分离的材料具有高纯度。纯度可以是100%或大于80%。纯度还可以取决于对于材料所意图用于的用途或应用中的每种所需的纯度。在一些实施方案中,本发明的分离的材料是如本领域中定义的医药级、化妆品级或农业级。
[0014]
未被分离的材料不构成本发明的一部分。
[0015]
本发明的材料是亲脂性活性材料或水不溶性活性材料的水溶性形式。这种水溶性形式是离子型的,其中抗衡离子是如下文所定义的。不希望受理论的束缚,本发明的材料在施用后在身体中被转化为相应的活性物质,即活性苯酚化合物,或者可以保持离子化。例如,在活性材料是大麻素的情况下,它以其离子化的苯酚盐形式在本发明的材料中呈现,所述离子化的苯酚盐形式在身体中可以被转化回到非未离子化的形式。
[0016]
如本领域中已知的,含苯酚的活性材料是具有芳族烃基团(苯环、苯基环(phenyl ring)、萘基基团、稠合的芳族环以及其他)的化合物,所述芳族烃基团具有与环碳原子中的一个或更多个结合的一个或更多个羟基基团(-oh)。含苯酚的活性材料通常是用于医药、化妆品、兽医或农业的生物活性化合物。活性材料可以是合成的或植物衍生的(phyto-derived)。盐形式是金属盐形式、铵盐形式、硝鎓盐形式或磷鎓盐形式。金属盐形式包含金属阳离子和苯酚盐化合物。换句话说,使金属阳离子和苯酚盐化合物的带电氧原子缔合的键是通常使阳离子和阴离子缔合的离子类型。与金属阳离子形成的离子键的数目取决于金属阳离子的化合价。
[0017]
金属阳离子可以选自单价、二价或更高的化合价的阳离子,其被用于医药中(人类和动物受试者的治疗和诊断两者)、化妆品领域中或农业中。因此,金属阳离子在用于医药应用时必须是药学上可接受的,在用于化妆品应用时必须是化妆品上可接受的,或者在用于农业应用时必须是农业上可接受的。通常,“药学上可接受的”意指适用于施用至人类和动物(特别是哺乳动物),而没有实质性的、主要的或致命的不良事件。“化妆品上可接受的”意指被认为在皮肤区域上使用的水平对人类和动物(特别是哺乳动物)无毒的和安全的,并且“农业上可接受的”暗示材料对植物或其环境是基本上没有损害或毒性的,并且对可能通过偶然的(被动的)暴露或接触或消费、直接地或间接地暴露于该材料的使用者或其他人不是不安全的。
[0018]
阳离子可以选自碱金属、碱土金属和过渡金属。非限制性的实例包括锂、钠、钾、钙、镁、锰、铝、锌、镍、铁、银、金、钡以及其他。
[0019]
在一些实施方案中,金属选自钠、钾和钙。
[0020]
在一些其他实施方案中,材料包含磷鎓阳离子和苯酚盐化合物。
[0021]
铵盐形式是包含铵阳离子和苯酚盐化合物的形式。铵阳离子可以选自任何具有中心带正电氮原子的铵阳离子。换句话说,铵可以具有nh
4+
、nh3r
+
、nh2rr'
+
、nhrr'r”+
或nrr'r”r”'
+
的形式,其中r、r'、r”和r”'中的每一个可以是与氮原子共价连接的官能团,例如烷基基团或芳基基团。例如,在材料包含单一的含苯酚的活性材料,即单一的苯酚盐基团的情况下,阳离子可以是nh
4+
或者可以是nh3r
+
、nh2rr'
+
、nhrr'r”+
和nrr'r”r”'
+
中的任一种,其中r基团中的每一个可以相同或不同,并且独立地选自烷基和芳基。
[0022]
在上文的r基团中的一个或更多个(或每一个)是“烷基”的情况下,它通常包含在1个至20个之间的碳原子,前提是材料保持水溶性。因此,在一些实施方案中,烷基包含呈直链形式或支链形式的在1个和6个之间的碳原子。示例性的烷基基团包括但不限于甲基、乙
基、丙基、异丙基、异丁基、正丁基、仲丁基、叔丁基、异己基以及其他。
[0023]
烷基基团还涵盖“环烷基”基团,即饱和的单环或多环的环体系,在某些实施方案中为3个至10个碳原子,在其他实施方案中为3个至6个碳原子。环烷基的环体系可以包括可以以稠合、桥接或螺环的方式连接在一起的一个环或两个或更多个环。
[0024]
作为“芳基”基团的是包含从6个至20个碳原子的芳族的单环或多环基团。芳族基团包括但不限于未被取代的或被取代的芴基、未被取代的或被取代的苯基和未被取代的或被取代的萘基。
[0025]
芳基基团还涵盖“杂芳基”基团,其在某些实施方案中包括从3个至15个原子的单环或多环的芳族环体系,其中环体系中的原子中的一个或更多个,在一些实施方案中1个至3个,是选自氮、氧、硫和其他非碳原子的杂原子。杂芳基基团可以任选地被稠合至苯基环。
[0026]
在一些实施方案中,铵阳离子可以是nh
4+
或四烷基铵阳离子。四烷基铵阳离子的非限制性实例包括四甲基铵、四乙基铵、四丁基铵、四戊基铵和四己基铵,或混合烷基形式。另一个实例是胆碱,或胆碱衍生物,或多胆碱(poly choline),诸如二胆碱或三胆碱,如本领域中已知的。
[0027]
在一些实施方案中,铵阳离子可以是具有多个胺基团(包括例如伯胺、仲胺和叔胺)的多胺的一部分,所述多胺可以通过使其与多于一种活性苯酚材料缔合而被转化为如本文描述的材料。
[0028]
在材料包含两个或更多个苯酚盐基团(在单一的含苯酚的材料上)的情况下,苯酚盐基团中的每一个苯酚盐基团可以与不同的阳离子离子缔合,例如,一个苯酚盐基团可以与铵阳离子缔合,而另一个苯酚盐基团可以与金属阳离子缔合,或者苯酚盐中的每一个苯酚盐可以与不同或相同的金属阳离子缔合。
[0029]
包含苯酚盐实体的材料是用于医药、化妆品、兽医或农业的任何含苯酚的活性材料(其可以是药物成分、化妆品成分或农业成分),该含苯酚的活性材料已经通过从苯酚-oh基团中摘取氢原子而转换为苯酚盐形式。如本文陈述的,苯酚盐化合物可以在芳族碳环基团(苯环或苯基环)上包含一个或两个或更多个-oh基团。
[0030]
呈金属络合物的形式的材料可以包含一种呈带电形式(阳离子)的金属,该金属与一个或更多个苯酚盐基团离子缔合,其中苯酚盐基团的数目由金属离子的化合价确定。例如,在使用碱金属原子诸如锂的情况下,材料可以包含锂离子和单一苯酚盐化合物。该单一苯酚盐化合物可以是具有一个或更多个-oh基团的化合物;然而,在金属离子和苯酚盐化合物之间的缔合可以是经由单个苯酚盐基团。
[0031]
类似地,金属络合物可以包含二价金属离子和两种苯酚盐化合物,这两种苯酚盐化合物可以相同或不同,即取代苯环的-oh可以在相同的苯环上或在不同的苯环上,或者可选择地,这两种苯酚盐化合物是两种单独的含苯酚的化合物,它们可以是相同的或不同的分子。换句话说,与苯酚盐化合物形成离子键的金属离子、铵离子或磷鎓离子可以作为以下中的一种或更多种被提供:
[0032]
(1)取代单个苯环的-oh基团与一个金属离子、铵离子或磷鎓离子形成离子键。
[0033]
(2)取代不同的苯环但在相同的分子(诸如多酚化合物或杂环化合物)上的-oh基团与一个或更多个金属离子、铵离子或磷鎓离子形成离子键。
[0034]
(3)在两个分开的分子(相同或不同)上取代相同或不同的苯环的-oh基团与一个
或更多个金属离子、铵离子或磷鎓离子形成离子键。
[0035]
(4)以上的任何组合。
[0036]
在金属离子和苯酚盐基团之间的离子键可以采用一种金属离子或多于一种金属离子,这取决于其化合价,如本文描述的。例如,二价离子可以形成一个或两个呈上文(1)至(4)的形式中的任何形式的离子键。
[0037]
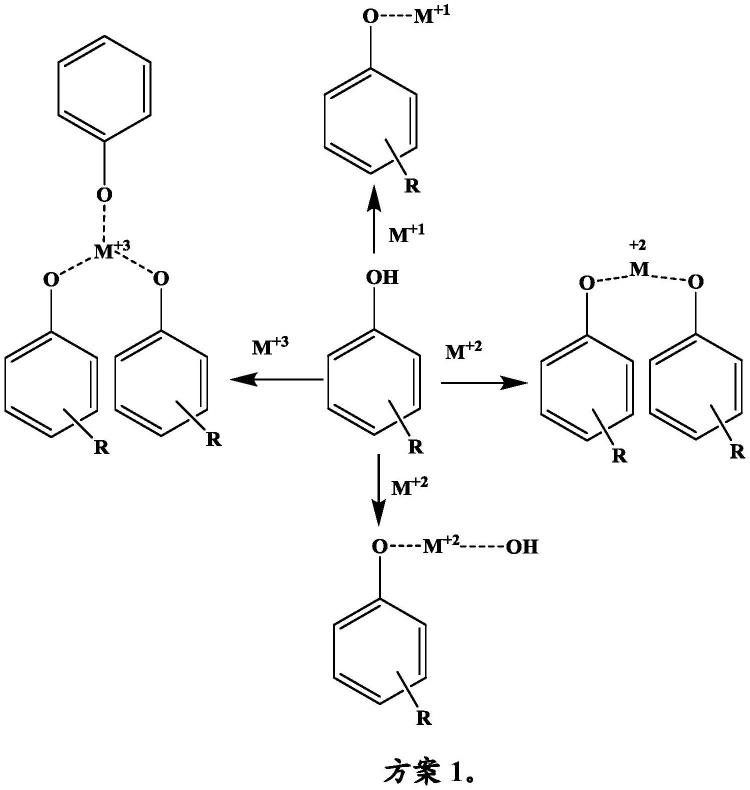
下文的方案1呈现了本发明的金属络合物中的一些的一般描绘,其中r=表示在含苯酚的活性材料上的取代基,该取代基可以是如上文定义的烷基、芳基或杂环基团,或者与苯酚稠合的环;并且m是单价金属离子、二价金属离子和三价金属离子或铵阳离子:
[0038][0039]
在上文的方案1中,描绘了本发明的四个不同的示例性实施方案。在每种情况下,通常描绘苯酚,其中r是取代基基团、与苯酚稠合的环或含苯酚的材料的任何衍生物。所描绘的苯酚可以包含单个酚类-oh基团或多于一个酚类-oh基团。换句话说,所指示的苯酚中的每一个可以包含一个、两个或更多个与芳族碳环直接缔合的羟基基团。
[0040]
在方案1中描绘的第一种情况下,含苯酚的材料与单价金属m
+1
(呈碱的形式,所述碱可以是碱金属氢化物、碱金属醇盐等,通常呈m-x的形式,其中m是金属阳离子,并且x代表阴离子)或与氨(或其等效物)反应,以分别在苯酚盐化合物(在与碱相互作用时形成的苯酚的阴离子)和金属阳离子或铵阳离子之间形成络合物。
[0041]
在方案1中描绘的第二种情况和第三种情况下,含苯酚的材料与二价金属m
+2
(呈碱
的形式,所述碱可以是碱土金属氢化物、碱土金属醇盐等,通常呈m-x的形式,其中m是二价金属阳离子,并且x代表二价阴离子(dianion))反应,以与两个苯酚盐基团形成金属盐或者在单个苯酚盐基团和另一个阴离子(不是苯酚盐)之间形成金属盐。这两个苯酚盐基团可以是相同的或不同的。
[0042]
在方案1中描绘的第四种情况下,含苯酚的材料与三价金属m
+3
(呈碱的形式,所述碱可以是三价金属三氢化物、三价金属三醇盐等,通常呈m-x的形式,其中m是三价金属阳离子,并且x代表三阴离子)反应,以在三个苯酚盐基团和三价金属阳离子之间形成金属络合物。在一些实施方案中,三个苯酚盐化合物是相同的,并且在其他情况下它们是不同的。在一些实施方案中,这三个中的两个是相同的,并且第三个是不同的。在一些实施方案中,这三个中的每一个彼此不同,全部是关于上文描述的。苯酚盐的抗衡离子可以通过经由在溶液中的置换交换抗衡离子来制备,例如,苯酚钠盐可以通过以下被转化为钙盐或胆碱盐:浸泡在钙盐或胆碱盐的溶液中,使得钠抗衡离子被溶液中的离子替换。
[0043]
在两个酚类-oh基团在相同的芳族环上是邻位的或以其他方式彼此靠近的儿茶酚分子的情况下,它们可以与一个二价金属离子或三价金属离子诸如钙、镁或铁形成离子络合物,如下文方案2中描绘的,其中r和m是如关于方案1所定义的:
[0044][0045]
在二价金属离子或三价金属离子诸如铁或氧化铝的情况下,可以形成与其他官能团的混合相互作用。例如,钙离子可以与苯酚盐阴离子和羧酸盐阴离子结合(如下文方案3中示出的),或者在铁的情况下,一个或两个苯酚盐阴离子与三价铁离子结合,并且第三化合价可以形成配位加合物。
[0046][0047]
在本发明的材料中,其中两个或更多个苯酚盐基团与金属阳离子缔合,苯酚盐基团中的每一个可以是活性材料,苯酚盐基团中的至少一个可以是活性药物材料,苯酚盐基
团中的仅一个是活性材料,或者苯酚盐基团中的大多数是活性材料。在一些实施方案中,其中两个或更多个苯酚盐化合物与金属阳离子缔合,苯酚盐化合物中的一种是活性材料并且苯酚盐化合物中的另一种是不同的活性材料或非活性药物材料。
[0048]
如本文使用的,活性材料是典型地分别用于以下的任何治疗材料、化妆品材料或兽医材料:用于治疗或预防紊乱或疾病状态的方法中,或者用于改善皮肤区域的状态或状况的方法(或通常用于化妆品的使用方法)或改善与人类或动物身体有关的任何状况的方法中,或者用于治疗或促进植物疾病和紊乱的方法或通常用于植物保护或其他农业方法中。
[0049]
在农业中使用的含苯酚的化合物是被用作除草剂、杀真菌剂、杀昆虫剂、灭鼠剂、植物生长调节剂、激素、引诱剂、驱避剂、营养物、肥料以及其他的任何这样的化合物。
[0050]
苯酚盐化合物或含苯酚的生物活性物不是苯酚(c6h6oh)或其任何被取代的形式。
[0051]
在一些实施方案中,含苯酚的生物活性物是包含一个酚类-oh基团和至少一个其他官能团的活性材料。在一些实施方案中,活性材料是包含两个或更多个酚类-oh基团和至少一个其他官能团的材料。在一些实施方案中,活性材料是包含多个酚类-oh基团和任选地至少一个其他官能团的材料。在一些情况下,带有-oh基团的芳族环可以包含一个、有时2个、有时3个并且有时4个、有时5个并且有时6个-oh基团。
[0052]“至少一个其他官能团”是与-oh不同的基团,其可以是取代芳族环上不是羟基基团(-oh)并且不是氢原子(-h)的位置的任何官能团。官能团可以选自卤原子、卤代烷基、硝基、烷氧基、烷基硫基、芳氧基、芳基硫基、卤代烷氧基、羧酸、酯、醚、酰胺、烷基氨基羰基、羧酰胺、芳基烷基氨基羰基、芳基氨基羰基、芳氧基羰基、烷基、烯基、炔基、氮杂亚烷基、环烷基、环烯基、环炔基、芳基、杂芳基、杂环基、亚烷基、亚烯基、亚炔基、亚环烷基、亚环烯基、亚环炔基、亚芳基、亚杂芳基、亚杂环基、亚磺酰基、磺基、胺以及其他。
[0053]
在一些实施方案中,含苯酚的活性材料包括稠合的芳族基团,即被羟基基团直接取代的苯环或苯基环。稠合的环体系可以呈与环状碳环或杂环(还包含一个或更多个杂原子诸如n、s、o)稠合的苯酚基团的形式,所述环状碳环或杂环可以是芳族的或非芳族的。这样的体系通常被称为杂环体系。
[0054]
在一些实施方案中,含苯酚的活性材料包含单个苯酚基团,诸如简单酚类(然而,不包括苯酚及其简单衍生物)和苯醌类。这样的化合物的非限制性实例包括儿茶酚、羟基醌和2,6-二甲氧基苯醌。
[0055]
在一些实施方案中,含苯酚的活性材料是酚酸或酚醛,诸如没食子酸和水杨酸。
[0056]
在一些实施方案中,含苯酚的活性材料选自苯乙酮类、酪氨酸衍生物和苯乙酸类。非限制性的实例包括3-乙酰基-6-甲氧基苯甲醛、酪醇和对羟基苯乙酸。
[0057]
在一些实施方案中,含苯酚的活性材料选自羟基肉桂酸类、苯基丙烯类和香豆素类。非限制性的实例包括咖啡酸、阿魏酸、肉豆蔻醚、丁子香酚、伞形酮、秦皮乙素、岩白菜素和丁子香色酮。
[0058]
在一些实施方案中,含苯酚的活性材料选自萘醌类。非限制性的实例包括胡桃醌和石苁蓉萘醌。
[0059]
在一些实施方案中,含苯酚的活性材料选自呫吨酮类(xanthonoids),诸如芒果苷。
[0060]
在一些实施方案中,含苯酚的活性材料选自芪类(stilbenoids)和蒽醌类。非限制性的实例包括白藜芦醇和大黄素。
[0061]
在一些实施方案中,含苯酚的活性材料选自查尔酮类化合物(chalconoids)、黄酮类化合物、异黄酮类化合物(isoflavonoids)和新黄酮类化合物(neoflavonoids)。非限制性的实例包括槲皮素、花青素和染料木黄酮。
[0062]
在一些实施方案中,含苯酚的活性材料选自木脂素类和新木脂素类。非限制性的实例包括松脂醇和优西得灵(eusiderin)。
[0063]
在一些实施方案中,含苯酚的活性材料选自生物黄酮类化合物(bioflavonoids)。非限制性的实例包括阿曼托黄素(amentoflavone)。
[0064]
在一些实施方案中,含苯酚的活性材料选自木质素类、儿茶酚黑色素类(catechol melanins)、聚黄烷类(flavolans)(诸如缩合丹宁)、多酚蛋白类和多酚类。非限制性的实例包括覆盆子鞣花单宁和鞣酸。
[0065]
在一些实施方案中,含苯酚的活性材料是选自雌二醇、甲状腺素、左旋甲状腺素、三碘甲状腺原氨酸、肾上腺素以及其他的激素。
[0066]
这样的含苯酚的活性材料的另外的非限制性实例包括大麻素、非诺多泮、酪氨酸、二甲酚、百里酚、丙泊酚、阿朴吗啡、吗啡及其衍生物、米托蒽醌、多柔比星、六氯酚、对乙酰氨基酚、对香豆酸、3,4-二羟基苯甲酸、4-羟基苯甲酸、对羟基苯甲酸丁酯、香草酸、愈创木酚、咖啡酸、托特罗定、雷洛昔芬、东莨菪亭、紫花前胡醇、多巴胺、左旋多巴、姜黄素、多酚、天碱(tianine)以及其他。
[0067]
在一些实施方案中,含苯酚的活性材料是沙波立沙钠(snac)。
[0068]
在一些实施方案中,含苯酚的活性材料是塔匹那洛夫(tapinarof)。
[0069]
在一些实施方案中,含苯酚的活性材料是肉桂提取物(cinnamon extract)。
[0070]
在一些实施方案中,含苯酚的活性材料是以下中的任一种或更多种:美沙拉嗪、沙丁胺醇、吡布特罗、辣椒素、沙美特罗、维兰特罗、巴柳氮、拉贝洛尔、霉酚酸、吡哆醇、苯肾上腺素、依酚氯铵(edrophonium)、扑热息痛、莫诺苯宗、他喷他多、间羟胺、甲酪氨酸、羟甲唑啉、大麻隆(nabilone)、二氟尼柳、奥沙拉嗪、碘塞罗宁钠(liothyronine sodium)、去甲文拉法辛(desvenlafaxine)、罗替戈汀、酚妥拉明、羟保泰松、阿莫地喹、奥达特罗(olodaterol)、曲格列酮、艾曲波帕(eltrombopag)、依伐卡托(ivacaftor)、茚达特罗、头孢羟氨苄、头孢丙烯、四氢大麻酚、雌二醇、戊酸雌二醇、环戊丙酸雌二醇、左洛啡烷(levallorphan)、羟吗啡酮、纳布啡、丁丙诺啡、布托啡诺、纳洛酮、左啡诺(levorphanol)、纳曲酮、地佐辛、吗啡、纳洛塞醇(naloxegol)、甲基纳曲酮、纳美芬、美他环素、萨瑞环素(sarecycline)、奥马环素(omadacycline)、伊拉瓦环素(eravacycline)、马烯雌酮、氟美他酚(flutemetamol)、己烯雌酚、双烯雌酚、普罗布考、米托蒽醌、巴多昔芬、雷洛昔芬、阿布他明(arbutamine)、多巴酚丁胺、马索罗酚、大麻二酚、特布他林、奥西那林、非诺多泮、去甲肾上腺素、异肾上腺素、异丙肾上腺素、异他林、屈昔多巴、卡比多巴、普罗托醇(protokylol)、阿朴吗啡、恩他卡朋、托卡朋、伊达比星、道诺霉素、多柔比星、表柔比星和戊柔比星(valrubicin)。
[0071]
在一些实施方案中,含苯酚的活性材料是大麻素材料。大麻素材料可以选自四氢大麻酚(thc)、四氢大麻酚酸(thca)、大麻二酚(cbd)、大麻二酚酸(cbda)、大麻酚(cbn)、大
麻萜酚(cbg)、大麻环萜酚(cbc)、大麻环醇(cbl)、次大麻酚(cannabivarin)(cbv)、四氢次大麻酚(thcv)、次大麻二酚(cbdv)、次大麻环萜酚(cannabichromevarin)(cbcv)、次大麻萜酚(cannabigerovarin)(cbgv)、大麻萜酚单甲醚(cbgm)、大麻艾尔松(cannabielsoin)(cbe)、大麻二吡喃环烷(cannabicitran)(cbt)以及其他。
[0072]
在一些实施方案中,大麻素是thc或cbd。
[0073]
在一些实施方案中,材料包含至少一种大麻素的苯酚盐形式以及选自钠、钾和钙的一种或更多种金属阳离子。
[0074]
与含苯酚的活性材料例如cbd的金属络合物通过由于从芳族环上移除电子密度而防止氧化或硫醇取代来改进化学稳定性;由于离子性质的存在而改进水溶性,这在添加稀酸以重整苯酚类的情况下可以是容易地可逆的。
[0075]
大麻素材料可以是合成大麻素或天然大麻素、印度大麻提取物(cannabis extract)或其级分。在最广泛的意义上,该术语指的是对内源性大麻素受体(cb1和cb2)起多种亲和力的作用的整个类别的化合物,大麻素/大麻素激动剂/大麻素相关的化合物。这组配体包括内源性大麻素(由人类和动物天然地产生)、植物大麻素(在印度大麻和一些其他的植物中发现)和合成大麻素(人工制造)。这样的术语还指的是源自或模仿在印度大麻植物中的天然大麻素的经典大麻素。
[0076]
在一些实施方案中,大麻素材料(包括酚类基团)可以选自上文的材料中的任一种。
[0077]
基于印度大麻的组合物的另一个优点在于其增加的萜烯类、倍半萜烯类、胡萝卜素类、黄酮类化合物的含量,这些化合物以多种组合和比存在,并且在更用户友好的意义上,有助于吸收、活性并且另外有助于赋予风味、气味和颜色的性质。因此,含苯酚的活性材料可以是存在于印度大麻植物中并且其中包含酚类基团的萜烯类、倍半萜烯类、胡萝卜素类、黄酮类化合物中的任一种。
[0078]
已经通过在环境条件下的简单的化学反应制备了多种药物实体的酚类盐。方案1描绘了用于其准备的一般方法。在形成的络合物中是大麻素,诸如cbd,如下文的方案4、方案5和方案6中示出的。
[0079]
如下文的方案4中描绘的,cbd金属络合物从在碱性条件下cbd与对应的金属氢氧化物或氯化物盐的反应而容易地制备。cbd络合物通过添加稀hcl被转化回到苯酚形式。每个m是如上文关于方案1所定义的。
[0080][0081]
本发明的金属络合物显示出显著的水溶性(10mg/ml,比cbd更可溶,接近800x),如下文的表1中总结的。
[0082]
多价金属中心可以用于与多个cbd分子形成盐(方案5)。经由cbd的苯酚类的金属盐还可以提高cbd的化学稳定性和物理性质。方案5示出了cbd二聚体或三聚体金属盐,并且描绘了它们从在碱性条件下cbd与对应的二价金属硫酸盐或氯化物盐的反应的简单合成。cbd盐通过添加稀hcl被转化回到苯酚形式。每个m是如关于方案1所定义的。
[0083][0084][0085]
cbd还连同其他酚类生物活性化合物诸如左旋多巴、多巴胺和姜黄素一起反应(方案6)。cbd异二聚体金属盐(heterodimer metal salt)从在碱性条件下cbd和其他酚类化合物(左旋多巴、多巴胺、姜黄素)与对应的二价金属硫酸盐或氯化物盐的反应来合成。
[0086][0087][0088]
本发明还提供了具有钠或钙的cbd盐。因此金属络合物是钠-cbd和钙双-cbd。
[0089]
本发明还提供了至少一种如所定义的金属阳离子和至少一种大麻素的金属盐,其中所述至少一种大麻素是分离的大麻素或存在于组合物中,例如印度大麻植物提取物,所述组合物具有至少一种不是呈本发明的金属络合物的形式的其他大麻素。这样的混合形式可以通过以下来形成:在碱性条件下使印度大麻提取物与金属阳离子反应以产生苯酚盐离子,以形成金属阳离子并且与金属阳离子缔合。由于并非所有天然存在的大麻素或存在于大麻素提取物中的天然存在的植物组分都包含苯酚基团,因此应发生在苯酚生物活性物和金属阳离子之间的选择性缔合。在一些情况下,可以在大麻素诸如cbd和印度大麻植物中包含的另一种材料诸如萜烯、倍半萜烯、胡萝卜素或黄酮类化合物之间形成离子键。
[0090]
一般说来,本发明的含苯酚的活性材料以分离的形式、以提取物形式、以纯化的形式以及作为药物活性物在用于医药用途、化妆品用途、兽医用途或农业用途的制剂中被提供。在活性物被分离的情况下,它们可以呈基本上纯的形式,即以在95%和100%之间的纯
度。
[0091]
本发明还提供了包含至少一种根据本发明的材料的药物制剂或组合物、化妆品制剂或组合物、农业制剂或组合物、食品制剂或组合物或者兽医制剂或组合物。
[0092]
在许多实施方案中,本发明的组合物还可以包括以多种组合的抗氧化剂、吸收促进剂、赋予颜色和风味的剂、防腐剂、稳定剂、盐。本领域中熟知的多种甜味剂、味道改良剂(taste modifier)、抗氧化剂、防腐剂包括味道改良剂诸如人工甜味剂,调味剂如草莓和薄荷油,例如植物甜味剂、糖、蜂蜜、甜叶菊(stevia)、甜菊醇(steviol)、糖苷、柠檬酸盐、酸类、薄荷醇、大茴香(anise)、桉树油、小茴香(fennel),天然抗微生物物质和天然抗氧化剂(例如murta、牛至、迷迭香、琉璃苣的提取物),抗氧化剂诸如维生素e(生育酚)和维生素c及其衍生物,丁基化羟基茴香醚(bha),被识别为gras的丁基化羟基甲苯(bht),以及硫化物;允许口服施用的任何甜味剂,诸如糖、葡萄糖、三氯蔗糖(sucralose)、甘氨酸、甜蜜素(cyclamate)、蔗糖、糖精、果糖、麦芽糖、甜叶菊提取物、糖精钠;盐类,诸如nacl、柠檬酸盐以及其他。
[0093]
应当注意,本发明的组合物适用于如医药科学或兽医科学中已知的任何类型的施用。本发明的制剂可以被配置用于以下或被工程化用于以下或适于以下或被选择用于以下或用于以下:局部施用(例如,以乳霜(cream)和软膏(ointment)的形式)、肠内施用(例如,包括所有涉及经由胃肠道施用诸如口服施用的全身施用途径)或肠胃外施用(例如,包括所有不涉及经由胃肠道施用的全身施用途径)。对本发明的制剂的施用有效的非限制性施用途径包括口服施用、舌下施用、粘膜施用、气溶胶施用、吸入施用、肠胃外施用、皮下施用、静脉内施用、肌肉内施用、腹膜内施用、直肠施用和阴道施用。
[0094]
如已经提及的,本发明的另一个方面是提供包括先前描述的制剂的用于口服施用或舌下施用的剂型。
[0095]
另外,本发明的材料可以通过与多种碱诸如乳化碱或水溶性碱混合而被制成栓剂。适合于阴道施用的制剂可以作为阴道栓剂、卫生棉条、乳霜、凝胶、糊剂、泡沫或喷雾制剂呈现,包含除了活性成分以外的如本领域中已知的适于这样的用途的这样的载体。
[0096]
在许多实施方案中,本发明的口服剂型可以包含至少一种合成或天然的大麻素金属盐或铵盐,其是作为衍生物或前体的thc、cbd、cbn、cbg、cbc、cbl、cbv、thcv、cbdv、cbcv、cbgv、cbgm。
[0097]
在又其他实施方案中,本发明的口服剂型可以包括印度大麻提取物或从菌株火麻(c.sativa)、印度大麻(c.indica)、莠草大麻(c.ruderalis)或其组合获得的级分,其中,在其中的一些材料呈其酚类金属盐形式。
[0098]
如上文提到的,本发明的口服剂型还可以包含以多种浓度和组合的另外的治疗剂、矿物、营养物、维生素。
[0099]
特别感兴趣的是具有控制释放性质的口服剂型。术语“控制释放”指的是使得能够实现药物的时间依赖性释放、持续释放、延长释放以及还有脉冲释放即延迟释放的性质或改性。该术语还涉及抗胃性(gastro-resistance),即使得能够实现ph控制的药物释放、胃肠靶向、结肠递送、保护酸敏感性活性物、保护胃粘膜免受侵蚀性活性物的性质或改性。在这个意义上,抗胃性也是靶向的药物释放。还已知抗胃性包衣和改性改进储存稳定性。
[0100]
改进的抗胃性和/或控制释放可以通过使用多种药理学技术的改性和/或包衣来
实现,所述药理学技术诸如使用聚(甲基)丙烯酸酯或分层。已经广泛用于制药工业以实现靶向的药物释放和控制的药物释放的聚(甲基)丙烯酸酯包衣的熟知的实例是聚(甲基)丙烯酸酯包衣的另一个重要特征是保护免于外部影响(水分)或味道/气味掩盖以增加患者依从性。
[0101]
可以添加某些固体油以促进控制释放,诸如通常的单酸甘油酯油、甘油二酯油、甘油三酯油,以及特别地,三月桂精、三癸精、三棕榈精、三肉豆蔻精、甘油基、氢化棕榈油二硬脂酸酯、氢化蓖麻油、氢化植物油。
[0102]
在某些实施方案中,本发明的口服剂型可以包含基质改性/控制释放材料,其包括但不限于甘油酯、蜡、脂肪酸、丙烯酸甲酯或甲基丙烯酸甲酯的聚合物、乙基纤维素、聚(乙烯醇)(pva)、聚(乙烯基吡咯烷酮)(pvp)、淀粉、壳聚糖以及其他。
[0103]
在其他实施方案中,本发明的口服剂型可以用以下来包衣:羟丙基甲基纤维素、聚丙烯酸酯、丙烯酸甲酯-甲基丙烯酸共聚物、乙酸纤维素、聚乙酸乙烯邻苯二甲酸酯和其他类型的包衣。
[0104]
在许多实施方案中,本发明的口服剂型可以以片剂或胶囊的形式被提供,这两种形式都是口服药物递送的最流行和最方便的方法。胶囊可以被提供有包衣,该包衣是使用基于gras的材料的抗胃性包衣。
[0105]
在另外的实施方案中,剂型可以使用二级包装(secondary package),诸如泡罩(pvc/pvdc-alufile)、瓶子、铝袋以及其他。
[0106]
在另一个方面中,本发明的制剂和组合物可以应用于治疗和减轻(alleviation)若干种疾病和医疗状况。这样的医疗状况可以是当施用组合物时展示出有益的治疗效果的那些医疗状况。本发明的所有组合物或制剂包含具有酚类基团和与其离子键合的金属阳离子、铵阳离子或磷鎓阳离子的至少一种活性材料。
[0107]
在又一个方面中,本发明的上文描述的制剂、组合物和剂型可以应用于治疗和减轻若干种疾病和医疗状况,特别是其中先前已经展示出大麻素或基于印度大麻的药物的有益效果的那些疾病和医疗状况。换句话说,本发明提供了一系列的治疗方法,用于通过应用当前描述的制剂、组合物或剂型来治疗与大麻素或印度大麻的有益效果有关的疾病或医疗状况。应当注意的是,本发明的治疗方法可以应用于广泛的人类状况,包括炎性紊乱、神经紊乱、精神紊乱,恶性肿瘤和另外的免疫紊乱、代谢紊乱,营养缺乏,感染性疾病,以及胃肠紊乱、心血管紊乱的类型和多种类型的疼痛,包括慢性疼痛和神经性疼痛。
[0108]
考虑到当前关于大麻素在年轻患者和老年患者中的临床应用的知识水平,预计当前描述的制品和方法可以应用于,但不限于,抑郁、睡眠障碍、进食障碍(eating disorder)、癌症、多发性硬化症、移植物抗宿主病(gvhd)、帕金森病、癫痫、自闭症、肺结核、溃疡性结肠炎、克罗恩病(morbus crohn)、炎性肠病(ibd)、肠易激综合征(ibs)、食欲刺激物(appetite stimulant)、食欲抑制物、肥胖、恶心、神经性疼痛、焦虑、阿尔茨海默病、肌萎缩性侧索硬化症(als)、胃肠紊乱、高血压、失禁、瘙痒、关节炎、关节病、风湿性炎症、失眠、真菌病、局部和/或慢性疼痛、炎症、注意力缺陷和多动障碍(addh)、呕吐、特应性皮炎、纤维肌痛、aids、情绪紊乱、勃起功能障碍、早泄、营养缺乏。
[0109]
应当理解,当前描述的制品和方法适用于是婴幼儿、青少年或成人的受试者。
[0110]
还应当注意,本发明的制剂、组合物和剂型以治疗有效量来应用。一般而言,“治疗
有效量”(也是药理学有效量或药学有效量或生理学有效量)表示实现预期的或期望的生理响应所需的本发明的材料的量。精确的量取决于许多因素,例如,剂的类型、活性和预期用途(例如,每天的剂量数),这些因素可以通过本领域已知的技术和方法来确定。应理解,有效量可以是对治疗健康护理专业人员和/或个人的部分的经验和/或个体化(个案(case-by-case))确定的结果。较大治疗剂量的施用可能涉及每天多次施用。
[0111]
术语“治疗(treating)”、“治疗(treatment)”或“治疗(therapy)”或其任何语言变体,同样地指的是治愈性治疗、改善性治疗(ameliorating therapy)或预防性治疗。这些术语涵盖用于获得有益的或期望的治疗效果的任何方法,这些方法可以借助于生理参数、代谢参数或生化参数在临床上建立。有益的或期望的临床结果包括但不限于症状的减轻、疾病程度的减小、症状的稳定、进展的延迟或减慢、状况或症状的改善或缓和(palliation)以及缓解(remission)(无论是部分的还是全部的)。术语“缓和”在本文中涵盖与相同但未经治疗的状况相比被减轻的生理状况或症状和/或被减慢或延长的进展的不合意的表现。这些术语还可以与疾病或紊乱的预防有关。
[0112]
还另外,在某些实施方案中,本发明的制剂和方法涉及与其他方法和药物(也是治疗剂)同时地或连续地施用的组合治疗。
[0113]
相关的治疗剂可以是,但不限于,由fda根据其临床效果和对常见人类紊乱的适用性被分类的一般药物类别:镇痛药、抗酸药、抗焦虑药、抗心律失常药、抗菌药、抗生素、抗霉菌药、抗凝血药和血栓溶解药、抗惊厥药、抗抑郁药、止泻药、止吐药、抗真菌药、抗组胺药、抗高血压药、抗炎药、抗肿瘤药、抗精神病药、退烧药、抗病毒药、巴比妥类药物、β阻断剂(beta-blocker)、支气管扩张剂、感冒药、降胆固醇药、皮质类固醇、止咳药、细胞毒性剂、减充血剂、利尿药、祛痰剂、激素类、降血糖药、免疫抑制剂、泻药、肌肉松弛剂、镇静剂、性激素、安眠药、镇定剂和维生素补充剂。
[0114]
就治疗效果而言,如果存在至少约5%的改进、或10%的改进、或至少25%、或至少50%、或至少75%或至少100%的改进或更多,则作为治疗的结果的改进被确定。本文中的改进可以在个体改进以及群体改进的意义上解释。
[0115]
文本中在所有其出现的术语“约”表示与指定的值和/或范围的高达
±
10%偏差,更具体地说,与指定的值和/或范围的高达1%、
±
2%、3%、4%、5%、6%、7%、8%、9%或
±
10%的偏差。
[0116]
本发明还提供了至少一种本发明的金属盐或铵盐在制备制剂中或在预防或治疗至少一种如本文描述的紊乱或疾病状态的方法中的用途。
[0117]
还提供了至少一种如本文描述的金属盐或铵盐用于治疗疾病或紊乱的用途,所述疾病或紊乱被发现通过使用含苯酚的材料(例如,cbd)可治疗。
[0118]
还提供了至少一种如本文描述的材料用于制备用于治疗、预防或改善疾病或紊乱的制剂、药物组合物或药物的用途,所述疾病或紊乱被发现通过这样的材料组合物可治疗。
[0119]
还提供了本发明的组合物或制剂用于保护和治疗植物和作物免受细菌感染和/或真菌感染的用途。
[0120]
在一些实施方案中,制剂中包含的材料选自亚氯酸钠、水杨酸铜、溴氯二甲基海因(bromochlorodimethylhydantoin)、百里酚铜和聚胍。然而,可以采用许多其他含苯酚的活性材料用于生产适合于治疗植物和作物的材料。
[0121]
在一些实施方案中,细菌感染/真菌感染选自欧文氏菌、腐霉、菜豆壳球孢菌、罗耳阿太菌(athelia rolfsii)和马铃薯疮痂病。然而,可以通过利用本发明的材料可治疗的涉及植物或作物的任何其他感染也是本文的发明的实施方案。
[0122]
本文还提供了一种包含材料的组合物或制剂,其中所述材料包含具有至少(
2+
)的化合价的多价金属阳离子以及至少一种包含至少一种与多价金属阳离子离子键合的苯酚盐部分的活性材料,其中所述组合物或制剂用于治疗通过向受试者施用治疗有效量的所述组合物或制剂可治疗的疾病或紊乱。
[0123]
还提供了一种包含材料的组合物或制剂,其中所述材料包含如本文定义的铵阳离子或聚铵以及至少一种活性材料,该至少一种活性材料包含至少一种与铵或聚铵阳离子离子键合的苯酚盐部分,其中所述组合物或制剂用于治疗通过向受试者施用治疗有效量的所述组合物或制剂可治疗的疾病或紊乱。
[0124]
还另外,提供了一种用于治疗通过向受试者施用治疗有效量的包含材料的组合物或制剂可治疗的疾病或紊乱的方法,其中所述材料包含如本文定义的铵阳离子或聚铵或具有至少(
2+
)的化合价的多价金属阳离子以及至少一种活性材料,该至少一种活性材料包含至少一种与铵阳离子或聚铵或多价金属阳离子离子键合的苯酚盐部分。
[0125]
本发明还提供了一种制备根据本发明的材料的方法,该方法包括用碱处理至少一种含苯酚的活性材料,所述碱例如包括金属阳离子或氨或其等效物,如本文定义的。碱可以选自金属氢化物、金属醇盐、碱金属氢氧化物、碱土金属氢氧化物、金属碳酸盐、金属碳负离子(metal carbanion)、金属酰胺(metal amide)、氨(如本文描述的,例如胆碱)以及其他。
[0126]
如本文描述的,改变和改进含苯酚的活性材料的物理化学性质的另一种方法是通过形成其铵盐,从而产生包括含苯酚的材料和任选地被取代的铵阳离子的盐。因此,在本发明的方面中的另一个方面中,本发明提供了至少一种酚类(或含苯酚的)材料和至少一种任选地被取代的铵离子的铵盐,其中含苯酚的活性材料不是苯酚(c6h6o)、甲基苯酚、溴苯酚、二溴苯酚、三溴苯酚、五氯苯酚、双酚a、四溴双酚a、间苯二酚、氢醌、氢醌和萘酚。
[0127]
合成任选地被取代的铵-cbd材料的非限制性示意性实例在方案7中展示:
[0128]
[0129]
反应可以在氨或铵等效物的存在下进行,如上文解释的。在方案7中,使用铵型氢氧化物(ammonium hydroxide)。它可以选自氢氧化铵(ammonium hydroxide)(其中每个r是h)、单烷基化的氢氧化铵(其中r基团中的一个是烷基,并且其他r基团各自是h)、或多(二、三或四)烷基化的氢氧化铵。
[0130]
以类似的方式,如本文所公开的、用于诸如医药、化妆品、兽医和农业的多个领域中的其他含苯酚的活性材料还可以转换为它们的苯酚铵形式。
[0131]
非诺多泮的非限制性实例在方案8中示意性地展示:
[0132][0133]
方案8描绘了使用铵型氢氧化物合成铵-非诺多泮络合物。反应可以在氨或铵等效物的存在下进行,如上文解释的。在这个方案中,使用铵型氢氧化物。它可以选自氢氧化铵(其中每个r是h)、单烷基化的氢氧化铵(其中r基团中的一个是烷基,并且其他r基团各自是h)、或多(二、三或四)烷基化的氢氧化铵。这样的铵型氢氧化物的实例在方案7中描述。
[0134]
如本领域技术人员将知道的,烷基化的氢氧化铵可以由简单的烷基或高度取代的烷基构成。这样的烷基化的氢氧化铵的合成是简单的,其可以用于与含苯酚的生物活性剂反应以形成对应的铵盐。在典型的合成中,三烷基胺与烷基卤化物在有机溶剂中反应以形成四烷基卤化物,然后该四烷基卤化物与koh反应以用oh部分替换卤化物。
[0135]
本文提及的任何任选地被取代的铵盐还可以通过文献中已知的任何其他方法来获得或合成,并且在方案中使用的试剂可以交换为本领域中已知的其他合适的试剂。
[0136]
本发明的材料可以作为活性成分用于多种应用和领域(医药、化妆品、农业、化学和生物工业、染料等)。当抗衡离子呈具有一个或多于一个阳离子中心的聚合物的形式时,诸如在多胺的情况下,这样的材料还可以用于制备用于递送其他剂的纳米胶囊和微胶囊。
[0137]
如本文使用的,术语“纳米颗粒”、“纳米球”、“纳米胶囊”和“颗粒”全部指的是颗粒载体,该颗粒载体是生物相容性的,并且对化学破坏和/或物理破坏是有足够抗性的,使得足够量的纳米颗粒在施用到人类或动物身体之后基本上保持完整,并且持续足够的时间以能够到达期望的靶组织(器官)。通常,纳米颗粒的形状是球形的,具有高达1000nm并且在大多数情况下甚至高达500nm的平均直径。在颗粒的形状不是球形的情况下,直径指的是颗粒的最长尺寸。
[0138]
当提及“微胶囊”时,含义是具有在约1μm和1,000μm之间的直径的如上文描述的颗粒。
[0139]
本发明还提供了与多于一种苯酚盐活性剂缔合的聚阳离子,以及包含所述聚阳离子的组合物。
[0140]
本发明还提供了根据本发明的材料,其呈多分子材料的形式,该多分子材料包含两种或更多种含苯酚的活性材料,每种含苯酚的活性材料与阳离子离子缔合;其中所述阳离子是多价金属阳离子或聚铵。
[0141]
术语“多胺”和“聚铵”通常指的是任何具有多于一个(通常至少三个,但有时超过两个)的氨基官能团的化合物。在一些情况下,这样的化合物经由蛋白质的分解形成。在一些实施方案中,这样的多胺是烷基多胺。在一些实施方案中,多胺是天然多胺,并且在其他实施方案中,多胺是合成的。
[0142]
如果多胺是天然的,它们可以不受限制地选自亚精胺和精胺或在自然界中并且特别是在人体中天然出现的任何其他三胺、四胺或多胺。
[0143]
在一些实施方案中,多胺是选自以下的合成多胺:二亚乙基三胺,五甲基二亚乙基三胺,三亚乙基四胺,四亚乙基五胺,五亚乙基六胺,大环多胺诸如1,4,7-三氮杂环壬烷、1,4,7,10-四氮杂环十二烷(cyclen)和1,4,8,11-四氮杂环十四烷(cyclam),支链多胺诸如三(2-氨基乙基)胺和三脚架多胺(tripodal polyamine)诸如1,1,1-三(氨基甲基)乙烷。
[0144]
在一些特定的实施方案中,多胺是二胆碱或三胆碱。
[0145]
在一些实施方案中,多胺是包含至少三个胆碱部分的多胆碱聚合物。根据本发明的“多胆碱”是具有多个(至少两个)胆碱部分的聚合物分子。
[0146]
在一些实施方案中,多价金属阳离子是二价阳离子、三价阳离子或四价阳离子,或更高价的金属阳离子。
[0147]
在一些实施方案中,材料通过多酚与多价阳离子或聚铵的反应来形成。
[0148]
在一些实施方案中,材料用于制备纳米颗粒或微米颗粒。在一些实施方案中,材料用于制备包衣材料。
[0149]
本发明还提供了一种包含根据本发明的材料的颗粒。
[0150]
在含苯酚的剂不是药学活性剂或其他的活性剂的情况下,本发明提供了一种呈多分子材料的形式的材料,该材料包括两种或更多种含苯酚的材料,每种含苯酚的材料与阳离子离子缔合;其中所述阳离子是多价金属阳离子或聚铵。在一些实施方案中,多价金属阳离子是二价阳离子、三价阳离子或四价阳离子,或更高价的金属阳离子。
[0151]
在一些实施方案中,材料通过多酚与多价阳离子或聚铵的反应来形成。
[0152]
在一些实施方案中,材料用于制备纳米颗粒或微米颗粒,或者用于制备包衣材料。
[0153]
因此,本发明的特定的方面和实施方案包括:
[0154]
一种分离的材料,其包含至少一种含苯酚的活性材料和金属盐抗衡离子、磷鎓抗衡离子或铵盐抗衡离子,其中所述活性材料不是苯酚(c6h6o)、甲基苯酚、溴苯酚、二溴苯酚、三溴苯酚、五氯苯酚、双酚a、四溴双酚a、间苯二酚、氢醌、氢醌或萘酚。
[0155]
所述材料,其包含金属阳离子和苯酚盐活性材料,其中使所述金属阳离子和在所述苯酚盐材料上的带电氧原子缔合的键是离子键。
[0156]
所述材料,其中所述金属阳离子选自单价阳离子、二价阳离子和多价阳离子。
[0157]
所述材料,其中所述金属阳离子是二价金属阳离子或三价金属阳离子。
[0158]
所述材料,其中所述金属阳离子不是单价阳离子。
[0159]
所述材料,其中所述金属阳离子选自碱金属、碱土金属和过渡金属。
[0160]
所述材料,其中所述金属阳离子不是碱金属。
[0161]
所述材料,其中所述金属阳离子选自锂金属阳离子、钠金属阳离子、钾金属阳离子、钙金属阳离子、镁金属阳离子、锰金属阳离子、铝金属阳离子、锌金属阳离子、镍金属阳离子、铁金属阳离子、银金属阳离子、金金属阳离子和钡金属阳离子。
[0162]
所述材料,其中所述材料呈铵盐的形式。
[0163]
所述材料,其中所述材料呈磷鎓盐的形式。
[0164]
所述材料,其中酚类活性材料是用于医药、化妆品、兽医或农业的含苯酚的活性材料,所述含苯酚的活性材料已经通过从苯酚-oh基团中摘取氢原子而转换为苯酚盐形式。
[0165]
所述材料,其中含苯酚的活性材料包含一个或更多个-oh基团。
[0166]
所述材料,其中所述材料包含二价金属和两种苯酚盐活性材料。
[0167]
所述材料,其中所述材料包含两种或更多种酚类活性材料,每种活性材料具有一个或更多个苯酚盐基团,所述苯酚盐基团中的每一个苯酚盐基团与选自金属阳离子、磷鎓阳离子和铵阳离子的不同阳离子缔合。
[0168]
所述材料,其中所述两种苯酚盐活性材料是相同的或不同的。
[0169]
所述材料,其包含三价金属和三种苯酚盐活性材料。
[0170]
所述材料,其中所述三种苯酚盐活性材料中的每一种苯酚盐活性材料彼此不同。
[0171]
所述材料,其中所述三种苯酚盐活性材料中的两种苯酚盐活性材料不同于所述苯酚盐活性材料中的第三种苯酚盐活性材料。
[0172]
所述材料,其中所述三种苯酚盐活性材料中的每一种苯酚盐活性材料是相同的。
[0173]
所述材料,其中所述苯酚盐活性材料中的一种或更多种苯酚盐活性材料是治疗材料、化妆品材料或兽医材料。
[0174]
所述材料,其中所述苯酚盐活性材料中的一种或更多种苯酚盐活性材料是农业材料。
[0175]
所述材料,其中所述材料包含一种或更多种苯酚盐活性材料和一种或更多种非活性苯酚盐材料。
[0176]
所述材料,其中所述苯酚盐活性材料选自大麻素、非诺多泮、酪氨酸、二甲酚、百里酚、丙泊酚、阿朴吗啡、吗啡及其衍生物、米托蒽醌、多柔比星、六氯酚、对乙酰氨基酚、对香豆酸、3,4-二羟基苯甲酸、4-羟基苯甲酸、对羟基苯甲酸丁酯、香草酸、愈创木酚、咖啡酸、托特罗定、雷洛昔芬、东莨菪亭、紫花前胡醇、多巴胺、左旋多巴、姜黄素、天碱和多酚。
[0177]
所述材料,其中活性药物实体是大麻素材料。
[0178]
所述材料,其中所述大麻素材料选自四氢大麻酚(thc)、四氢大麻酚酸(thca)、大麻二酚(cbd)、大麻二酚酸(cbda)、大麻酚(cbn)、大麻萜酚(cbg)、大麻环萜酚(cbc)、大麻环醇(cbl)、次大麻酚(cbv)、四氢次大麻酚(thcv)、次大麻二酚(cbdv)、次大麻环萜酚(cbcv)、次大麻萜酚(cbgv)、大麻萜酚单甲醚(cbgm)、大麻艾尔松(cbe)和大麻二吡喃环烷(cbt)。
[0179]
所述材料,其中所述大麻素是thc或cbd及其化学衍生物。
[0180]
所述材料,其中所述苯酚活性材料选自沙波立沙钠(snac)、塔匹那洛夫和来自肉桂提取物的材料。
[0181]
所述材料,其中所述苯酚活性材料是植物材料(phytomaterial)。
[0182]
所述材料,其中所述苯酚活性材料选自美沙拉嗪、沙丁胺醇、吡布特罗、辣椒素、沙美特罗、维兰特罗、巴柳氮、拉贝洛尔、霉酚酸、吡哆醇、苯肾上腺素、依酚氯铵、扑热息痛、莫
诺苯宗、他喷他多、间羟胺、甲酪氨酸、羟甲唑啉、大麻隆、二氟尼柳、奥沙拉嗪、碘塞罗宁钠、去甲文拉法辛、罗替戈汀、酚妥拉明、羟保泰松、阿莫地喹、奥达特罗、曲格列酮、艾曲波帕、依伐卡托、茚达特罗、头孢羟氨苄、头孢丙烯、四氢大麻酚、雌二醇、戊酸雌二醇、环戊丙酸雌二醇、左洛啡烷、羟吗啡酮、纳布啡、丁丙诺啡、布托啡诺、纳洛酮、左啡诺、纳曲酮、地佐辛、吗啡、纳洛塞醇、甲基纳曲酮、纳美芬、美他环素、萨瑞环素、奥马环素、伊拉瓦环素、马烯雌酮、氟美他酚、己烯雌酚、双烯雌酚、普罗布考、米托蒽醌、巴多昔芬、雷洛昔芬、阿布他明、多巴酚丁胺、马索罗酚、大麻二酚、特布他林、奥西那林、非诺多泮、去甲肾上腺素、异肾上腺素、异丙肾上腺素、异他林、屈昔多巴、卡比多巴、普罗托醇、阿朴吗啡、恩他卡朋、托卡朋、伊达比星、道诺霉素、多柔比星、表柔比星和戊柔比星。
[0183]
一种材料,包含至少一种呈苯酚盐的形式的大麻素和选自金属阳离子、磷鎓和铵的阳离子。
[0184]
所述材料,其中所述金属阳离子是单价金属原子、二价金属原子或三价金属原子。
[0185]
所述材料,其中所述金属阳离子是二价阳离子或三价阳离子。
[0186]
所述材料,其中所述金属阳离子不是单价阳离子。
[0187]
所述材料,其中所述材料包含二价金属原子和一种或两种大麻素。
[0188]
所述材料,其中所述材料包含一种大麻素和一种非大麻素。
[0189]
所述材料,其中所述材料包含三价金属原子和一种或两种或三种大麻素。
[0190]
所述材料,其中所述材料包含一种大麻素和两种非大麻素。
[0191]
所述材料,其中所述材料包含两种大麻素和一种非大麻素。
[0192]
所述材料,其中所述大麻素是cbd或thc。
[0193]
所述材料,其中所述金属阳离子选自锂金属阳离子、钠金属阳离子、钾金属阳离子、钙金属阳离子、镁金属阳离子、锰金属阳离子、铝金属阳离子、锌金属阳离子、镍金属阳离子、铁金属阳离子、银金属阳离子、金金属阳离子和钡金属阳离子。
[0194]
所述材料,其中所述金属阳离子是钠或钙。
[0195]
一种钙cbd盐,包括一种cdb和任选地另一种非大麻素、或者两种cbd。
[0196]
所述材料,其中所述铵是任选地被取代的铵阳离子。
[0197]
所述材料,其中所述材料包含任选地被取代的铵阳离子和苯酚盐活性化合物。
[0198]
所述材料,其中所述铵阳离子是四烷基铵,所述四烷基铵任选地选自四甲基铵、四乙基铵、四丁基铵、四戊基铵、四己基铵、胆碱和胆碱衍生物。
[0199]
一种材料,是至少一种苯酚活性材料的胆酸盐。
[0200]
所述材料,其中所述活性材料选自大麻素、非诺多泮、酪氨酸、二甲酚、百里酚、丙泊酚、阿朴吗啡、吗啡及其衍生物、米托蒽醌、多柔比星、六氯酚、对乙酰氨基酚、对香豆酸、3,4-二羟基苯甲酸、4-羟基苯甲酸、对羟基苯甲酸丁酯、香草酸、愈创木酚、咖啡酸、托特罗定、雷洛昔芬、东莨菪亭、紫花前胡醇、多巴胺、左旋多巴、姜黄素、天碱和多酚。
[0201]
所述材料,其中活性药物实体是大麻素材料。
[0202]
所述材料,其中所述大麻素材料选自四氢大麻酚(thc)、四氢大麻酚酸(thca)、大麻二酚(cbd)、大麻二酚酸(cbda)、大麻酚(cbn)、大麻萜酚(cbg)、大麻环萜酚(cbc)、大麻环醇(cbl)、次大麻酚(cbv)、四氢次大麻酚(thcv)、次大麻二酚(cbdv)、次大麻环萜酚(cbcv)、次大麻萜酚(cbgv)、大麻萜酚单甲醚(cbgm)、大麻艾尔松(cbe)和大麻二吡喃环烷(cbt)。
[0203]
所述材料,其中所述大麻素是thc或cbd及其化学衍生物。
[0204]
所述材料,其中所述苯酚活性材料选自沙波立沙钠(snac)、塔匹那洛夫和来自肉桂提取物的材料。
[0205]
所述材料,其中所述苯酚活性材料是植物材料。
[0206]
所述材料,其中所述苯酚活性材料选自美沙拉嗪、沙丁胺醇、吡布特罗、辣椒素、沙美特罗、维兰特罗、巴柳氮、拉贝洛尔、霉酚酸、吡哆醇、苯肾上腺素、依酚氯铵、扑热息痛、莫诺苯宗、他喷他多、间羟胺、甲酪氨酸、羟甲唑啉、大麻隆、二氟尼柳、奥沙拉嗪、碘塞罗宁钠、去甲文拉法辛、罗替戈汀、酚妥拉明、羟保泰松、阿莫地喹、奥达特罗、曲格列酮、艾曲波帕、依伐卡托、茚达特罗、头孢羟氨苄、头孢丙烯、四氢大麻酚、雌二醇、戊酸雌二醇、环戊丙酸雌二醇、左洛啡烷、羟吗啡酮、纳布啡、丁丙诺啡、布托啡诺、纳洛酮、左啡诺、纳曲酮、地佐辛、吗啡、纳洛塞醇、甲基纳曲酮、纳美芬、美他环素、萨瑞环素、奥马环素、伊拉瓦环素、马烯雌酮、氟美他酚、己烯雌酚、双烯雌酚、普罗布考、米托蒽醌、巴多昔芬、雷洛昔芬、阿布他明、多巴酚丁胺、马索罗酚、大麻二酚、特布他林、奥西那林、非诺多泮、去甲肾上腺素、异肾上腺素、异丙肾上腺素、异他林、屈昔多巴、卡比多巴、普罗托醇、阿朴吗啡、恩他卡朋、托卡朋、伊达比星、道诺霉素、多柔比星、表柔比星和戊柔比星。
[0207]
一种制剂,选自药物制剂、化妆品制剂、兽医制剂或农业制剂,所述制剂包含根据本发明的分离的材料。
[0208]
所述制剂,作为适于局部施用、经皮施用、口服施用、气溶胶施用、肠胃外施用、皮下施用、静脉内施用、肌肉内施用、腹膜内施用、直肠施用或阴道施用的药物制剂。
[0209]
所述制剂,其是包含在0.1%和99%之间的所述分离的材料。
[0210]
至少一种根据本发明的材料在制备制剂中的用途。
[0211]
至少一种根据本发明的材料在预防或治疗至少一种紊乱或疾病状态的方法中的用途。
[0212]
一种制备根据本发明的材料的方法,所述方法包括用碱处理至少一种含苯酚的活性材料,所述碱选自含金属的碱和氨或其等效物。
[0213]
所述方法,其中所述碱选自金属氢化物、金属醇盐、碱金属氢氧化物、碱土金属氢氧化物、金属碳酸盐、金属碳负离子或金属酰胺。
[0214]
所述方法,其中所述碱选自naoh、ca(oh)2、cu(oh)2、cacl2、fecl3、zn(oh)2和nah。
[0215]
一种制备根据本发明的材料的方法,所述方法包括用碱处理至少一种含苯酚的活性材料以获得所述材料,所述碱选自含金属的碱和氨或其等效物;以及在使得能够交换抗衡离子的条件下处理所述材料。
[0216]
根据本发明的材料,通过本发明的方法制备。
[0217]
一种材料,呈多分子材料的形式,所述多分子材料包含两种或更多种含苯酚的活性材料,每种含苯酚的活性材料与阳离子离子缔合;其中所述阳离子是多价金属阳离子或聚铵。
[0218]
所述材料,其中所述多价金属阳离子是二价阳离子、三价阳离子或四价阳离子。
[0219]
所述材料,其中所述材料通过多酚与所述多价阳离子或聚铵的反应形成。
[0220]
所述材料,用于制备纳米颗粒或微米颗粒。
[0221]
所述材料,用于制备包衣材料。
[0222]
一种颗粒,包含根据本发明的材料。
[0223]
所述颗粒是微米颗粒或纳米颗粒。
[0224]
所述颗粒,其中所述材料包含来自绿茶提取物的多酚。
[0225]
所述颗粒,其中所述材料包含鞣酸。
[0226]
一种材料,呈多分子材料的形式,所述多分子材料包含两种或更多种含苯酚的材料,每种含苯酚的材料与阳离子离子缔合;其中所述阳离子是多价金属阳离子或聚铵。
[0227]
所述材料,其中所述多价金属阳离子是二价阳离子、三价阳离子或四价阳离子。
[0228]
所述材料,其中所述材料通过多酚与所述多价阳离子或聚铵的反应形成。
[0229]
所述材料,用于制备纳米颗粒或微米颗粒。
[0230]
所述材料,用于制备包衣材料。
[0231]
一种颗粒,包含根据本发明的材料。
[0232]
根据本发明的材料用于治疗、改善或预防疾病或紊乱的用途,所述材料呈至少一种含苯酚的材料的金属或铵材料的形式,所述疾病或紊乱被发现通过所述材料可治疗。
[0233]
至少一种根据本发明的材料用于制备用于治疗、预防或改善疾病或紊乱的制剂、药物组合物或药物的用途,所述疾病或紊乱被发现通过这样的制剂/组合物/药物可治疗。
[0234]
至少一种根据本发明的材料用于制备用于保护和治疗植物和作物免受细菌感染和/或真菌感染的制剂的用途。
[0235]
所述用途,其中所述材料选自亚氯酸钠、水杨酸铜、溴氯二甲基海因、百里酚铜和聚胍。
[0236]
所述用途,其中所述细菌感染和/或真菌感染选自欧文氏菌、腐霉、菜豆壳球孢菌、罗耳阿太菌和马铃薯疮痂病。
[0237]
一种组合物或制剂,包含材料,其中所述材料包含具有至少(
2+
)的化合价的多价金属阳离子以及至少一种呈苯酚盐的形式的、与所述多价金属阳离子离子键合的活性含苯酚的材料,其中所述组合物或所述制剂用于治疗通过向受试者施用治疗有效量的所述组合物或所述制剂可治疗的疾病或紊乱。
[0238]
一种组合物或制剂,包含材料,其中所述材料包含铵阳离子或聚铵以及至少一种活性材料,所述至少一种活性材料包含至少一种与所述铵或所述聚铵阳离子离子键合的苯酚盐部分,其中所述组合物或所述制剂用于治疗通过向受试者施用治疗有效量的所述组合物或所述制剂可治疗的疾病或紊乱。
[0239]
实施方案的详细描述
[0240]
cbd金属络合物根据多种程序被形成。在这里,为了防止不必要的副反应,特别是cbd的化学改性,我们仅应用最大程度的环境反应条件。基于金属化合价调整金属试剂的相对量,始终保持该相对量以确保存在足够的金属离子来络合cbd中的两个可用的苯酚。
[0241]
实施例1:cbd-盐的合成与分析
[0242]
na盐:将naoh(9.4mg,0.24mmol)在乙醇(3.0ml)中的溶液加入到cbd(35mg,0.11mmol)在乙醇(5.0ml)中的搅拌的溶液中。将混合物在室温搅拌持续1h,并且蒸发溶剂,以定量收率提供cbd-na。
[0243]
通过以下分析技术来表征cbd金属络合物:
[0244]
1)1h和
13
c核磁共振(nmr)
[0245]
2)红外光谱法(ir)
[0246]
3)薄层色谱法(tlc)
[0247]
4)差示扫描量热法(dsc)
[0248]
5)熔点(mp)
[0249]
6)元素分析
[0250]
7)紫外吸收光谱法(uv)。
[0251]
每种表征的方法都证实了成功合成的方面:
[0252]
nmr光谱法证实了反应条件没有破坏分子的任何其他部分,并且cbd结构被维持,
[0253]
ir光谱法鉴定了新的官能团,特别是苯酚-金属络合物,
[0254]
基于极性的差异,tlc证实了起始材料的消耗以及新材料的存在,
[0255]
熔点证实了新物质的纯度,
[0256]
元素分析证实了,产物中的ch%由于金属含量的重量贡献而被降低。
[0257]
uv显示出苯酚环的吸收光谱的变化
[0258]
结果:已经合成了具有单价金属中心、二价金属中心和三价金属中心的多种cbd-金属络合物。络合物通过与金属氢氧化物或金属盐诸如氯化物或硫酸盐的反应(方法1)或与金属氢化物在无水条件下的反应(方法2)形成。
[0259]
cbd-na的合成
[0260]
将naoh(9.4mg,0.24mmol)在乙醇(3.0ml)中的溶液加入到cbd(35mg,0.11mmol)在乙醇(5.0ml)中的搅拌的溶液中。将混合物在rt搅拌持续1h,并且蒸发溶剂,以定量收率提供cbd-na。
[0261]
cbd-k的合成
[0262]
将koh(2.5mg,0.045mmol)在甲醇(2.5ml)中的溶液加入到cbd(7.1mg,0.023mmol)在甲醇(2.5ml)中的搅拌的溶液中。将混合物在rt搅拌持续1天,并且蒸发溶剂,以定量收率提供cbd-k。
[0263]
cbd-ca的合成
[0264]
将cacl2(5.2mg,0.046mmol)在甲醇(2.5ml)中的溶液加入到cbd(7.3mg,0.023mmol)在甲醇(2.5ml)中的搅拌的溶液中。将混合物在rt搅拌持续1天,并且蒸发溶剂,提供cbd-ca。
[0265]
cbd-cu的合成
[0266]
将caso4(7.2mg,0.045mmol)在甲醇(2.5ml)中的溶液加入到cbd(9.2mg,0.029mmol)在甲醇(2.5ml)中的搅拌的溶液中。将混合物在rt搅拌持续1天,并且蒸发溶剂,提供cbd-cu。
[0267]
cbd-zn的合成
[0268]
将zncl2的溶液(2.5ml的0.8mg ml-1
溶液)加入到cbd(5.2mg,0.017mmol)在甲醇(2.5ml)中的搅拌的溶液中。将混合物在rt搅拌持续1天,并且蒸发溶剂,以定量收率提供cbd-zn。
[0269]
cbd-al的合成
[0270]
将alcl3(3.1mg,0.011mmol)在甲醇(5ml)中的溶液加入到cbd(5mg,0.016mmol)在甲醇(2.5ml)中的搅拌的溶液中。将混合物在rt搅拌持续1天,并且蒸发溶剂,以定量收率提
供cbd-al。
[0271]
cbd-fe的合成
[0272]
将fecl3(9.2mg,0.057mmol)在乙醇(3ml)中的溶液加入到cbd(24.1mg,0.077mmol)在乙醇(5ml)中的搅拌的溶液中。将混合物在rt搅拌持续1天,并且蒸发溶剂,以定量收率提供cbd-fe。
[0273]
实施例2:通过与金属氢化物或金属乙醇盐的反应制备cbd盐
[0274]
将氢化钠(50%在油中,2.3mg,0.096mmol)洗涤(3
×
1ml),并且最后在氩气下用thf(5ml)覆盖。然后加入cbd(15mg,0.048mmol),并且允许混合物搅拌持续0.5h。将混合物用乙醚(10ml)稀释,并且用10%naoh(3
×
5ml)洗涤。合并的有机提取物经na2so4干燥,并且蒸发溶剂,以定量收率提供cbd-na。
[0275]
cbd与乙醇钠反应以获得cbd-na
[0276]
cbd-na络合物还可以采用在乙醇中的金属钠来合成。将钠(2.2mg,0.096mmol)加入到cbd(15mg,0.048mmol)在乙醇(5ml)中的溶液。将混合物搅拌持续0.5h,用乙醚(10ml)稀释,并且用10% naoh(3
×
5ml)洗涤。合并的有机提取物经na2so4干燥,并且蒸发溶剂,以定量收率提供cbd-na。
[0277]
cbd金属络合物的分析
[0278]
cbd金属络合物的合成首先通过薄层色谱法(tlc)来指示,薄层色谱法采用二氧化硅作为固定相以及己烷:乙酸乙酯的9:1混合物作为流动相。板在uv光(254nm)下可视化。将金属络合物与cbd(rf=0.69)进行比较,并且显示出增加的极性(rf=0)。
[0279]
nmr研究表明,cbd经过这些反应条件保持了其结构完整性,在合成之前和之后显示出几乎相同的1h nmr光谱。酚类质子消失,并且苯酚的芳族质子位移。
[0280]
测定了cbd金属络合物的水溶性(表1)。值得注意地,钠络合物、钙络合物和铜络合物在10mg/ml时全部是可溶的。cbd植物提取物仅在0.0126mg/ml时是可溶的。
[0281]
表1.cbd金属络合物的水溶性。
[0282]
cbd金属络合物在水中的溶解度cbd-na10mg/mlcbd-k2mg/mlcbd-ca10mg/mlcbd-cu10mg/mlcbd-zn1.7mg/mlcbd-al2mg/mlcbd-fe1.4mg/ml
[0283]
可逆性
[0284]
将1m hcl溶液逐滴加入到cbd-na的水溶液中,直到达到ph为3。在混合物中观察到不溶性棕色油的立即出现,其通过1h nmr被证实为cbd。可逆性的容易程度对这项工作的成功至关重要,因为原始化合物可以通过简单的化学过程被容易地重构。
[0285]
热性质
[0286]
通过差示扫描量热法(dsc)比较了cbd-na与cbd的热性质。在60ml min-1
的氮气流量下以10℃min-1
的扫描速率从25℃至100℃进行dsc,示出了金属络合物和母体分子在整个
该温度范围内的热行为。发现cbd-na金属络合物的熔点为55℃,这与母体cbd分子的观察到的66℃的熔点相比较。
[0287]
使用金属氢化物制备金属络合物
[0288]
采用金属氢化物(nah、cah2)实现了cbd离子络合物,以便提供没有副产物的络合物。在干燥的氩气下将cbd(35mg,0.11mmol)在dcm(1ml)中的溶液加入到过量nah/cah2在dcm(1ml)中的分散体中。使混合物在室温混合过夜。在反应混合物中观察到鼓泡,这与氢气的形成和释放有关。将溶剂蒸发以提供cbd-na/ca。产物通过以9:1己烷/乙酸乙酯的tlc(rf=0)和光谱分析来证实。产物的外观类似于通过与金属氯化物或金属氢氧化物的反应形成的离子络合物的外观。
[0289]
使用乙醇钠制备金属络合物
[0290]
采用乙醇钠实现了cbd离子络合物,以便提供没有副产物的络合物。在干燥的氩气下将cbd(35mg,0.11mmol)在乙醇(1ml)中的溶液加入到乙醇钠溶液中,该乙醇钠溶液通过将钠金属加入到乙醇(1ml)中来制备。使混合物在室温混合持续30分钟。将溶剂蒸发以提供cbd-na。产物通过以9:1己烷/乙酸乙酯的tlc(rf=0)和光谱分析来证实。产物的外观类似于通过与金属氯化物或金属氢氧化物的反应形成的离子络合物的外观。
[0291]
氧化稳定性
[0292]
将cbd在乙醇中的溶液暴露于通过氧化和太阳光鼓泡的空气。在24小时的暴露之后,识别到cbd含量下降。当cbd金属络合物暴露于通过氧化和太阳光鼓泡的空气持续24小时时,未发现cbd含量的变化,如通过hplc确定的。
[0293]
cbd金属材料形成
[0294]
使用不同的二价金属氢氧化物诸如ca(oh)2和ba(oh)2使用以下程序合成了二价金属-cbd络合物。
[0295]
将cbd(1当量)和二价金属氢氧化物(1当量)放入带有磁棒的反应小瓶中。将反应小瓶保持在氩气气氛下。然后加入乙醇:水(1:1)混合物,并且在rt搅拌反应混合物。通过tlc监测反应的完成。在反应完成之后,使用真空去除乙醇。使用冻干机去除水,这得到作为固体的二价金属-cbd络合物。
[0296]
ca-cbd络合物:将cbd(100mg,0.32mmol,1当量)和氢氧化钙(24mg,0.32mmol,1当量)放入带有磁棒的4ml反应小瓶中。随后,加入3ml的乙醇:水(1:1)混合物。将反应小瓶保持在氩气气氛下。然后在rt搅拌反应混合物。使用10%乙酸乙酯/正己烷溶液作为洗脱剂检查tlc。在通过tlc确认的cbd的消耗(~48h)之后,使用真空去除乙醇。将所得到的络合物冻干过夜,这得到作为深紫色固体的产物。使用ft-ir和nmr分析所获得的固体产物。
[0297]
ba-cbd络合物:将cbd(100mg,0.32mmol,1当量)和五水合氢氧化钡(101mg,0.32mmol,1当量)放入在带有磁棒的4ml反应小瓶中。随后,加入3ml的乙醇:水(1:1)混合物。将反应小瓶保持在ar气氛下。然后在rt搅拌反应混合物。使用10%乙酸乙酯/正己烷溶液作为洗脱剂检查tlc。在通过tlc确认的cbd的消耗(~48h)之后,使用真空去除乙醇。将所得到的络合物冻干过夜,这得到作为深紫色固体的产物。使用ft-ir和nmr分析所获得的固体产物。
[0298]
通常,cbd可溶于乙醇但不溶于水。然而,ca(oh)2和ba(oh)2不溶于乙醇,并且微溶于水。因此,乙醇:水(1:1)混合物被用于二价金属-cbd反应的反应。当cbd和ca(oh)2/ba
(oh)2在室温(rt)在乙醇:水(1:1)混合物中搅拌时,观察到白色部分溶解的溶液。当反应进行时,溶液的颜色逐渐变为深紫色,并且最后,在溶剂的蒸发之后获得深紫色固体。
[0299]
二价金属-cbd络合物的nmr分析
[0300]
获得了cbd和ba-cbd络合物的1h nmr光谱。在6.01ppm处的对应于cbd的两个芳族c-h质子的峰的强度几乎消失,并且在ca-cbd络合物中,在6.15ppm处出现了新的峰。另外,在ca-cbd络合物中,在8.66ppm处的归属于cbd的两个oh的峰消失。
[0301]
类似地,对于ca-cbd盐,在6.01ppm处的对应于cbd的两个芳族c-h质子的峰的强度降低,并且在ca-cbd络合物中,在附近观察到新的峰。另外,在8.66ppm处的归属于cbd的两个oh的峰的强度降低,并且在ca-cbd络合物中,在附近注意到新的峰。
[0302]
cbd-na衍生物的表征
[0303]
通过1h nmr光谱和ftir光谱表征所合成的cbd-na衍生物。在(使用nah合成的)cbd-na的情况下,在6.99ppm处出现新的峰,这支持了cbd中苯酚盐阴离子的形成。由于cbd包含两个苯酚基团,因此可以形成单钠盐或二钠盐。
[0304]
ftir光谱还支持cbd的钠衍生物的形成。由于cbd的金属衍生物的形成,对于(使用nah合成的)cbd-na,cbd的在3425cm-1
处的-oh基团的氢键峰被抑制,并且位移至3420cm-1
。在cbd-na中,cbd的在1628cm-1
和1582cm-1
处的峰也位移至1643cm-1
和1514cm-1
。
[0305]
cbd-ca衍生物的表征
[0306]
通过1h nmr光谱和ftir光谱表征所合成的cbd-ca衍生物。在cbd-ca的情况下,在6.97ppm处出现新的峰,这支持了cbd中苯酚盐阴离子的形成。ftir光谱还支持cbd的钙衍生物的形成。由于cbd的金属衍生物的形成以及cbd的分子间氢键的断裂,对于cbd-ca,cbd的在3425cm-1
处的-oh基团的氢键峰被抑制,并且在3640cm-1
处出现尖锐的非氢键的-oh基团的峰。
[0307]
实施例3:cbd-fe衍生物的合成和表征
[0308]
合成:在氮气吹扫的圆底烧瓶中,将200mg的cbd(0.63mmol)溶解在20ml的乙醇中。将溶解在10ml脱水乙醇中的fecl3(34mg,0.21mmol)加入到cbd溶液中。将溶液保持在室温搅拌持续24h。
[0309]
cbd-fe的edx光谱证实了元素c、o、fe和cl的存在,并且原子%对于碳为82,并且对于fe为2.17。因此,c和fe的比是~38:1。在二氯化物离子被cbd的酚类-oh基团完全地替换的情况下,此比应当是~42:1。这个结果证实了cbd2fecl的形成。
[0310]
ftir光谱还支持cbd的铁衍生物的形成。由于cbd的金属衍生物的形成,对于cbd-fe,cbd的在3425cm-1
处的-oh基团的氢键峰被抑制,并且位移至3300cm-1
。在cbd-fe中,cbd的在1628cm-1
处的归属于-oh基团的弯曲振动模式的峰也被抑制,并且位移至1621cm-1
。在cbd-fe盐中,cbd的在1582cm-1
和1442cm-1
处的强芳族带位移至1576cm-1
和1426cm-1
指示芳族c=c键的变化,这表明cbd向cbd-fe的转化。
[0311]
dsc研究表明,纯的cbd在69℃处具有峰,而cbd-fe示出在132.69℃处的主峰,以及还有在64℃处的小峰。
[0312]
对于所有的盐,加入酸性溶液,诸如hcl,转化回到天然的cbd。
[0313]
实施例4:cbd与多种单价金属盐的反应
[0314]
cbd与naoh、na2co3或nahco3的反应:在氮气吹扫的圆底烧瓶中,将25mg的cbd溶解
在3ml的甲醇中。然后将在2ml乙醇中的17mg的na2co3或等当量的碳酸氢钠和naoh加入到cbd溶液中。将溶液保持在室温搅拌持续24h。最终的干燥的混合物包含两部分。一部分可溶于水;另一部分不溶于水。检查这两部分两者的nmr、ftir和ms。水溶性的部分包含钠盐cbd和可以是氧化的cbd的化合物。水不溶性的部分包含未反应的cbd或单一的钠盐。
[0315]
cbd与nacl的反应;将20mg的cbd溶解在3ml的脱水乙醇中。然后将溶解在2ml脱水乙醇中的8.8mg的nacl加入到cbd溶液中。将溶液在室温搅拌持续72h。没有反应,cbd保持原样,没有水溶性化合物。
[0316]
当在类似的条件下使用koh、lioh、licl、kcl、khco3和k2co3时获得了类似的结果,其中kcl和licl不与cbd反应,而碱化合物形成对应的cbd盐,但是作为与cbd的混合物,并且可能作为氧化的cbd。
[0317]
cbd与zncl2的反应:将cbd(100mg,0.32mmol,1当量)和氯化锌(44mg,0.32mmol,1当量)放入带有磁棒的4ml反应小瓶中。随后,加入3ml的乙醇:水(1:1)混合物。将反应小瓶保持在ar气氛下。然后在rt搅拌反应混合物。使用10%乙酸乙酯/正己烷溶液作为洗脱剂检查tlc。在如通过tlc确认的cbd的消耗之后,使用真空去除乙醇。将所得到的盐冻干过夜,以得到作为固体的产物。
[0318]
cbd与mgcl2的反应:将cbd(100mg,0.32mmol,1当量)和氯化镁(31mg,0.32mmol,1当量)放入带有磁棒的4ml反应小瓶中。随后,加入3ml的乙醇:水(1:1)混合物。将反应小瓶保持在ar气氛下。然后在rt搅拌反应混合物。使用10%乙酸乙酯/正己烷溶液作为洗脱剂检查tlc。在如通过tlc确认的cbd的消耗之后,使用真空去除乙醇。将所得到的盐冻干过夜,以得到作为固体的产物。
[0319]
检查tlc以确认反应的完成。在24h-48h的反应之后,观察到cbd的完全消耗。在tlc的基线处观察到二价金属-cbd络合物。tlc和h nmr光谱指示,获得了金属盐,并且形成了一些副产物(单金属或二聚体)。
[0320]
cbd与fecl3的反应:将cbd(58.7mg,0.187mmol)和氯化铁(iii)(20.4mg,0.126mmol)放入无水乙醇(6ml)中,并且在rt搅拌过夜。将溶剂蒸发至接近干燥,并且然后沉淀到戊烷中。戊烷级分仅包含cbd,并且收集沉淀物。由于顺磁铁(iii)的存在,1h nmr不可获得,并且因此通过凝胶渗透色谱法(gpc)表征样品。观察到的1950的mw指示在络合物中存在若干个(~6个)cbd单元。然后将产物溶解在乙醇/0.1m hcl(2:1的比)中,并且所得到的沉淀物是起始的cbd。在thf中进行相同的反应,该反应形成fe络合物,该fe络合物在用hcl酸化时转化回到cbd。当使fecl3与cbd以不同的比和反应条件反应时获得不同的络合物。
[0321]
cbd与al(cl)3的反应:将cbd(106mg,0.337mmol)和无水氯化铝(35.2mg,0.264mmol)放入无水乙醇(15ml)中,并且在rt搅拌过夜。将溶剂蒸发至干燥,并且将粗产物用氯仿洗涤以去除未反应的cbd。1h nmr显示出芳族质子峰的位移,这指示络合物的形成。然后将产物溶解在乙醇/0.1m hcl(2:1的比)中,并且所得到的沉淀物被示出为cbd。
[0322]
实施例5:使用铵型氢氧化物合成铵-cbd络合物
[0323]
使用一系列的铵型氢氧化物诸如四甲基氢氧化铵、四乙基氢氧化铵、四丁基氢氧化铵、四戊基氢氧化铵或四己基氢氧化铵使用以下程序(方案7)来合成铵-cbd络合物:
[0324]
将cbd(1当量)在乙醇中的溶液与四丁基氢氧化铵(2当量)在乙醇中的溶液混合。
将反应混合物在室温搅拌过夜,其中乙醇被蒸发以得到铵-cbd络合物。
[0325]
实施例6:使用铵型氢氧化物合成铵-非诺多泮
[0326]
使用包含四乙基铵、四丁基铵和胆碱的一系列铵型氢氧化物来合成铵-非诺多泮络合物。将在乙醇:水(1:1)混合物中的非诺多泮甲磺酸盐(1当量)与四烷基氢氧化铵(3当量)在乙醇:水(1:1)中的溶液混合,并且在室温搅拌。通过tlc监测反应的完成。在溶剂蒸发之后,获得铵-非诺多泮络合物。
[0327]
实施例7:使用铵型氢氧化物合成铵-cbd络合物:
[0328]
铵型氢氧化物的合成:将多种胺诸如三甲胺、三乙胺、三丁胺、三己胺、n-甲基吡咯烷和n-甲基哌啶使用以下程序转化为氢氧化铵衍生物。这允许合成不同系列的苯酚铵络合物。
[0329]
在第一步中,通过胺(1当量)与在乙腈中的对应的烷基卤化物(1当量)的烷基化来合成四烷基取代的铵溴化物/碘化物。将反应混合物在75℃的温度剧烈搅拌持续12h以形成固体产物,将该固体产物过滤并且用己烷彻底洗涤,以获得纯的四烷基取代的卤化铵。
[0330]
这些合成的卤化铵衍生物和其他可商购的卤化铵衍生物使用以下程序被转化为氢氧化铵衍生物。
[0331]
将在乙醇中的四铵溴化物/碘化物(1当量)与koh(1当量)在乙醇中的溶液混合,并且逐渐加热至60℃过夜。通过过滤去除kbr沉淀物,以得到四烷基氢氧化铵溶液。通过蒸发去除溶剂,以得到四烷基氢氧化铵。
[0332]
cbd-铵盐的制备:将cbd(100mg,0.32mmol,1当量)放入带有磁棒的4ml的反应小瓶中。将反应小瓶保持在ar气氛下。然后加入1ml乙醇,并且搅拌以溶解cbd。之后,逐滴加入溶解在1ml乙醇中的四甲基氢氧化铵五水合物(115mg,0.64mmol,2当量)或四丁基氢氧化铵(40%水溶液,414μl,0.64mmol,2当量)的溶液。然后在rt搅拌反应混合物。使用10%乙酸乙酯/正己烷溶液作为洗脱剂检查tlc。在通过tlc确认的cbd的消耗之后,使用真空去除乙醇。将所得到的盐冻干过夜,这得到作为固体的产物。使用ft-ir、uv和nmr分析所获得的固体产物。
[0333]
使用卤化铵合成铵-cbd络合物:将四乙基溴化铵(3.15g,15mmol,1当量)或四戊基溴化铵(5.68g,15mmol,1当量)或四己基碘化铵(7.22g,15mmol,1当量)放入配备有磁棒的50ml圆底烧瓶中。加入溶剂乙醇(10ml),并且搅拌混合物以溶解四铵溴化物/碘化物。在观察到澄清溶液之后,加入溶解在乙醇中的koh(0.84g,15mmol,1当量)的溶液。然后将混合物逐渐加热至60℃。在24h之后,将混合物冷却至室温。之后通过过滤去除kbr沉淀物,这得到四烷基氢氧化铵溶液。通过蒸发去除溶剂,其给出四烷基氢氧化铵。
[0334]
将cbd(100mg,0.318mmol,1当量)放入带有磁棒的4ml的反应小瓶中。将反应小瓶保持在ar气氛下。然后加入1ml乙醇,并且搅拌以溶解cbd。之后,逐滴加入溶解在1ml乙醇中的四乙基氢氧化铵(94mg,0.636mmol,2当量)或四戊基氢氧化铵(201mg,0.636mmol,2当量)或四己基氢氧化铵(236mg,0.636mmol,2当量)的溶液。然后在rt搅拌反应混合物。使用10%乙酸乙酯/正己烷溶液作为洗脱剂检查tlc。在通过tlc确认的cbd的消耗之后,使用真空去除乙醇。将所得到的盐冻干过夜,这得到作为固体的产物。使用ft-ir、uv和nmr分析所获得的固体产物。
[0335]
铵-cbd络合物的合成:cbd在乙醇中表现出淡棕色的澄清溶液。当该溶液加入有四
丁基氢氧化铵和四甲基氢氧化铵在乙醇中的溶液时,当反应进行时,溶液的颜色逐渐变为深紫色。检查tlc以确认反应的完成。在48h的反应之后,观察到cbd的完全消耗。在底部处观察到铵-cbd络合物。然而,在tlc中观察到另外三个点。
[0336]
铵-cbd络合物的ft-ir分析:cbd及其四甲基铵和四丁基铵络合物的ft-ir光谱表现出其频率的显著变化。cbd的在1582cm-1
和1442cm-1
处的强带至在铵-cbd络合物中的1514cm-1
的变化指示芳族c=c键的变化,这示出cbd的转化。另外,在1628cm-1
处的频率的消失指示cbd的双键的变化。
[0337]
铵-cbd络合物的nmr分析:比较了cbd、铵型氢氧化物和铵-cbd络合物的1h nmr光谱。在铵-cbd络合物中,对应于cbd的两个oh的在6.18ppm和5.97ppm处的峰消失。另外,相应的铵型氢氧化物的甲基质子和丁基质子被并入铵-cbd络合物中。然而,由于涉及cbd的双键,在nmr光谱中观察到副反应。还观察到cbd的氧化产物。该化合物可以通过用合适的溶剂洗涤或柱色谱法被进一步纯化。
[0338]
铵-cbd络合物的uv分析:进行了铵-cbd络合物与cbd比较的uv分析。分析了在乙醇中的cbd或铵-cbd络合物(20μg/ml)的uv。cbd在229nm-235nm和274nm-281nm处示出吸收。在铵-cbd络合物中,这些吸收加宽并且位移。
[0339]
实施例8:使用铵型氢氧化物合成铵-非诺多泮络合物
[0340]
将非诺多泮甲磺酸盐(50mg,0.12mmol,1当量)放入带有磁棒的4ml的反应小瓶中。将反应小瓶保持在氩气气氛下。然后,加入1ml乙醇:水(1:1)混合物,并且搅拌以溶解非诺多泮甲磺酸盐。之后,逐滴加入溶解在1ml乙醇:水(1:1)混合物中的四甲基氢氧化铵五水合物(68mg,0.36mmol,3当量)或四丁基氢氧化铵(40%水溶液,243μl,0.36mmol,3当量)的溶液。然后在rt搅拌反应混合物。在通过tlc确认的cbd的消耗之后,使用真空去除乙醇。水使用冻干机过夜去除,这得到作为固体的产物。使用ft-ir、uv和nmr分析所获得的固体产物。
[0341]
铵-非诺多泮络合物的合成:非诺多泮甲磺酸盐在乙醇:水(1:1)混合物中表现出无色溶液。当该溶液加入有四甲基氢氧化铵或四丁基氢氧化铵在乙醇:水(1:1)中的溶液时变为淡绿色溶液。在反应完成之后,四甲基氢氧化铵和四丁基氢氧化铵的反应分别转化为深棕色和深棕色固体。
[0342]
比较了非诺多泮甲磺酸盐和四丁基铵-非诺多泮络合物的1h nmr光谱。在四丁基铵-非诺多泮络合物中,对应于非诺多泮的三个oh的在9.01ppm、8.94ppm和8.81ppm处的峰消失。另外,四丁基氢氧化铵的丁基质子被并入四丁基铵-非诺多泮络合物中。
[0343]
铵-非诺多泮络合物的uv分析:进行了铵-非诺多泮络合物与非诺多泮甲磺酸盐比较的uv分析。通过uv分析了在乙醇中的非诺多泮或铵-非诺多泮络合物(20μg/ml)。非诺多泮在218nm和284nm处示出吸收。在铵-非诺多泮络合物中,这些吸收加宽并且位移。在四甲基铵-非诺多泮络合物中,在218nm和284nm处的吸收分别向229nm和280nm位移。此外,在306nm和338nm处观察到新的吸收。在四丁基铵-非诺多泮络合物中,在218nm和284nm处的吸收分别向226nm和279nm位移。此外,在308nm和331nm处观察到新的吸收。
[0344]
实施例9:cbd-胆碱的合成、制剂及药代动力学
[0345]
cbd-胆碱盐作为深紫色固体获得,其可溶于甲醇、乙醇和dmso,不溶于甲苯、己烷和氯仿,并且微溶于水。cbd-胆碱使用nmr和ft-ir分析来确认。cbd在ph~3从cbd-胆碱盐中成功地再生。cbd-胆碱盐的方法学扩展到诸如l-α-磷脂酰胆碱的其他胆碱衍生物。使用自
纳米乳化递送系统制剂(self nano-emulsifying drug delivery system formulation)将cbd-胆碱分散在水性介质中,其中颗粒具有流体动力学直径85nm以及ζ电位-3.7mv。在辛醇/水中确定logp 1.71。使用用于cbd的类似方法、通过hplc-ms检测cbd-胆碱盐。从酸化的等离子体中以高收率提取盐。
[0346]
使用氢氧化胆碱(46%在水溶液中)合成了cbd-胆碱盐。使用甲醇作为溶剂、在氮气气氛下在rt搅拌cbd(1.0当量)和氢氧化胆碱(2.2当量)。通过tlc监测反应的完成。在反应完成之后,在真空下去除甲醇,并且冻干以得到作为深紫色固体的cbd-胆碱。
[0347]
cbd可溶于甲醇,并且形成淡棕色澄清溶液。当cbd在甲醇中的溶液被逐滴加入到氢氧化胆碱在甲醇中的溶液中时,观察到深紫色的澄清溶液。当反应进行时,溶液的颜色增强为深紫色。盐作为深紫色固体被分离,其在溶剂的蒸发之后获得。
[0348]
表征:比较了cbd-胆碱盐与在甲醇-d4中的cbd的1h nmr光谱。在cbd-胆碱盐中,在6.08ppm处的对应于cbd的两个芳族c-h质子的峰位移至6.00ppm。另外,出现在5.28ppm、4.47ppm和4.43ppm处的cbd双键的其他质子在cbd-胆碱中位移至5.33ppm、4.51ppm和4.43ppm。nmr研究揭示了cbd-胆碱的形成。比较了cbd-胆碱和cbd的ir光谱。在cbd-胆碱中,在1643cm-1
、1622cm-1
和1581cm-1
中观察到的芳族频率位移到与cbd相比较的1640cm-1
和1561cm-1
,这证实了cbd-胆碱盐的形成。
[0349]
从cbd-胆碱盐中再生cbd。为了检查cbd从cbd-胆碱盐中的再生,将cbd-胆碱加入到ph=3的溶液中并且用cdcl3提取。cdcl3层清楚地示出cbd从cbd-胆碱盐中的再生,这通过了1h nmr证实。
[0350]
已经开发了用于通过hplc-ms检测cbd-胆碱的方法。对于范围从5ng/ml-1μg/ml的浓度,已经确定了线性曲线。已经确定了在峰auc(y轴)与cbd-胆碱浓度(x轴)之间的线性关系。
[0351]
cbd-胆碱盐的合成:使用氢氧化胆碱(46%在水溶液中)合成了cbd-胆碱盐,如方案7中示出的。使用甲醇作为溶剂、在氮气气氛下在rt搅拌cbd(1.0当量)和氢氧化胆碱(2.2当量)。通过tlc监测反应的完成。在反应完成之后,使用真空去除甲醇,并且使用冻干机干燥,这得到作为深紫色固体的cbd-胆碱。
[0352][0353]
高再生为cbd的纯cbd盐的合成:
[0354]
使用可容易地去除的副产物合成纯cbd盐。
[0355]
cbd-na盐:将cbd(300mg,0.953mmol,1当量)溶解在3ml干燥的thf中,然后在n2气氛下在10分钟的时间段内逐滴加入溶解在3ml干燥的thf中的nah(83mg,2.09mmol,2当量)。之后,允许反应混合物在rt、在n2气氛下搅拌持续24h。在反应结束之后,在反应混合物中形
成黑色沉淀物,收集该黑色沉淀物并且通过用正庚烷洗涤若干次(10ml
×
3)来纯化。收集沉淀物并且在温度控制的热风烘箱中干燥。收率:260mg(76%)。使用ft-ir和nmr分析所得到的cbd-na盐。
[0356]
cbd-胆碱(1:1)盐:将氢氧化胆碱溶液(46%水溶液,391μl,1.59mmol,1当量)放入20ml反应小瓶中。加入10ml甲醇并且涡旋以溶解。然后,在氮气气氛下逐滴加入溶解在10ml的甲醇中的cbd(500mg,1.59mmol,1当量)。将反应小瓶在氮气气氛下紧密地封闭,并且用铝箔覆盖。然后在rt搅拌反应混合物。使用10%乙酸乙酯/正己烷溶液作为洗脱剂检查tlc。在通过tlc确认的cbd的消耗(~48h)之后,使用真空去除甲醇。将所得到的盐冻干过夜,这提供作为深紫色固体的cbd-胆碱盐,其使用ft-ir和nmr来分析。
[0357]
cbd盐的修改的合成以及cbd回收:通过使用cbd和nah代替如方案1中示出的naoh的修改的程序合成了cbd-na盐。使用干燥的thf作为溶剂、在氮气气氛下在rt搅拌cbd(1.0当量)和nah(2.0当量)。在反应完成之后,收集cbd-na盐并且用正庚烷洗涤,以去除未反应的cbd和其他杂质。
[0358][0359]
nmr分析:在1h-nmr光谱中,与纯的cbd不同,cbd-na的nmr光谱在8.6ppm附近没有示出任何酚类oh共振。这代表了cbd-na的形成。此外,cbd-na的其他芳族和脂族的共振与cbd相比发生了位移。
[0360]
ir分析:分析了cbd-na的ft-ir光谱,并且与cbd进行了比较。cbd示出了关于芳族羟基基团在3518cm-1
和3406cm-1
附近的ft-ir伸缩频率。这些频率在其类似物cbd-na盐中消失,这证实了芳族羟基基团向其对应的cbd-na盐的转化。这些化合物的烷烃和烯烃c-h伸缩频率分别在2931cm-1
和2857cm-1
处示出。最重要地,这些化合物分别表现出在1643cm-1-1428cm-1
范围内和1251cm-1
的不同的芳族c=c和c-o伸缩频率。
[0361]
cbd-ch盐的合成、结果与分析:使用cbd和氢氧化胆碱(46%在水溶液中)合成了cbd-胆碱盐(1:1),如方案10中示出的。使用甲醇作为溶剂、在氮气气氛下在rt搅拌cbd(1.0当量)和氢氧化胆碱(1.0当量)。通过tlc监测反应的完成。在反应完成之后,使用真空去除甲醇,并且使用冻干机干燥,这得到作为深紫色固体的cbd-胆碱。
[0362]
cbd-胆碱盐的nmr分析:在dmso-d6中获得了1h nmr光谱。在cbd-胆碱盐中,在8.64ppm处的对应于酚类oh的峰消失。其他芳族质子和双键质子发生位移。与胆碱部分相关的峰对于三个ch3质子在3.11ppm处观察到,对于两个ch2质子在3.38ppm和3.81ppm处观察到,并且对于oh质子在5.52ppm处观察到。
[0363]
从cbd-胆碱(1:1)再生的cbd的hplc分析:对在hcl溶液(ph=1.0)中的cbd-胆碱盐(1:1)进行再生研究,随后使用氯仿提取游离的cbd。将适量的cbd-胆碱盐(详情请参见表1)
放入5ml小瓶中,向该小瓶中加入2ml的hcl溶液(ph=1.0)。加入几滴浓hcl以使溶液ph=1.0。将所得到的混合物保持在振荡器中持续30min。在30min的酸处理之后,向反应混合物中加入2ml的氯仿(重复提取3次,3
×
2ml),并且涡旋持续15min。将有机层和水层分离并且使用旋转蒸发仪干燥,随后冻干过夜。通过使用称重法和hplc研究测量了在酸处理之后从cbd-胆碱盐中产生的cbd的重量。结果在表2中给出。有趣地,根据hplc分析,cbd-胆碱(1:1)以约67.0% hplc再生为cbd。
[0364]
表2.在ph=1.0,从cbd-胆碱(1:1)盐再生的cbd的hplc研究。
[0365][0366]
概述:cbd-胆碱(1:1)盐以67.01%再生为cbd;使用cbd和nah成功地制备了cbd-na;这个方法适用于采用新鲜的cah2和mgh2来制备cbd-ca和cbd-mg。
[0367]
大麻二酚(cbd)-胆碱(ch)盐的药代动力学(pk):在对自由活动的大鼠施用cbd和ch-cbd盐之后cbd pk概况的比较。
[0368]
制剂性质
[0369]
将cbd和cbd-胆碱盐溶解在表3中示出的脂质和表面活性剂的混合物中。并且分散在水中以形成约30纳米的纳米分散体。该分散体通过iv或口服施用于大鼠。
[0370]
表3.用于将cbd及其盐类分散在水性介质中的pnl制剂。
[0371][0372]
用于iv施用:将每种分子溶解(2%w/w)在前纳米脂球(pro-nano liposphere)(pnl)制剂中(表3)。加入双蒸水(ddw)至在纳米悬浮液中的最终cbd浓度为0.2%w/w。
[0373]
对于口服施用:将每种分子溶解(5%w/w)在前纳米脂球(pnl)制剂中(表2)。加入双蒸水(ddw)至在纳米悬浮液中的最终cbd浓度为0.5%w/w。
[0374]
上文的百分比指的是盐的实际重量,对此量的校正在下文的表4和表5中提及:
[0375]
表4.通过mw差异比率(mw difference ratio)的剂量调节
[0376][0377]
表5.通过ms表征的校正因子和剂量调节
[0378][0379]
在iv施用和口服施用之后大麻二酚(cbd)-胆碱(ch)盐在大鼠中的药代动力学(pk):使用自由活动的大鼠模型进行药代动力学研究。对于静脉内施用,给予1mg/kg的剂量(对于盐的校正在表4和表5中)。两个组被随机分配为施用cbd(n=5)或ch-cbd(n=4)。在给药前5min以及在给药后5分钟、15分钟、30分钟和1h、1.5h、2h、4h、6h和8h取全身血样(0.35ml)。
[0380]
口服制剂通过采用15mg/kg的剂量的口服灌胃被施用于动物(对于盐的校正在表4和表5中)。两个组被随机分配为施用cbd(n=5)或ch-cbd(n=3)。在给药前5min以及在给药后0.33h、0.66h、1h、1.5h、2h、4h、6h、8h和10h取全身血样(0.35ml)。为了防止脱水,在血样的每次取出之后,施用等体积的生理溶液。血浆通过离心来分离(4000rpm,10min),并且在-20℃储存,等待分析。将150μl的血浆等分试样掺入10μl的内标大麻萜酚(cbg;1μg/ml)。将acn(200μl)加入到每个试管(管a)中并且涡旋混合持续1min。cbd和cbg的提取通过加入到每个试管(管a)中的正己烷(3ml)随后1min的涡旋混合来进行。在以4000rpm离心持续10min之后,将正己烷有机层转移到新的玻璃试管(管b)中并且蒸发至干燥(vacuum evaporation system(真空蒸发系统),labconco,kansas city,mo)。然后,管b在80μl的acn:水(80:20)中重构。将所得到的溶液(80μl)注入hplc-ms系统中。使用的柱:xterra ms c18柱3.5μm 2.1mm
×
100mm柱(milford,ma)。流动相:20:80(v/v)2mm乙酸铵/乙腈的等度流动相。稀释剂:20:80(v/v)水/乙腈。流量:0.2ml/min。柱温度:35℃
±
5℃。样品温度:20℃
±
5℃。检测质量(m/z):cbg-312.2和cbd-313.2,采用负的电喷射。
[0381]
cbd与ch-cbd盐药代动力学概况比较
[0382]
在血浆中发现的盐的量用表6中提及的0.17校正因子调节,以类似于用于iv施用的1mg/kg的剂量以及用于口服施用的15mg/kg的剂量。
[0383]
表6:在iv施用1mg/kg(或调节到该剂量)的cbd和cbd-胆碱之后的cbd的pk参数,所述cbd和cbd-胆碱以在水中悬浮(x10)直到8小时的pnl制剂给予。
[0384][0385]
*在p值《0.05的情况下发现的统计差异,#auc
0-∞
通过使用末端斜率对于无穷大来计算
[0386]
使用在p值为《0.05的情况下的t检验,同时将每个盐参数与cbd自身进行比较,在auc、cl和分布的体积之间在两个校正因子下均发现统计差异。
[0387]
表7:在po施用15mg/kg(或调节到该剂量)的cbd和cbd-胆碱之后的cbd的pk参数,所述cbd和cbd-胆碱以在水中悬浮(x10)直到10小时的pnl制剂给予。
[0388][0389]
*在p值《0.05的情况下发现的统计差异,#auc0-∞通过使用末端斜率对于无穷大来计算,$fabs=使用在通过iv施用的pnl中的cbd的数据来计算绝对生物利用度的%
[0390]
使用在p值为《0.05的情况下的t检验,未发现统计差异。在将cbd的实际量校正为ch-cbd盐(两者均通过mw和ms)之后,在cbd和ch-cbd盐之间的比较。在iv之后的cbd和cbd-胆碱盐的血液水平是相当的,然而胆碱盐的口服生物利用度显著更高。
[0391]
实施例10:氧苯酮和水杨酸辛酯的钠盐防晒霜的合成
[0392]
实施例44-实施例49的目的是氧苯酮和水杨酸辛酯的钠盐和胆碱盐的合成、表征和分析。氧苯酮(sigma-aldrich,usa)和水杨酸辛酯sigma-aldrich,usa)按原样使用。在varian 300mhz nmr光谱仪上使用dmso-d6作为溶剂获得了1h nmr光谱。
[0393]
在100ml圆底烧瓶中,将500mg的氧苯酮(2.19mmol)在氮气气氛下溶解在10ml的ch3oh中。然后,将溶解在10ml的甲醇中的87.6mg的naoh(2.19mmol)经长的时间段(1h)逐滴加入到氧苯酮溶液中。将溶液在室温保持搅拌过夜。然后在真空下去除甲醇,并且沉淀物在空气中干燥,以得到作为纯的氧苯酮钠盐的固体微黄色粉尘。
[0394]
在100ml圆底烧瓶中,将500mg的水杨酸辛酯(2.0mmol)在氮气气氛下溶解在10ml的ch3oh中。然后,将溶解在10ml的甲醇中的80mg的naoh(2.0mmol)经长的时间段(1h)逐滴加入到水杨酸辛酯溶液中。将溶液在室温保持搅拌过夜。然后将甲醇蒸发,并且沉淀物在空气中干燥,以得到作为纯的水杨酸辛酯钠盐的固体白色粉末。
[0395]
通过1h nmr对合成的氧苯酮和水杨酸辛酯的钠盐进行表征。在12.02ppm处的对应
于氧苯酮的-oh基团的峰在用naoh处理之后完全消失。在氧苯酮钠盐的情况下,氧苯酮的所有其他余下的峰都是原样的,具有一定的向高磁场的位移(up field shifting),这支持了氧苯酮苯酚盐阴离子的形成。类似地,在水杨酸辛酯钠盐的情况下,在10.60ppm处的对应于水杨酸辛酯的-oh基团的峰在盐形成之后完全消失。在水杨酸辛酯钠盐的情况下,水杨酸辛酯的所有其他余下的峰也是原样的,具有一定的向高磁场的位移,这支持了水杨酸辛酯苯酚盐阴离子的形成。
[0396]
胆碱-氧苯酮/水杨酸辛酯的盐分别使用氧苯酮或水杨酸辛酯以及用氢氧化胆碱、使用以下程序(方案11)来合成。
[0397][0398]
将氧苯酮/水杨酸辛酯(1.0当量)放入带有磁棒的反应小瓶中。将反应小瓶保持在氮气气氛下。然后加入甲醇,并且搅拌以溶解氧苯酮/水杨酸辛酯。之后,逐滴加入氢氧化胆碱(1.1当量)水溶液。然后在rt搅拌反应混合物。通过tlc监测反应的完成。在反应完成之后,使用真空在rt去除甲醇。所得到的盐使用冻干机干燥,这得到胆碱-氧苯酮/水杨酸辛酯的盐。
[0399]
实验程序:将氧苯酮(500mg,4.38mmol,1.0当量)或水杨酸辛酯(548mg,4.38mmol,1.0当量)放入带有磁棒的20ml反应小瓶中。将反应小瓶保持在氮气气氛下。然后加入10ml甲醇,并且搅拌以溶解氧苯酮/水杨酸辛酯。之后,逐滴加入氢氧化胆碱溶液(46%水溶液,592μl,4.82mmol,1.1当量)。然后在rt搅拌反应混合物。在通过tlc确认的氧苯酮/水杨酸辛酯的消耗之后,使用真空去除甲醇。将所得到的产物冻干过夜,这得到作为粘性液体的产物(胆碱-氧苯酮-淡棕色澄清粘性液体;胆碱-水杨酸辛酯-淡黄色澄清粘性液体),所述产物使用ft-ir和nmr来分析。
[0400]
结果与讨论:氧苯酮/水杨酸辛酯在甲醇中表现出淡黄色/无色的澄清溶液。在向此溶液中加入氢氧化胆碱水溶液之后,当反应进行时,反应混合物逐渐变为黄色/淡黄色的澄清溶液。在反应完成之后,去除甲醇,并且获得作为粘性液体的产物(胆碱-氧苯酮-淡棕色澄清粘性液体;胆碱-水杨酸辛酯-淡黄色澄清粘性液体)。
[0401]
对氧苯酮和胆碱-氧苯酮盐的1h nmr光谱进行了比较。在胆碱-氧苯酮盐中,在12.03ppm处的对应于氧苯酮的oh的峰消失。此外,在胆碱-氧苯酮盐中,由于苯酚盐离子的较大电子密度,氧苯酮的芳族质子发生位移(向高磁场)。另外,氢氧化胆碱的三个甲基(ch3)基团的九个质子在3.59ppm处并入。此外,氢氧化胆碱的两个亚甲基(ch2)基团在3.73ppm和3.28ppm处被观察到。
[0402]
氧苯酮或水杨酸2-乙基己酯的铁(iii)盐。将fecl3(1.2当量)在乙醇中的溶液加入到氧苯酮或水杨酸2-乙基己酯(1.0当量)在乙醇中的搅拌的混合物中。紫色立即可见。用铝箔覆盖混合物,并且使其在rt搅拌过夜。将溶剂蒸发,并且将粗产物溶解在氯仿中,并且
滴入己烷中。然后将混浊的混合物离心,并且将上清液蒸发,以定量收率提供具有紫色颜色的粉末。
[0403]
实施例11:沙波立沙钠(snac)盐的合成
[0404]
沙波立沙钠是沙波立沙的羧酸钠盐形式,一种口服吸收促进剂。沙波立沙钠被用作促进维生素b、胰岛素、肝素和最近的塞马鲁肽(semaglutide)的口服吸收的递送剂(delivery agent)。在本实施例中,已经制备了金属和铵的苯酚盐,目的是改进制剂以及改进口服生物利用度。使用上文描述的方法中的一种制备了钠、钾、钙和铁的苯酚盐。已经使用上文描述的方法制备了具有胆碱和四乙基铵离子的铵盐。这些盐已经用于改进塞马鲁肽和胰岛素的口服生物利用度。
[0405]
下文呈现的是snac盐的化学结构。1-snac;2-snac-钠;3-snac-胆碱;4-snac-磷脂酰胆碱。
[0406][0407]
制剂制备:通过逐步混合下文表8中呈现的组分来制备制剂。所有的化合物都是粉末,并且每个材料比率是按质量计。
[0408]
表8:制剂的组成。所有的化合物都是粉末,并且每个比率是按质量计。
[0409]
成分%w/w塞马鲁肽2.4snac或snac-盐70.7pvp k901.9avicel ph10123.6硬脂酸镁1.4
[0410]
体内研究:
[0411]
对于每种snac或盐制剂使用4只雄性wistar大鼠(harlan,israel)。
[0412]
药代动力学研究方案:称重为275g-300g的雄性wistar大鼠(harlan,israel)在试验前保持在12h光/暗循环下,并且自由获得食物(标准大鼠食物)和水。动物在手术阶段被麻醉。在每只动物的右颈静脉中放置留置插管,用于全身血液采样。插管在皮肤下方打开通
道,并且在颈的背部外置。在手术程序完成之后,动物被转移到单独的笼子以过夜恢复(12h-18h)。在所述恢复时间段期间,如果进行口服吸收实验,则剥夺食物,而不剥夺水。在整个实验中,口服施用后4h可以自由获得食物。
[0413]
动物被随机分配到不同的实验组。将口服snac或盐制剂分散在蒸馏水中,然后通过口服灌胃施用(对于大鼠为~1.2ml,以得到12mg/kg的塞马鲁肽剂量)。全身血样(0.36ml)由放置在颈静脉中的静脉插管获得。在口服施用的情况下,根据药代动力学概况,在给药前5分钟以及在给药后不同时间点取样(将抽取不超过大鼠血容量的10%的血液)。为了防止脱水,在每次血液采样后向大鼠施用等体积的生理溶液。血浆通过离心来分离(4000rpm,7分钟,4℃),并且在-20℃储存,等待分析。
[0414]
通过对塞马鲁肽开发的lc-ms方法来分析血浆样品。
[0415]
结果:
[0416]
盐合成表征:
[0417]
溶解度:snac及其盐可溶于水,除了snac-pc,其是水不溶性的。当降低ph至酸性ph(1.2)时,如在肠环境中存在的那样,snac和盐沉淀。nmr证实,在此条件下,盐再生为snac。
[0418]
药代动力学吸收概况:
[0419]
在snac和盐制剂的口服施用之后长的时间以大鼠血浆中塞马鲁肽浓度来检查了塞马鲁肽在大鼠中的吸收。从半对数标度中的数据中提取pk参数。从表9中描述的图中提取pk参数。清楚地看出,与snac相比,snac-na制剂对塞马鲁肽吸收没有贡献,因为所有的pk参数与其他制剂相比都呈现较低的值:该制剂的auc、c最大和绝对生物利用度。snac-ch呈现与原始snac相似的pk值,但通过pc盐制剂对塞马鲁肽的吸收显示出最高的auc、c最大和生物利用度。%f(绝对生物利用度)在所有情况下是非常低的,然而,snac-pc的该值比其他的高一个数量级。塞马鲁肽的口服制剂的商业公司报告,其产品在犬中的生物利用度是1%。此外,与采用snac的概况相比,使用snac-pc的塞马鲁肽浓度在血液中随时间的降低慢得多,这指示一种用于经口施用塞马鲁肽的良好的潜在的和改进的制剂。对于pc盐是表现出改进的吸收概况的制剂的原因可能在于以下的事实:pc具有可以有助于制剂的疏水性的两个亲脂性的长链以及穿过肠壁进入血流的能力。还重要的是提及,这些制剂是通过质量比来制备。pc的分子量比snac大,所以在pc盐制剂的情况下,当前snac的量比snac制剂低得多。因此,如果将pc盐制剂中的snac量增加到与原始比相等,则可以获得甚至更好的结果。
[0420]
在以snac、snac-na、snac-ch或snac-pc制剂po施用12mg/kg的塞马鲁肽之后塞马鲁肽的pk参数在表9中示出。auc
0-∞#
通过使用末端斜率对于无穷大来计算。f
abs$
=使用在通过iv施用的snac制剂中的塞马鲁肽的数据来计算绝对生物利用度的%,如表10中呈现的。
[0421]
表9:在以snac、snac-na、snac-ch或snac-pc制剂po施用12mg/kg的塞马鲁肽之后塞马鲁肽的pk参数
[0422][0423][0424]
表10:在iv施用0.043mg/kg的塞马鲁肽之后的塞马鲁肽的pk参数,所述塞马鲁肽以在水中经过8小时的悬浮(x10)的snac制剂给予。
[0425]
平均值
±
sd在snac iv中的塞马鲁肽(n=2)auc
0-∞
(μg*h/l)7199
±
2527t
0.5
(h)3.91
±
0.26
[0426]
实施例12:二胆碱分子和三胆碱分子的合成
[0427]
发现氢氧化胆碱适合于制备活性含苯酚的分子的胆碱苯酚盐。为了获得具有多于一个铵部分的分子,胆碱通过使用光气或光气衍生物形成醚键或形成碳酸酯键而二聚化。柠檬酸和诸如草酸、丙二酸、癸二酸和富马酸的二元酸的胆碱酯已经使用酯化催化剂通过缩合而被制备。在聚羧酸分子诸如聚丙烯酸上的酯化可以形成具有多胆碱季铵位点的聚合物,所述多胆碱季铵位点可以用于形成苯酚盐。
[0428]
实施例13:微球和递送系统的制备
[0429]
姜黄素盐与铁或钙以1:1摩尔比形成一种材料,该材料可以被配制成负载有药物或活性剂的纳米颗粒和微米颗粒以用于递送到人类或动物身体,或者被用于控制递送农业物质诸如肥料和农药。这些化合物可以被用作用于将药物或化妆品递送到皮肤的载体。
[0430]
实施例14:用于治疗化脓性汗腺炎的塔匹那洛夫的制备
[0431]
塔匹那洛夫是通常用于治疗皮肤紊乱,特别是化脓性汗腺炎的酚类分子。该药物通过在患病部位处的皮肤下的注入来递送,或者使用软膏或乳霜被局部应用。本实施例的目的是制备塔匹那洛夫的金属盐或铵盐,以增加皮肤渗透性和/或允许该药物的延长释放。制备银、铜、锌和胆碱的盐,如上文描述的。这些盐在水性介质中释放药物持续从几天至4周的时间段。这些盐被配制成软膏和乳霜局部载体以及被配制在脂质或plga微球中用于在应用后的延长释放。
[0432]
实施例15:绿茶多酚纳米颗粒的制备
[0433]
从在水中以10mg/ml的浓度的绿茶中提取的多酚以1:100至1:10的摩尔比在fecl3的水溶液中混合以形成纳米颗粒。在室温4小时的混合之后,获得200纳米-400纳米的纳米颗粒。通过冻干来分离纳米颗粒。类似地,制备了ca盐和zn盐纳米颗粒。影响粒度的因素是金属离子与酚类基团的比。每个酚类基团的金属离子的量的增加以及水溶液中苯酚和金属离子的浓度的减小,降低了纳米颗粒的尺寸。
[0434]
类似地,通过将多酚溶解在naoh中并且向溶液中加入金属盐并混合持续30分钟直
到获得良好的沉淀物,制备了鞣酸与不同金属离子的纳米颗粒和微米颗粒。颗粒通过离心或过滤分离以形成酚类分子的自由流动的水不溶性纳米颗粒和微米颗粒。使用天然多酚和不同来源的多酚的混合物来制备具有金属离子的纳米颗粒和微米颗粒,用作用于控制药物递送的控制释放系统和药物包埋或者用作食品添加剂。合成的聚多巴胺与金属离子反应以形成离子型聚多巴胺。na、k和li的单价金属盐形成相对可溶性聚合物,而二价金属离子和三价金属离子形成不溶性材料。采用ca、mn、mg、zn、cu、al和fe制备了不溶性盐。
[0435]
在另外的实施例中,由与铵离子的氢氧化物的直接反应制备胆碱和四丁基铵的铵盐。
[0436]
实施例16:肉桂提取物的金属盐
[0437]
如在food sci.biotechnol.24(4):1201-1207(2015)中描述的水溶性肉桂提取物与钠盐和钾盐在水中反应以形成可溶性苯酚盐或者水不溶性肉桂提取物与ca、mn、mg、zn、cu、al和fe反应。盐的制备如上文描述。分子量在1000至20000的范围内的或呈纳米颗粒形式的高分子量肉桂提取物被用于制备金属盐或铵盐。这些盐在针对一系列微生物剂测试时已经示出抗病毒活性和抗微生物活性。
[0438]
实施例17:百里酚(2-异丙基-5-甲基苯酚)盐
[0439]
目的是在与百里酚比较的情况下2-异丙基-5-甲基苯酚金属盐的合成、表征和再生研究(方案11)。
[0440][0441]
通过在水/甲醇混合物的存在下,在rt或60℃使百里酚与其对应的碱金属氢氧化物即koh、lioh、naoh和ba(oh)2反应持续24h,分别合成了百里酚的k盐、li盐、na盐和ba盐。类似地,通过在水/甲醇混合物的存在下,在70℃使等摩尔量的百里酚与四丁基氢氧化铵(tba.oh)反应持续24h,制备了2-异丙基-5-甲基苯酚四丁基铵(参见方案11)。而通过在水的存在下,在rt搅拌2-异丙基-5-甲基苯酚钠与对应的过渡金属硫酸盐/氯化物cuso4、zncl2、fecl2和mncl2持续24h,分别制备了百里酚的cu盐、zn盐、fe盐和mn盐(参见方案12)。通过使用包括nmr、ir、uv-可见、dsc、edx和元素分析的多种分析和光谱技术对所有合成的百里酚盐进行充分地表征。
[0442][0443]
如下描述了多种2-异丙基-5-甲基苯酚金属盐的合成和表征细节:
[0444]
2-异丙基-5-甲基苯酚钾的合成:将溶解在5ml meoh中的百里酚(1g,6.656mmol)逐滴加入到在4ml水中的koh(0.373g,6.656mmol)中。之后,将反应混合物在rt搅拌持续24h。在反应完成之后,反应混合物在旋转蒸发仪下蒸发。将所得到的黑色粘性固体用正庚烷洗涤(10ml
×
3),并且在50℃的热风烘箱中干燥持续24h。收率:60%(0.760g)。ft-ir:ν最大/cm-1 2956-2865(νc-h伸缩),1589-1556-1487-1447-1397(νc=c伸缩),1292-1270-1242-1193-1165-1150(νc-o伸缩),1111,1085-1056-1006(νc-h平面内弯曲),951(νc=c弯曲),857-794-738(νc-h平面外弯曲)cm-1
。1h-nmr(300mhz,dmso-d6):δ=6.57(d,1h,j=9hz,ar-h),6.02(s,1h,ar-h),5.76(d,1h,j=6hz,ar-h),3.25-3.16(m,1h,ipr-h),1.98(s,3h,ar-ch3),1.01(d,6h,j=9hz,ipr-(ch3)2)。
[0445]
2-异丙基-5-甲基苯酚锂的合成:将溶解在6ml meoh中的百里酚(1g,6.656mmol)逐滴加入到在6ml水中的lioh(0.279g,6.656mmol)中。之后,将反应混合物在70℃搅拌持续24h。在反应完成之后,反应混合物在旋转蒸发仪下蒸发。将所得到的深棕色沉淀物用水(10ml
×
3)和正庚烷(10ml
×
3)洗涤若干次,并且在50℃的温度控制的热风烘箱中干燥持续24h。收率:87%(0.900g)。ft-ir:ν最大/cm-1 2957-2922(νc-h芳族),2865(νc-h脂族),
1596-1577-1562-1508-1447-1401(νc=c),1292-1274-1233-1169-1157-1112(νc-o),1086-1054-1037-1000(νc-h平面内弯曲),953-946(νc=c弯曲),867-859-803-745(νc-h平面外弯曲)cm-1
。1h-nmr(300mhz,dmso-d6):δ=6.69(d,1h,j=6hz,ar-h),6.31(s,1h,ar-h),5.97(d,1h,j=6hz,ar-h),3.48-3.26(m,1h,ipr-h),2.03(s,3h,ar-ch3),1.06(d,6h,j=6hz,ipr-(ch3)2)。
[0446]
2-异丙基-5-甲基苯酚四丁基铵的合成:将溶解在5ml meoh中的百里酚(1.00g,6.65mmol)逐滴加入到在5ml水中的tba.oh.30h2o(5.32g,6.65mmol)中。之后,将反应混合物在50℃搅拌持续24h。在反应完成之后,反应混合物在旋转蒸发仪下蒸发。在加入蒸馏水(40ml)之后,所得到的粘性固体给出小麦色沉淀物,该沉淀物使用布氏漏斗进一步收集,并且随后用蒸馏水(40ml
×
3)和正庚烷(20ml
×
3)洗涤若干次,然后在50℃的温度控制的热风烘箱中干燥持续24h。收率:1.20g(46%)。1h-nmr(300mhz,dmso-d6):δ=6.79(d,2h,j=6hz,arh),6.51(s,2h,arh),6.21(d,2h,j=6hz,arh),3.27-3.20(m,2h,ipr-h),3.18-3.13(m,8h,tba),2.06(s,3h,ar-ch3),1.61-1.51(m,8h,tba),1.36-1.24(m,8h,tba),1.11-1.09(m,6h,ipr-(ch3)2),0.95-0.91(m,12h,tba)。ft-ir:ν最大/cm-1 2960(νc-h芳族),2872(νc-h脂族),1586-1480-1458-1380(νc=c),1283-1235-1148(νc-o),1087-1050-1004(νc-h平面内弯曲),948(νc=c弯曲),883-864-798-738(νc-h平面外弯曲)cm-1
。
[0447]
2-异丙基-5-甲基苯酚钠的合成:将溶解在30ml meoh的百里酚(20g,133.13mmol)逐滴加入到在40ml水中的naoh(5.857g,146.451mmol)中。之后,将反应混合物在rt搅拌持续24h。在反应完成之后,反应混合物在旋转蒸发仪下蒸发。将所得到的黑色粘性固体用正庚烷洗涤若干次(50ml
×
3),并且然后在50℃的热风烘箱中干燥持续24h。ft-ir:ν最大/cm-1
2952(νc-h芳族),2864(νc-h脂族),1635-1595-1557-1491-1396(νc=c),1289-1263-1196-1166-1151(νc-o),1086-1054-1008(νc-h平面内弯曲),952(νc=c弯曲),860-795-739(νc-h平面外弯曲)cm-1
。1h-nmr(300mhz,dmso-d6):δ=6.66(d,1h,j=6hz,ar-h),6.22(s,1h,ar-h),5.93(d,1h,j=9hz,ar-h),3.25-3.16(m,1h,ipr-h),2.00(s,3h,ar-ch3),1.05(d,6h,j=9hz,ipr-(ch3)2)。
[0448]
2na(2-异丙基-5-甲基苯酚)4zn的合成:将溶解在70ml的干燥的thf中的na-百里酚(5g,29.03mmol)逐滴加入到在20ml干燥的thf中的zn(ii)cl2(1.009g,7.404mmol)中。之后,允许反应混合物在rt搅拌持续36h。在反应完成之后,通过离心将nacl从反应混合物中去除。此后,所得到的澄清溶液在减压下蒸发,随后在50℃的热风烘箱中干燥持续24h。收率:71%(3.742g)。ft-ir:ν最大/cm-1 2959(νc-h芳族),2868(νc-h脂族),1581-1506-1493-1456-1418(νc=c),1287-1230-1181-1152(νc-o),1087-1058(νc-h平面内弯曲),945(νc=c弯曲),861-805-738(νc-h平面外弯曲)cm-1
。1h-nmr(300mhz,dmso-d6):δ=6.93(d,1h,j=6hz,ar-h),6.57(s,1h,ar-h),6.48(d,1h,j=9hz,ar-h),3.22-3.08(m,1h,ipr-h),2.14(s,3h,ar-ch3),1.11(d,6h,j=9hz,ipr-(ch3)2)。
[0449]
所得到的结构如下:
[0450][0451]
2-异丙基-5-甲基苯酚钡的合成:将溶解在5ml meoh中的百里酚(0.5g,3.28mmol)逐滴加入到在10ml水中的ba(oh)2.8h2o(0.5g,1.64mmol)中。之后,允许反应混合物在70℃搅拌持续24h。在反应完成之后,反应混合物在旋转蒸发仪下蒸发。将所得到的黑色沉淀物用正庚烷洗涤若干次(10ml
×
3),并且在50℃的热风烘箱中干燥持续24h。收率:0.520g(36%)。ft-ir:ν最大/cm-1 2961(νc-h芳族),2872(νc-h脂族),1578-1423(νc=c),1290-1246-1177(νc-o),1089-1059(νc-h平面内弯曲),947(νc=c弯曲),855-807-768(νc-h平面外弯曲)cm-1
。1h-nmr(300mhz,dmso-d6):δ=6.95(d,1h,j=9hz,ar-h),6.56(s,1h,ar-h),6.53(d,1h,j=9hz,ar-h),3.32-3.08(m,1h,ipr-h),2.16(s,3h,ar-ch3),1.11(d,6h,j=9hz,ipr-(ch3)2)。
[0452]
所得到的结构如下:
[0453][0454]
2-异丙基-5-甲基苯酚铜(ii)的合成:将溶解在500ml蒸馏水中的cu(ii)so4.5h2o(139.199g,557.49mmol)缓慢加入到在1l蒸馏水中的na-百里酚(192g,1114.9mmol)中。在加入之后,观察到绿色沉淀物的立即形成。之后,允许反应混合物搅拌持续24h。在反应完成之后,反应混合物通过布氏漏斗过滤,随后用过量的蒸馏水洗涤(200ml
×
3)。将所得到的浅绿色沉淀物在50℃的温度控制的热风烘箱中干燥持续24h。收率:80%(160.3g)。由于cu(ii)离子的顺磁性质,该化合物对nmr分析是无活性的。ft-ir:ν最大/cm-1 2958(νc-h芳族),2868(νc-h脂族),1583-1491-1455-1417(νc=c),1338-1286-1243-1177-1155(νc-o),1088-1058-1004(νc-h平面内弯曲),943(νc=c弯曲),894-805-737(νc-h平面外弯曲)cm-1
。
[0455]
所得到的结构如下:
[0456][0457]
2-异丙基-5-甲基苯酚锌(ii)的合成:将溶解在5ml蒸馏水中的zn(ii)cl2(0.451g,3.31mmol)逐滴加入到在10ml蒸馏水中的na-百里酚(1.14g,6.62mmol)中。在加入
之后,观察到小麦色沉淀物的立即形成。之后,允许反应混合物在rt搅拌持续24h。在反应完成之后,反应混合物通过布氏漏斗过滤,随后用过量的蒸馏水洗涤(20ml
×
3)。将所得到的小麦色沉淀物在50℃的温度控制的热风烘箱中干燥持续24h。收率:71.3%(0.85g)。ft-ir:ν最大/cm-1 2962(νc-h芳族),2868(νc-h脂族),1551-1457-1418(νc=c),1289-1260-1155(νc-o),855-829-807(νc-h平面外弯曲)cm-1
。
[0458]
所得到的结构如下:
[0459][0460]
2-异丙基-5-甲基苯酚亚铁(ii)的合成:将溶解在5ml蒸馏水中的fe(ii)cl2(0.367g,2.903mmol)逐滴加入到在10ml蒸馏水中的na-百里酚(1g,5.87mmol)中。在加入之后,观察到小麦色沉淀物的立即形成。之后,允许反应混合物在rt搅拌持续24h。在反应完成之后,反应混合物通过布氏漏斗过滤,随后用过量的蒸馏水洗涤(20ml
×
3)。将所得到的小麦色沉淀物在50℃的温度控制的热风烘箱中干燥持续24h。收率:37%(0.380g)。ft-ir:ν最大/cm-1 2960(νc-h芳族),2869(νc-h脂族),1642-1611-1599-1506-1455(νc=c),1288-1255-1220-1154(νc-o),890-855-809(νc-h平面外弯曲)cm-1
。
[0461]
所得到的结构如下:
[0462][0463]
2-异丙基-5-甲基苯酚银的合成:将溶解在4ml干燥的乙腈中的agno3(0.986g,5.807mmol)逐滴加入到在10ml干燥的乙腈中的na-百里酚(1g,5.87mmol)中。之后,允许反应混合物在rt搅拌持续24h。在反应完成之后,反应混合物通过whatman滤纸过滤,随后使用旋转蒸发仪蒸发。将所得到的固体在50℃的温度控制的热风烘箱中干燥持续24h。收率:55%(0.820g)。ft-ir:ν最大/cm-1 2961(νc-h芳族),2870(νc-h脂族),1652-1537-1485-1412(νc=c),1289-1223-1175(νc-o),855-829-807(νc-h平面外弯曲)cm-1
。1h-nmr(300mhz,dmso-d6):δ=7.31(s,1h,ar-h),7.13(s,1h,ar-h),6.96(s,1h,ar-h),3.18-3.10(m,1h,ipr-h),1.85(s,3h,ar-ch3),1.17-1.10(m,6h,ipr-(ch3)2)。
[0464]
所得到的结构如下:
[0465][0466]
2-异丙基-5-甲基苯酚盐的表征
[0467]
nmr分析:通过使用nmr分析表征了在一系列的2-异丙基-5-甲基苯酚盐中有机单元的存在。所有样品在室温在氘代dmso-d6中记录。与百里酚形成对照,在这些样品中约9ppm处不存在芳族羟基共振表明百里酚是去质子化的,并且这一观察结果揭示了它们的盐特性。此外,与百里酚中的芳族质子相比,百里酚盐的芳族质子被屏蔽,这进一步揭示了它们的盐特性。由于其顺磁性质,2-异丙基-5-甲基苯酚的cu盐、fe盐、mn盐对nmr分析是无活性的。
[0468]
ir分析:记录了所有百里酚盐的ft-ir光谱,并且与百里酚进行了比较。百里酚示出羟基基团在3177.48cm-1
处的ft-ir伸缩频率,而此峰在其类似物百里酚盐中消失,这证实了它们的盐特性。然而,百里酚-li和百里酚-fe分别示出羟基基团在3436cm-1
和3387cm-1
处的伸缩频率。设想的是,这可能是由于在它的分子结构中配位的溶剂水分子,因为反应是在水中进行的。这些化合物的芳族和烷基(甲基/异丙基)c-h伸缩频率分别在2922cm-1-2961cm-1
和2865cm-1-2872cm-1
的范围内示出。最重要地,这些化合物分别表现出在1635cm-1-1418cm-1
和1290cm-1-1155cm-1
的范围内的芳族c=c和c-o伸缩频率。
[0469]
dsc分析:使用差示扫描量热法(dsc)在与百里酚比较的情况下确定了多种百里酚盐的熔化温度。dsc研究揭示了,百里酚盐示出与母体百里酚不同的熔化温度(参见表11)。
[0470]
表11:在与百里酚比较的情况下多种百里酚金属盐的熔化温度和焓。
[0471][0472]
uv-可见光谱法:为了研究电子跃迁,使多种2-异丙基-5-甲基苯酚盐经历uv-可见分析,并且与稀释浓度的在dmf中的百里酚进行比较。结果揭示了2-异丙基-5-甲基苯酚的zn盐、co盐、li盐、fe盐、ba盐和ag盐的吸收是蓝移的。而2-异丙基-5-甲基苯酚的tba盐、mn
盐和cu盐的吸收在百里酚的范围内。多种百里酚盐的吸收与百里酚的比较如下描述:百里酚:277nm,283nm;2-异丙基-5-甲基苯酚锌(ii):266nm;2-异丙基-5-甲基苯酚钴(ii):265nm;2-异丙基-5-甲基苯酚锂:269nm;2-异丙基-5-甲基苯酚亚铁(ii):264nm;2-异丙基-5-甲基苯酚四丁基铵:277nm,283nm;2-异丙基-5-甲基苯酚锰(ii):282nm;2-异丙基-5-甲基苯酚铜(ii):282nm;2-异丙基-5-甲基苯酚钡:268nm;2-异丙基-5-甲基苯酚银:268nm;2na(2-异丙基-5-甲基苯酚)4zn:277nm,282nm。分析清楚地证明了在2-异丙基-5-甲基苯酚盐中百里酚的存在。
[0473]
edx分析:对2-异丙基-5-甲基苯酚金属盐进行了edx分析,以便确定其中金属离子的组成百分比。具有不同原子组成和重量组成的包括ag、na、k、fe、zn、fe和mn的不同金属的存在证实了它们对应的2-异丙基-5-甲基苯酚金属盐的形成。
[0474]
百里酚从在酸性水性介质中的2-异丙基-5-甲基苯酚金属盐中的再生
[0475]
用于再生研究的实验程序:对在0.1n hcl溶液(1ph)中的2-异丙基-5-甲基苯酚盐进行再生研究,随后用庚烷提取游离的百里酚。将适量的对应的百里酚盐(表12)放入20ml玻璃小瓶中。向该玻璃小瓶中加入10ml的0.1n。将所得到的混合物保持在振荡器中持续30min。在30min的酸处理之后,向反应混合物中加入10ml的庚烷。将收集的有机层与水层分离,并且使用旋转蒸发仪小心地干燥,然后在35℃的温度控制的热烘箱中干燥持续12h。在多种百里酚盐的酸处理之后再生的百里酚的百分比在表12中呈现。
[0476]
表12:百里酚从在0.1n hcl溶液(ph=1)中的百里酚盐的再生研究。
[0477]
再生的百里酚的表征
[0478][0479][0480]
通过使用nmr和ir分析表征再生的百里酚。表征数据揭示,所有合成的百里酚盐均具有在酸处理之后转化回百里酚的能力。例如,从tba-异丙基-5-甲基苯酚盐获得的再生的百里酚示出在9.06ppm处的共振,这对应于芳族羟基基团。这指示tba-异丙基-5-甲基苯酚盐在酸性水性介质中转化为百里酚和tba-cl。另外,在相同的光谱中消失的tba共振证实了从tba-异丙基-5-甲基苯酚盐中返回的百里酚的再生。类似地,2-异丙基-5-甲基苯酚钠也通过在酸处理之后在9.06ppm处表现出羟基共振证实了百里酚的再生。此外,再生的百里酚的ft-ir光谱示出了在3424cm-1
处芳族羟基基团的伸缩频率,这进一步证实了从2-异丙基-5-甲基苯酚钠中返回的百里酚的再生。
[0481]
溶解度研究
[0482]
在不同ph的水溶液(ph=4、7、10)和包括乙醇、丙酮和丙二醇的有机溶剂中进行了多种百里酚盐的溶解度研究。除了可溶于水的钠盐,这些盐在任何ph都不溶于水。一些盐在有机溶剂中具有一定的溶解度。
[0483]
上文实验的总结
[0484]
在与母体百里酚分子进行比较的情况下已经制备了百里酚的na盐、k盐、tba盐、mn盐、fe盐、ag盐、cu盐、li盐、ba盐和zn盐,并且进行分析。通过在1h-nmr光谱中约9ppm处的盐的消失的羟基共振证实了对应的盐的形成。与百里酚相比,所有这些化合物在3177.48cm-1
处没有ft-ir伸缩频率进一步证实了它们的盐特性。dsc研究揭示了,与百里酚相比,这些样品具有高熔化温度。在酸性水性介质(ph=1)中研究了百里酚从2-异丙基-5-甲基苯酚盐中的再生,并且表征研究揭示了从所有样品中以其纯的形式返回的百里酚的再生。苯酚盐的熔点高于原始的苯酚,这允许在高温用塑料挤出或蒸汽发泡来加工盐。
[0485]
实施例18:苯酚盐的抗微生物活性
[0486]
苯酚盐展示了在保护作物免受细菌感染和真菌感染中的活性。测试了以下材料:水杨酸铜、水杨酸、百里酚铜和百里酚。
[0487]
针对以下污染物测试了材料:欧文氏菌(细菌)、腐霉(真菌)、菜豆壳球孢菌(真菌)、罗耳阿太菌(真菌)和马铃薯疮痂病(细菌)。
[0488]
收集被上文的病原菌感染的根并且进行去污染。欧文氏菌感染的根暴露于两个测试。在第一个测试中,在30天之后测量腐烂的马铃薯的百分比。在第二个测试中,测量了在120天之后的作物收率。
[0489]
针对欧文氏菌、腐霉、菜豆壳球孢菌和罗耳阿太菌进行了实验室测试。
[0490]
马铃薯疮痂病在2020年春季110天的生长之后的作物中进行了评价。测定了结痂水平和作物收率两者。
[0491]
腐霉在2020年春季的田间试验中被进一步评价。在110天的生长之后测定腐霉水平。
[0492]
结果
[0493]
·
欧文氏菌和疮痂病两者在实验室条件下用所有剂被根除。
[0494]
·
水杨酸铜是针对腐霉感染最有活性的化合物。
[0495]
·
水杨酸铜相对于水杨酸示出改进的活性。
[0496]
·
对菜豆壳球孢菌的活性比其他真菌大。
[0497]
将铜和锌的百里酚盐在它们的制造期间并入聚苯乙烯泡沫中、并入水凝胶中,以及并入马铃薯种子的包衣材料和干草包装材料中。此外,在熔体挤出期间,将百里酚铜并入聚乙烯片材中。将盐并入这些制剂中,而百里酚由于其蒸发速率和低熔点而不能并入。在3周的时间段中,百里酚不断地释放到空气中,同时保护干草免受微生物污染。
[0498]
在典型的实验中,新鲜干草用百里酚包衣的包装和百里酚铜盐包衣的包装来打包。在2周之后,对照包变得发霉。包衣的包没有示出任何霉菌。
[0499]
实施例19:水杨酸/苯甲酸从包含6%的cu-水杨酸/苯甲酸的聚苯乙烯泡沫中的释放
[0500]
制备水杨酸铜、苯甲酸铜及其混合物,如上文的实施例中描述的。由于高熔点和水不溶性,这些盐能够在蒸汽吹塑(steam blowing)期间被有效地并入聚苯乙烯泡沫中。研究
了包含6%的cu-水杨酸或6%的cu-苯甲酸的聚苯乙烯泡沫托架(polystyrene foam tray)的水杨酸/苯甲酸释放性质。释放研究是在室温在ddw中采用150rpm的振荡进行的。对于每个托架取约10g的泡沫样品,并且加入到550ml的ddw中直到完全浸入。采用没有任何活性剂的聚苯乙烯泡沫作为参照物。在1h以及1天、2天、8天、16天和30天之后更换溶液。使用uv通过分别测定在298nm和225nm处的吸收,来分析溶液的水杨酸/苯甲酸的量。在可检测浓度非常低的情况下进行冻干过程。包含6%的cu-水杨酸的聚苯乙烯泡沫托架示出在30天内每个托架34.5mg的水杨酸释放,并且6%的cu-苯甲酸展示出在30天内每个托架30.7mg的水杨酸释放。水杨酸/苯甲酸从聚苯乙烯泡沫中的释放概况在下文表13中呈现。
[0501]
表13:水杨酸/苯甲酸从包含6%的cu-水杨酸/苯甲酸的聚苯乙烯泡沫中的释放(每个托盘以mg计的释放)
[0502][0503]
活性剂在整个聚苯乙烯泡沫中的分布的确定:在泡沫制备期间,由于不溶性问题,在最终产品中发生活性剂的不均匀分布是可能的。本文的方法是分析活性剂在泡沫中的分布。
[0504]
从托架的若干区段中取小部分并且分散在水中。使用uv吸收光谱法分析水杨酸(sa)和苯甲酸。金属离子,在这种情况下是铜(ii),使用锌试剂(zincon)进行分析。
[0505]
从样品泡沫的多个区段中取出小块,并且加入到水溶液中,该水溶液强烈搅拌持续24小时。此后,使用在296nm处的uv分析溶液的水杨酸含量。此外,该溶液使用锌试剂水溶液稀释,并且通过使用在620nm处的uv吸光度进行分析。对于所有样品获得相似的浓度,这指示泡沫中的均匀分布。
[0506]
结果揭示了,活性剂cu-sa和cu-苯甲酸盐两者在聚苯乙烯泡沫中均匀地分布。
[0507]
这些托架用于种植被多种病毒污染的番茄植物。在包含水杨酸铜或苯甲酸铜的托架中的植物生长良好,并且没有显示出任何污染的感染。在另一方面,空白聚苯乙烯托架中的植物受到严重感染。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1