用于多维基因组分析的装置和方法与流程
用于多维基因组分析的装置和方法
1.相关申请的交叉引用
2.本文件要求2020年8月10日提交的美国临时申请序号63/063,728和2020年10月2日提交的美国临时申请序号63/087,131的优先权权益,这些美国临时申请中的每一个通过引用以其整体特此并入。
3.背景
4.众所周知,离散和远距离的基因组序列元件可以远距离调控基因功能(https://www.genome.gov/funded-programs-projects/encode-project-ency clopedia-of-dna-elements)。近年来,变得明显的是基因组的空间组织是其功能的关键。基因组如何调控其功能不仅与线性序列信息的一级水平相关,还与基因组所在的物理构型相关。序列元件和其他细胞组分如何以空间和时间的方式以顺式或者反式彼此相互作用,影响它们如何发挥其作用。哺乳动物基因组在空间上被组织成亚核区室(subnuclear compartment)、区域、高级折叠复合物、拓扑关联结构域(tad)和环,以促进基因调控和其他重要的染色体功能,诸如复制。这些结构可能是许多异常基因组重组和具有病理后果或生物学影响的错误的来源。已提出,染色体区域、区室、拓扑关联结构域(tad)、染色质环和局部直接调控因子的结合、基因组dna聚合物的弯曲和弯折以涉及许多核和细胞组分(诸如转录因子、阻遏因子、绝缘子、反式激活因子和酶)的复杂和精巧的方式调控。这些三维区域、区室、tad和环究竟是如何产生或调控的,仍在深入研究中,并且尚不清楚。能够在其天然基因组、亚细胞和亚核背景下对这些复杂的动态相互作用直接可视化和作图的技术对于理解一级测序信息如何与基因组的3-d组织联系起来非常有价值,并且从而有助于更好地理解和表征基因的调控以及最终生物学和病理生理学功能和后果。
5.染色体构象捕获(3c)方法,在本文件中称为“接近性3d作图(proximity 3d mapping)”((dekker等人,science 295,1306-11,2002;zhao等人,nat.genet.38,1341-47,2006;dostie等人,genome research 16,1299-1309,2006;lieberman-aiden等人,science 326,289-93,2009)已被广泛用于研究不同物种和细胞类型中的染色质组织。这些方法及其变型采用甲醛介导的交联然后原位酶促消化和接近连接来推断基因组基因座之间的空间关系。它们有助于阐明染色质折叠的原理。使用这些技术的研究已经证实了多层基因组组织的存在,诸如染色体区域(chromosome territories)、染色体区室(chromosome compartments)(lieberman-aiden等人,science 326,289-93,2009)、拓扑关联结构域(tad)(dixon等人,nature 485,376-80,2012)、亚tad(phillips-cremins等人,cell 153,1281-95,2013)、绝缘邻域(insulated neighborhoods)(dowen等人,cell 159,374-87,2014)和染色质环(rao等人,cell 159,1665-80,2014)。
6.绝大多数接近性3d作图方法依赖于甲醛介导的交联,这在染色质中产生广泛的蛋白质-蛋白质和蛋白质-dna共价连接。这些交联可以掩蔽某些限制性位点,并且阻止它们完全消化。部分消化片段的连接导致对它们实际基因组接近性的不精确推断。需要可以理想地暴露所有潜在的限制性位点的新的交联策略,以在所有长度尺度上普遍捕获接近性接触(proximal contact)。此外,现有方法限于捕获特定时刻的接近信息,通常具有对精确时间
点的性质的最小控制。理想地,可以在特定的期望时间点收集接近信息,和/或在所监测的持续时间内收集特定持续时间内的接近关系变化。另外,包含长核酸分子的样品的消化将可能排除从样品阐明精确的长程结构变异信息的能力,特别是如果样品来源于不同的汇集细胞。
7.除了接近性3d作图方法之外,已经开发了另外一组3d物理作图方法,其包括“超分辨率显微术”方法[jerkovic,2021],以使得能够对样品的结构信息进行原位荧光探查,与需要消化样品的“接近性3d作图”方法不同,其通常采用允许同时并行探查多于一个位置的多重化技术,同时保持样品的物理完整性。然而,如果完全处于固定折叠构型,这样的3d物理作图方法具有有限的空间和时间分析能力。另外,接近关系的分辨率和多重化通量受到探查占据小体积(单微米)3d空间的样品,诸如中期染色体的挑战。
[0008]
对鉴定和评估各种分子复合物和实体在各种特定时间点和持续时间上的物理接近性和结构,同时保持样品的长程完整性的另外的组合物和方法仍然存在需求。此处,我们提出了动态探查和分析长核酸分子及其相关的高级核酸结构的新的装置和方法。
[0009]
发明概述
[0010]
本文提供了用于分析长核酸分子的高级核酸结构的方法和装置,其包括:(a)将具有高级结构的长核酸分子的至少一部分定位在微流体装置的狭缝通道内,使得所述部分中的至少一部分(at least portion of said portion)可以被拉长(elongate),(b)探查所述高级结构的动态状态。
[0011]
本文还提供了用于分析长核酸分子的动态高级核酸结构的方法和装置,其包括:(a)将具有高级结构的长核酸分子的至少一部分定位在微流体装置的狭缝通道内,使得所述部分中的至少一部分可以被拉长;(b)沿着所述拉长部分探查物理图谱并探查高级结构。
[0012]
本文还提供了用于分析长核酸分子的动态高级核酸结构的方法和装置,其包括:(a)将具有高级结构的长核酸分子的至少一部分定位在微流体装置的狭缝通道内,使得所述高级结构的至少一部分可以被拉长;(b)沿着所述拉长部分探查物理图谱并探查高级结构。
[0013]
本文还提供了用于分析长核酸分子的动态高级核酸结构的方法和装置,其包括:(a)将具有高级结构的长核酸分子的至少一部分固定在多孔凝胶材料内;(b)将所述结构暴露于至少一种试剂。
[0014]
本文还提供了用于分析长核酸分子的高级核酸结构的方法和装置,其包括:(a)通过使与所述分子关联的蛋白质消化或变性而使所述长核酸分子的至少一部分膨胀(swelling);(b)将所述膨胀的具有高级结构的长核酸分子的至少一部分固定在多孔凝胶材料内;(c)将所述结构暴露于至少一种试剂。
[0015]
本文还提供了用于分析长核酸分子的高级核酸结构的方法,其包括:(a)将构成fret对的两种标记体各自结合到包含高级核酸结构的长核酸分子上的不同位置;(b)用光学探查系统探查和监测由fret对的受体产生的fret信号。
[0016]
本文还提供了用于分析长核酸分子的高级核酸结构的方法,其包括:(a)将包含构成fret对的两种标记体各自结合到各自包含高级核酸结构的不同长核酸分子上的不同位置,(b)用光学探查系统探查和监测由fret对的受体产生的fret信号。
[0017]
此处还提供了用于分析长核酸分子的高级核酸结构的方法,其包括:(a)将实体引
入生物分子溶液中,所述实体包含至少两个捕获探针,其中每个捕获探针包含条形码和捕获结构域,并且至少一个捕获探针是通过至少一个可裂解接头与所述实体连接的可释放捕获探针;(b)允许至少两个捕获探针通过它们各自的捕获结构域与它们各自的靶生物分子结合;(c)通过裂解所述至少一个可释放捕获探针的至少一个可裂解接头从所述实体释放所述至少一个可释放捕获探针。
[0018]
通过参考本文实施方案的以下编号的方面进一步阐明本公开内容:
[0019]
1.本公开内容的方面包括一种流体装置,所述流体装置包括封闭的狭缝流体通道以及荧光探查系统,所述封闭的狭缝流体通道被配置为容纳包含至少一种高级核酸结构的长核酸分子,其中所述分子的至少一部分处于拉长状态,所述荧光探查系统被配置为探查所述长核酸分子。
[0020]
2.根据方面1所述的装置,所述装置还包含所述长核酸分子。3.根据方面2所述的装置,其中所述长核酸分子包含至少一个荧光标记体。4.根据方面1所述的装置,其中所述封闭的狭缝流体通道具有小于所述荧光探查系统的焦深的3倍的深度。5.根据方面1所述的装置,其中所述封闭的狭缝流体通道具有不超过1微米的深度。6.根据方面1所述的装置,其中所述封闭的狭缝流体通道具有不超过500nm的深度。7.根据方面1所述的装置,其中所述封闭的狭缝流体通道具有不超过100nm的深度。8.根据方面1所述的装置,其中所述封闭的狭缝流体通道具有不超过50nm的深度。9.根据方面1所述的装置,其中所述封闭的狭缝流体通道具有不超过25nm的深度。10.根据方面2所述的装置,其中处于拉长状态的所述分子的至少一部分指定物理图谱。11.根据方面10所述的装置,其中所述物理图谱是线性物理图谱。12.根据方面10所述的装置,其中所述物理图谱包含荧光标记体。13.根据方面2所述的装置,其中在第一时间点和第二时间点探查所述结构。14.根据方面13所述的装置,其中在所述第一时间点的探查指示第一物理构象,并且在所述第二时间点的探查指示第二物理构象。15.根据方面2所述的装置,其中所述长核酸分子在所述探查之前当在所述流体装置中时暴露于试剂。16.根据方面2所述的装置,其中所述长核酸分子在所述探查期间当在所述流体装置中时暴露于试剂。17.根据方面2所述的装置,其中所述高级核酸结构与性状相关。18.根据方面2所述的装置,其中所述高级核酸结构与状况相关。19.根据方面18所述的装置,其中所述状况是疾病。20.根据方面18所述的装置,其中在第一时间点探查所述高级核酸结构的状态产生与所述状况有关的信息。21.根据方面20所述的装置,其中在第二时间点探查所述高级核酸结构产生与所述状况有关的信息。22.根据方面2所述的装置,其中所述高级核酸结构包含核小体。23.根据方面2所述的装置,其中所述高级核酸结构包含核小体簇(nucleosome clutch)。24.根据方面2所述的装置,其中所述高级核酸结构包含染色质。25.根据方面2所述的装置,其中所述高级核酸结构包含染色质纳米结构域。26.根据方面2所述的装置,其中所述高级核酸结构包含ccctc结合因子。27.根据方面2所述的装置,其中所述高级核酸结构包含环。28.根据方面2所述的装置,其中所述高级核酸结构包含拓扑关联结构域。29.根据方面2所述的装置,其中所述高级核酸结构包含环结构域。30.根据方面2所述的装置,其中所述高级核酸结构包含区室a。31.根据方面2所述的装置,其中所述高级核酸结构包含区室b。32.根据方面2所述的装置,其中所述高级核酸结构包含增强子启动子复合物。33.根据方面2所述的装置,其中所述高级核酸结构包含绝缘子复合物。34.根据方面2所述的装置,其中所述高级核酸结构包含转录因子复合物。35.根据方面2所述的装置,
其中所述高级核酸结构包含ctcf蛋白。36.根据方面2所述的装置,其中所述高级核酸结构包含pds5蛋白。37.根据方面2所述的装置,其中所述高级核酸结构包含wapl蛋白。38.根据方面2所述的装置,其中所述高级核酸结构包含异染色质。39.根据方面2所述的装置,其中所述高级核酸结构包含常染色质。40.根据方面2所述的装置,其中所述高级核酸结构包含异染色质-常染色质边界。41.根据方面2所述的装置,其中所述高级核酸结构包含转录因子。42.根据方面2所述的装置,其中所述高级核酸结构包含甲基结合蛋白。43.根据方面2所述的装置,其中所述高级核酸结构包含外源核酸基因组整合复合物。44.根据方面43所述的装置,其中所述外源核酸基因组整合复合物包括病毒基因组整合复合物。45.根据方面43所述的装置,其中所述外源核酸基因组整合复合物包括重组核酸。46.根据方面2所述的装置,其中所述高级核酸结构包含染色体外附加体物理对接复合物(extrachromosomal episome physical docking complex)。47.根据方面46所述的装置,其中所述染色体外附加体物理对接复合物通过至少一个结合位点承载(hosts)染色体。48.根据方面2所述的装置,其中所述高级核酸结构包含源自宿主染色体的染色体外核酸。
[0021]
49.本公开内容的方面包括一种流体装置,所述流体装置包括流体通道和至少一种试剂,所述流体通道包含能够固定包含至少一种高级核酸结构的长核酸分子的至少一部分的多孔凝胶。
[0022]
50.根据方面49所述的流体装置,其中包含至少一种高级核酸结构的长核酸分子使所述分子的至少一部分固定在所述多孔凝胶中。51.根据方面50所述的流体装置,其中所述长核酸分子包含至少一个荧光标记体。52.根据方面50所述的流体装置,其中所述长核酸分子由荧光探查系统探查。53.根据方面49所述的流体装置,其中用于将所述长核酸分子固定至所述凝胶的时间点基于选择标准来选择。54.根据方面53所述的装置,其中用于固定所述长核酸分子的所述时间点的所述选择标准与事件有关。55.根据方面54所述的装置,其中所述事件包括引入试剂。56.根据方面54所述的装置,其中所述事件包括特定环境条件的改变。57.根据方面54所述的装置,其中所述事件包括对所述长核酸分子的光学探查的分析结果。58.根据方面54所述的装置,其中所述事件包括鉴定其中包含所述长核酸分子的细胞的状态。59.根据方面58所述的装置,其中所述状态是特定的细胞周期点。60.根据方面59所述的装置,其中所述细胞周期点是间期。61.根据方面59所述的装置,其中所述细胞周期点是前期。62.根据方面59所述的装置,其中所述细胞周期点是前中期。63.根据方面59所述的装置,其中所述细胞周期点是中期。64.根据方面59所述的装置,其中所述细胞周期点是后期。65.根据方面59所述的装置,其中所述细胞周期点是末期。66.根据方面58所述的装置,其中所述状态为处于细胞生命的特定点。67.根据方面66所述的装置,其中所述细胞生命的特定点是细胞凋亡。68.根据方面58所述的装置,其中所述状态表现出特定的细胞形态。69.根据方面68所述的装置,其中所述细胞形态包括起泡(blebbing)。70.根据方面68所述的装置,其中所述细胞形态包括质壁分离。71.根据方面68所述的装置,其中所述细胞形态包括核碎裂。72.根据方面68所述的装置,其中所述细胞形态包括核固缩。73.根据方面68所述的装置,其中所述细胞形态包括胞吞作用。74.根据方面68所述的装置,其中所述细胞形态包括吞噬作用。75.根据方面68所述的装置,其中所述细胞形态包括病毒出芽。76.根据方面68所述的装置,其中所述细胞形态包括分泌性裂解。77.根据方面66所述的装置,其中所述细胞正在经历核酸片段化。78.根据方面66所述的装置,其中所述状态是经历特定的酶促活性。
79.根据方面78所述的装置,其中所述酶促活性是转录。80.根据方面55所述的装置,其中所述试剂暴露在固定在所述凝胶中之前开始。81.根据方面49所述的装置,其中所述长核酸分子在暴露于所述试剂之前被固定在所述凝胶中。82.根据方面49所述的装置,其中所述长核酸分子的蛋白质在被固定之前至少部分被消化。83.根据方面49所述的装置,其中所述长核酸分子的蛋白质在被固定之前至少部分变性。84.根据方面82所述的装置,其中所述长核酸分子是染色体。85.根据方面82所述的装置,其中所述长核酸分子具有表现出膨胀的物理构象。86.根据方面85所述的装置,其中所述膨胀导致在所述流体装置内占据的体积的至少两倍增加。87.根据方面49所述的装置,其中用于接近性3d作图的至少一个处理步骤包括使用所述试剂。88.根据方面49所述的装置,其中用于3d物理作图的至少一个处理步骤包括使用所述试剂。89.根据方面49所述的装置,其中所述长核酸分子的至少一部分指定被探查的物理图谱。90.根据方面49所述的装置,其中所述高级核酸结构与性状相关。91.根据方面49所述的装置,其中所述高级核酸结构与状况相关。92.根据方面91所述的装置,其中所述状况是疾病。93.根据方面91所述的装置,其中在第一时间点探查所述高级核酸结构的状态产生与所述状况有关的信息。94.根据方面93所述的装置,其中在第二时间点探查所述高级核酸结构产生与所述状况有关的信息。95.根据方面49所述的装置,其中所述高级核酸结构包含核小体。96.根据方面49所述的装置,其中所述高级核酸结构包含核小体簇。97.根据方面49所述的装置,其中所述高级核酸结构包含染色质。98.根据方面49所述的装置,其中所述高级核酸结构包含染色质纳米结构域。99.根据方面49所述的装置,其中所述高级核酸结构包含ccctc结合因子。100.根据方面49所述的装置,其中所述高级核酸结构包含环。101.根据方面49所述的装置,其中所述高级核酸结构包含拓扑关联结构域。102.根据方面49所述的装置,其中所述高级核酸结构包含环结构域。103.根据方面49所述的装置,其中所述高级核酸结构包含区室a。104.根据方面49所述的装置,其中所述高级核酸结构包含区室b。105.根据方面49所述的装置,其中所述高级核酸结构包含增强子启动子复合物。106.根据方面49所述的装置,其中所述高级核酸结构包含绝缘子复合物。107.根据方面49所述的装置,其中所述高级核酸结构包含转录因子复合物。108.根据方面49所述的装置,其中所述高级核酸结构包含ctcf蛋白。109.根据方面49所述的装置,其中所述高级核酸结构包含pds5蛋白。110.根据方面49所述的装置,其中所述高级核酸结构包含wapl蛋白。111.根据方面49所述的装置,其中所述高级核酸结构包含异染色质、常染色质或异染色质-常染色质边界。112.根据方面49所述的装置,其中所述高级核酸结构包含转录因子。113.根据方面49所述的装置,其中所述高级核酸结构包含甲基结合蛋白。114.根据方面49所述的装置,其中所述高级核酸结构包含外源核酸基因组整合复合物。115.根据方面114所述的装置,其中所述外源核酸基因组整合复合物包括病毒基因组整合复合物。116.根据方面114所述的装置,其中所述外源核酸基因组整合复合物包括重组核酸。117.根据方面49所述的装置,其中所述高级核酸结构包含染色体外附加体物理对接复合物。118.根据方面117所述的装置,其中所述染色体外附加体物理对接复合物通过一个或更多个结合位点承载染色体。119.根据方面49所述的装置,其中所述高级核酸结构包含源自宿主染色体的染色体外核酸。
[0023]
120.本公开内容的方面包括分析两个区域的接近状态的方法,第一区域被第一标记体结合并位于包含高级核酸结构的第一核酸分子区段内,并且第二区域被第二标记体结合并位于包含高级核酸结构的第二核酸分子区段内,并且其中所述第一标记体和所述第二
标记体一起形成fret对,并且其中荧光成像探查系统监测并检测由所述fret对的受体发出的荧光信号。
[0024]
121.根据方面120所述的方法,其中所述第一核酸分子区段和所述第二核酸分子区段共有共同的磷酸二酯主链。122.根据方面120所述的方法,其中所述监测包括检测所述荧光信号的状态。123.根据方面122所述的方法,其中所述状态包括检测的持续时间。124.根据方面122所述的方法,其中所述状态包括信号幅度。125.根据方面122所述的方法,其中所述状态包括信号偏振。126.根据方面120所述的方法,其中同时探查接近对的群体,并且确定群体信号。127.根据方面120所述的装置,其中所述监测发生在一个时间段内。128.根据方面127所述的装置,其中所述时间段跨越对试剂的暴露。129.根据方面127所述的装置,其中所述时间段跨越对特定环境条件的暴露。130.根据方面127所述的装置,其中所述时间段跨越对细胞的特定状态的检测,其中所述长核酸分子包含在所述细胞中。131.根据方面130所述的装置,其中所述状态是特定的细胞周期点。132.根据方面131所述的装置,其中所述细胞周期点是间期。133.根据方面131所述的装置,其中所述细胞周期点是前期。134.根据方面131所述的装置,其中所述细胞周期点是前中期。135.根据方面131所述的装置,其中所述细胞周期点是中期。136.根据方面131所述的装置,其中所述细胞周期点是后期。137.根据方面131所述的装置,其中所述细胞周期点是末期。138.根据方面130所述的装置,其中所述状态是细胞生命的特定点。139.根据方面138所述的装置,其中所述特定点是细胞凋亡。140.根据方面139所述的装置,其中所述特定点包括核酸片段化。141.根据方面130所述的装置,其中所述状态表现出特定的细胞形态。142.根据方面141所述的装置,其中所述细胞形态包括起泡。143.根据方面141所述的装置,其中所述细胞形态包括质壁分离。144.根据方面141所述的装置,其中所述细胞形态包括核碎裂。145.根据方面141所述的装置,其中所述细胞形态包括核固缩。146.根据方面141所述的装置,其中所述细胞形态包括胞吞作用。147.根据方面141所述的装置,其中所述细胞形态包括吞噬作用。148.根据方面141所述的装置,其中所述细胞形态包括病毒出芽。149.根据方面141所述的装置,其中所述细胞形态包括分泌性裂解。150.根据方面130所述的装置,其中所述状态是经历特定的酶促活性。151.根据方面150所述的装置,其中所述酶促活性是转录。152.根据方面120所述的方法,其中所述高级核酸结构在微流体装置中被荧光探查。153.根据方面120所述的方法,其中所述核酸分子区段中的至少一个的至少一部分处于拉长状态。154.根据方面120所述的装置,其中所述高级核酸结构与性状相关。155.根据方面120所述的装置,其中所述高级核酸结构与状况相关。156.根据方面155所述的装置,其中所述状况是疾病。157.根据方面155所述的装置,其中在第一时间点结构探查所述高级核酸的状态的产生与所述状况有关的信息。158.根据方面156所述的装置,其中在第二时间点探查所述高级核酸的状态产生与所述状况有关的信息。159.根据方面120所述的装置,其中对所述高级核酸结构的状态的探查产生与疾病有关的信息。160.根据方面120所述的装置,其中对所述高级核酸结构的动力学的探查产生与疾病有关的信息。161.根据方面120所述的装置,其中所述高级核酸结构包含核小体。162.根据方面120所述的装置,其中所述高级核酸结构包含核小体簇。163.根据方面120所述的装置,其中所述高级核酸结构包含染色质。164.根据方面120所述的装置,其中所述高级核酸结构包含染色质纳米结构域。165.根据方面120所述的装置,其中所述高级核酸结构包含ccctc结合因子。166.根据方面120所述的装置,其中所述高级核酸结构包含环。167.根
据方面120所述的装置,其中所述高级核酸结构包含拓扑关联结构域。168.根据方面120所述的装置,其中所述高级核酸结构包含环结构域。169.根据方面120所述的装置,其中所述高级核酸结构包含区室a。170.根据方面120所述的装置,其中所述高级核酸结构包含区室b。171.根据方面120所述的装置,其中所述高级核酸结构包含增强子启动子复合物。172.根据方面120所述的装置,其中所述高级核酸结构包含绝缘子复合物。173.根据方面120所述的装置,其中所述高级核酸结构包含转录因子复合物。174.根据方面120所述的装置,其中所述高级核酸结构包含ctcf蛋白。175.根据方面120所述的装置,其中所述高级核酸结构包含pds5蛋白。176.根据方面120所述的装置,其中所述高级核酸结构包含wapl蛋白。177.根据方面120所述的装置,其中所述高级核酸结构包含异染色质、常染色质或异染色质-常染色质边界。178.根据方面120所述的装置,其中所述高级核酸结构包含转录因子。179.根据方面120所述的装置,其中所述高级核酸结构包含甲基结合蛋白。180.根据方面120所述的装置,其中所述高级核酸结构包含外源核酸基因组整合复合物。181.根据方面180所述的装置,其中所述外源核酸基因组整合复合物包括病毒基因组整合复合物。182.根据方面180所述的装置,其中所述外源核酸基因组整合复合物包括重组核酸。183.根据方面120所述的装置,其中所述高级核酸结构包含染色体外附加体物理对接复合物。184.根据方面183所述的装置,其中所述染色体外附加体物理对接复合物通过一个或更多个结合位点承载染色体。185.根据方面120所述的装置,其中所述高级核酸结构包含源自宿主染色体的染色体外核酸。
[0025]
186.本公开内容的方面包括追踪接近关系的方法,所述方法包括:(a)将实体引入生物分子溶液中,所述实体包含至少两个捕获探针,其中每个捕获探针包含条形码和捕获结构域,并且至少一个捕获探针是通过至少一个可裂解接头与所述实体连接的可释放捕获探针;(b)允许至少两个捕获探针通过它们各自的捕获结构域与它们各自的靶生物分子结合;(c)通过裂解所述至少一个可释放捕获探针的至少一个可裂解接头从所述实体释放所述至少一个可释放捕获探针。
[0026]
187.根据方面186所述的方法,其中单个生物分子被至少一个捕获结构域结合。188.根据方面186所述的方法,其中单个生物分子被至少两个捕获结构域结合。189.根据方面186所述的方法,其中所述实体内的至少一个捕获结构域与核酸非特异性结合。190.根据方面186所述的方法,其中所述实体内的至少一个捕获结构域与具有特定核酸序列的核酸特异性结合。191.根据方面186所述的方法,其中所述实体内的至少一个捕获结构域与特定蛋白质特异性结合。192.根据方面186所述的方法,其中所述实体内的至少一个捕获结构域与蛋白质非特异性结合。193.根据方面186所述的方法,其中所述实体内的至少一个捕获结构域在受到激活时与可用的靶生物分子结合。194.根据方面193所述的方法,其中所述激活包括光激活。195.根据方面186所述的方法,其中所述条形码属于关系集。196.根据方面195所述的方法,其中所述关系集包括相同的条形码。197.根据方面195所述的方法,其中每个实体具有独特的条形码关系集。198.根据方面195所述的方法,其中所述关系集包括关系子集。199.根据方面198所述的方法,其中关系子集用于鉴定捕获结构域的类型。200.根据方面198所述的方法,其中关系子集鉴定靶生物分子的类型。201.根据方面198所述的方法,其中关系子集鉴定所述捕获结构域在所述实体中相对于第二捕获结构域的物理位置。202.根据方面186所述的方法,其中至少一个捕获结构域被光不稳定保护基团加笼。203.根据方面
202所述的方法,其中所述光不稳定保护基团通过暴露于特定波长的光而降解。204.根据方面186所述的方法,其中所述实体包含可光裂解接头。205.根据方面186所述的方法,其中所述实体包含易被第一光波长裂解的第一可光裂解接头和易被第二光波长裂解的第二可光裂解接头。206.根据方面186所述的方法,其中所述实体包含可酶促裂解的可裂解接头。207.根据方面186所述的方法,其中所述实体包含可热裂解的可裂解接头。208.根据方面186所述的方法,其中所述实体包含可化学裂解的可裂解接头。209.根据方面186所述的方法,其中来自第一实体的至少一个捕获探针与所述生物分子结合,并且来自第二实体的至少一个捕获探针也与同一生物分子结合。210.根据方面186所述的方法,其中来自第一实体的至少一个可释放捕获探针与所述生物分子结合,并且在从所述第一实体释放所述可释放捕获探针之后,来自第二实体的至少一个捕获探针也与所述生物分子结合。211.根据方面186所述的方法,其中所述实体包括珠,所有捕获探针通过可裂解接头与所述珠连接。212.根据方面211所述的方法,其中所述珠是树枝状大分子。213.根据方面211所述的方法,其中所述珠是纳米颗粒。214.根据方面211所述的方法,其中所述珠具有基于待追踪的接近关系的所需空间体积而选择的尺寸。215.根据方面211所述的方法,其中所述珠的尺寸为至少5nm。216.根据方面211所述的方法,其中所述珠的尺寸为至少10nm。217.根据方面211所述的方法,其中所述珠的尺寸为至少25nm。218.根据方面211所述的方法,其中所述珠的尺寸为至少100nm。219.根据方面211所述的方法,其中所述珠的尺寸为至少500nm。220.根据方面211所述的方法,其中所述珠表现出荧光特性。221.根据方面211所述的方法,其中所述珠表现出光致发光特性。222.根据方面211所述的方法,其中所述珠表现出磁特性。223.根据方面186所述的方法,其中所述至少一个结合的生物分子是长核酸分子。224.根据方面223所述的方法,其中所述长核酸分子包含高级核酸结构。225.根据方面223所述的方法,其中所述长核酸分子的至少一部分被进一步处理。226.根据方面225所述的方法,其中所述长核酸分子与可释放捕获结构域结合。227.根据方面226所述的方法,其中所述处理在从所述实体释放所述可释放捕获结构域之后进行。228.根据方面225所述的方法,其中所述处理包括梳理。229.根据方面225所述的方法,其中所述处理包括在微流体装置中拉长。230.根据方面225所述的方法,其中所述处理包括产生物理图谱。231.根据方面225所述的方法,其中所述处理包括测序。232.根据方面225所述的方法,其中所述处理包括核酸分子的杂交。233.根据方面225所述的方法,其中所述处理包括荧光探针的结合。234.根据方面225所述的方法,其中所述处理包括扩增核酸的至少一部分。235.根据方面225所述的方法,其中所述处理包括酶促反应。236.根据方面225所述的方法,其中所述处理包括连接。237.根据方面225所述的方法,其中所述处理包括使用限制性内切酶进行消化。238.根据方面225所述的方法,其中所述处理包括使用切口酶产生切口。239.根据方面225所述的方法,其中所述处理包括使用聚合酶掺入核苷酸。
[0027]
240.本公开内容的方面包括测定长核酸的高级核酸复合物结构的方法,所述方法包括将高级核酸复合物结构定位在可视化区域中,确定所述高级核酸复合物的至少一部分的第一物理图谱,使所述高级核酸复合物经历反应,以及确定所述高级核酸复合物的至少一部分的第二物理图谱。
[0028]
241.根据方面240所述的方法,其中所述反应包括所述高级核酸复合物的部分降解。242.根据方面240所述的方法,其中所述反应包括向所述高级核酸复合物添加外源组
分。243.根据方面240所述的方法,所述方法包括使所述高级核酸复合物经历第二反应,并确定所述高级核酸复合物的至少一部分的第三物理图谱。244.根据方面240或243中任一项所述的方法,其中所述长核酸不经历磷酸二酯键的裂解。互联网上可得的和本说明书中提及的所有出版物、专利、专利申请和信息通过引用并入本文,其程度如同每个单独的出版物、专利、专利申请或信息项目被具体和单独地指明通过引用并入。就通过引用并入的出版物、专利、专利申请和信息项目与本说明书中包含的公开内容相矛盾的方面而言,本说明书旨在替代和/或优先于任何这样的矛盾材料。
[0029]
本说明书中使用的术语在本领域中、在本发明的上下文中以及在使用每个术语的特定上下文中通常具有它们的普通含义。某些术语在下文或本说明书中别处讨论,以在描述本发明的装置和方法以及如何制备和使用它们的方面向从业者提供另外的指导。应以该方式理解。因此,替代语言和同义词使同一事物通常可以用多于一种语言和同义词来描述,用于此处讨论的任何一个或更多个术语。提供了某些术语的同义词。然而,一个或更多个同义词的叙述并不排除其他同义词的使用,术语是否在本文中进行阐述或讨论也没有任何特殊意义。本文提及的所有出版物、专利申请、专利及其他参考文献均通过引用并入。在冲突的情况下,将以本说明书包括定义为准。另外,材料、方法和实例仅为说明性的而非意图限制。
[0030]
本发明还通过特定实例的方式进行描述。然而,这样的实例,包括本文讨论的任何术语的实例,在本说明书中任何地方的使用仅是说明性的,并不以任何方式限制本发明或任何示例性术语的范围和含义。同样地,本发明不限于本文描述的任何特定实施方案。事实上,在阅读本说明书后,对本发明的许多修改和变化对本领域技术人员将是明显的,并且可以在不脱离本发明的精神和范围的情况下进行。因此,本发明仅受所附权利要求书的条款以及所述权利要求所赋予的等同物的全部范围的限制。
[0031]
通过引用并入
[0032]
本说明书中提及的所有出版物、专利和专利申请通过引用并入本文,其程度如同每个单独的出版物、专利或专利申请被具体和单独地指明通过引用并入。
附图说明
[0033]
对于所有的附图,使用罗马数字:i)、ii)、iii)、iv)等来表示时间的推移。除非特别说明,否则图不是按比例绘制的。
[0034]
图1(a)展示了通过在已知识别位点处裂解分子产生有序的长度模式而产生沿着长核酸分子的长度的线性物理图谱的实施方案。
[0035]
图1(b)展示了通过在已知识别位点处附接标记体产生有序的区段模式而产生线性物理图谱的实施方案。
[0036]
图1(c)展示了通过以使得标记体的密度与潜在的at/cg比相关的方式沿着分子长度附接标记体而产生线性物理图谱的实施方案。
[0037]
图2展示了用于以并行方式产生经梳理的线性拉长核酸分子的封闭流体装置和方法,其中(i)示出分子流动到封闭通道中,并且其中(ii)示出将顶部从通道移除之后的所述分子。
[0038]
图3展示了流体装置内受限和非受限通道类型的不同的非限制性实施方案。
[0039]
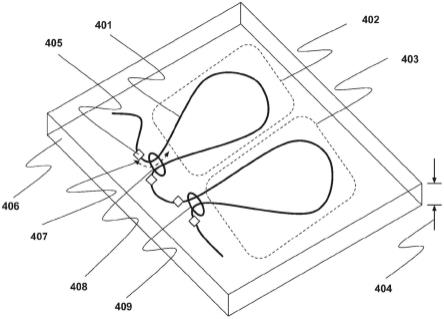
图4展示了一种具有流体狭缝通道的流体装置,其中长核酸分子可以在探查系统的焦深内被拉长,允许对高级核酸结构的动态探查,在该图中高级核酸结构是黏连蛋白复合物。
[0040]
图5展示了一种具有流体狭缝通道的流体装置,其中长核酸分子可以在探查系统的焦深内被拉长,允许对高级核酸结构进行动态探查,在该图中高级核酸结构是一对拓扑关联的结构域。
[0041]
图6展示了一种流体装置,该流体装置包括两个流体通道的交会部,使得含有染色体的细胞可以被鉴定、裂解,并使染色体固定在多孔凝胶材料中。流体装置使得能够在凝胶中固定之前和之后将染色体灵活地暴露于任意试剂。(i)细胞通过第一通道进入装置。(ii)细胞通过过滤特征保持在交会部中,并且周围的胶凝剂被胶凝成多孔凝胶材料。(iii)细胞通过从第二通道流出的裂解剂被裂解,留下固定在凝胶中的细胞染色体。
[0042]
图7(a)展示了被膨胀并且然后固定在多孔凝胶材料中的染色体,以使染色体能够以膨胀、固定的状态受控地暴露于试剂。(i)在存在蛋白质消化酶和含有胶凝剂的溶液的情况下的染色体。(ii)染色体随着蛋白质的消化而膨胀。(iii)使胶凝剂胶凝,使膨胀的染色体固定在凝胶中。
[0043]
图7(b)展示了图7(a)的放大示意图。(i)将来自染色体的染色质的一部分暴露于蛋白质消化酶,使得消化后(ii)染色体的该部分膨胀,并且然后(iii)固定在多孔凝胶材料中。
[0044]
图8展示了选择性提取固定在凝胶材料中的染色体的一部分的实例。
[0045]
图9展示了选择性提取固定在凝胶材料中的完整染色体的实例。
[0046]
图10(a)展示了具有用fret对标记的凝缩蛋白复合物的长核酸分子,使得可以探查fret对的接近动力学。
[0047]
图10(b)展示了具有用fret对标记的转录复合物的长核酸分子,使得可以探查fret对的接近动力学。
[0048]
图11(a)展示了包含由可裂解接头连接的两个捕获探针的可裂解接近条形码交联剂(cleavable proximity barcode crosslinker,cpbc)的实例。
[0049]
图11(b)展示了图11(a)中示出的cpbc的使用实例。此处(i)两个这样的具有不同条形码的cpbc与长核酸分子交联,并且然后(ii)使cpbc裂解,去除交联,并产生具有结合的捕获探针的长核酸分子,其中条形码保留先前接近关系的信息。
[0050]
图12展示了图11(a)中示出的cpbc的使用实例。此处(i)两个这样的具有不同条形码的cpbc与长核酸分子交联,并且然后随后被裂解,去除交联。接下来,在稍后的时间点(ii),第三个具有不同条形码的cpbc与长核酸分子交联,并且然后随后被裂解。该结果是(iii)具有结合的捕获探针的长核酸分子,其中条形码保留在不同时间点的先前接近关系的信息。
[0051]
图13(a)展示了一种实例cpbc,其中cpbc的可裂解接头包含可以在适当环境条件下解链的双链核酸。此处,(i)解链前的cpbc,和(ii)解链后的cpbc,产生两个分离的捕获探针。
[0052]
图13(b)展示了图13(a)中示出的cpbc的使用实例。此处(i)两个这样的具有不同条形码的cpbc与长核酸分子交联,并且然后(ii)使cpbc裂解(解链),去除交联,并产生具有
结合的捕获探针的长核酸分子,其中条形码保留了先前接近关系的信息。
[0053]
图14展示了一种实例cpbc,其中cpbc包含dna折纸结构,类似于霍利迪连接体(holliday junction)的结构,使得cpbc包含4个捕获探针。(i)与靶分子交联后,(ii)cpbc的可裂解接头然后解链,使4个捕获探针彼此释放。
[0054]
图15展示了包含为官能化树枝状大分子的球的cpbc的实例,4个捕获探针通过可裂解接头附接至该球。
[0055]
图16展示了图15中示出的cpbc的使用实例。此处(i)cpbc与长核酸分子和蛋白质交联。交联后(ii),捕获探针从cpbc释放,导致蛋白质和核酸与保留了生物分子先前接近关系的信息的捕获探针结合。
[0056]
图17展示了通过在基底上梳理分子来探查与来自cpbc的捕获探针结合的长核酸分子,使得探针相对于分子物理图谱的定位位置可以通过荧光探查来建立的实例。
[0057]
详述
[0058]
定义
[0059]
如本文使用的,“约”或“大约”在数字的上下文中应指跨该数字+/-10%的范围,或者在范围的上下文中应指跨所列出的范围的下限以下10%至所列出的范围的上限以上10%的扩展范围。
[0060]
尽管本公开内容支持仅指替代物和“和/或”的定义,但是除非明确指示为仅指替代物或替代物是相互排斥的,否则在权利要求书中使用的术语“或”用于意指“和/或”。
[0061]
在权利要求书或说明书中,当与词语“包含”一起使用时,词语“一(a)”和“一(an)”表示一个或更多个,除非特别指出。
[0062]
除非上下文另外明确要求,否则在整个说明书和权利要求书中,词语“包含(comprise)”、“包含(comprising)”等应以包括的意义而不是排他或穷尽的意义来解释;也就是说,意义为“包括但不限于”。使用单数或复数数字的词语也分别包括复数和单数数字。此外,在本技术中使用时,词语“本文”、“上文”、“下文”以及类似含义的词语应指本技术作为整体,而不是本技术的任何特定部分。
[0063]
使用术语“组合”用于意指从集合中选择项目,使得选择中的顺序并不重要,并且当明确说明时,空集(无)的选择也是有效选择。例如,可以选择的包括集合{a,b}的空集的独特组合为:空集、a、b、a和b。
[0064]
样品.如本文使用的术语“样品”通常指受试者的生物样品,其至少部分地含有来源于所述受试者的核酸。生物样品可以包含任何数量的大分子,例如细胞长核酸分子。样品可以是细胞样品。样品可以是细胞系或细胞培养样品。样品可以是ctc(循环肿瘤细胞)或cfc(循环胎儿细胞)样品。样品可以包含一个或更多个细胞。样品可以是一个或更多个包含生物材料的液滴。样品可以包含一种或更多种微生物。生物样品可以是核酸样品。生物样品可以源自另一个样品。样品可以是组织样品,诸如活检、核芯活检(core biopsy)、针抽吸物或细针抽吸物。样品可以是流体样品,诸如血液样品、尿液样品或唾液样品。样品可以是皮肤样品。样品可以是颊拭子。样品可以是血浆或血清样品。样品可以是无细胞(cell-free或cell free)样品。无细胞样品可以包括细胞外多核苷酸。细胞外多核苷酸可以从身体样品中分离,所述身体样品可以选自由血液、血浆、血清、尿液、唾液、粘膜分泌物、痰、粪便和眼泪组成的组。
[0065]
核酸.术语“核酸”、“核酸分子”、“寡核苷酸”和“多核苷酸”、“核酸聚合物”、“核酸片段”、“聚合物”可互换使用,并指任何长度的聚合形式的核苷酸(脱氧核糖核苷酸或核糖核苷酸或其类似物)。这些术语涵盖例如dna、rna及其修饰形式。多核苷酸可以具有任何三维结构,并且可以执行任何已知或未知的功能。多核苷酸的非限制性实例包括基因、基因片段、外显子、内含子、信使rna(mrna)、转运rna、核糖体rna、lncrna(长非编码rna)、lincrna(长基因间非编码rna)、核酶、cdna、ecdna(染色体外dna)、人工微型染色体、cfdna(循环游离dna)、ctdna(循环肿瘤dna)、cffdna(无细胞胎儿dna)、重组多核苷酸、支链多核苷酸、质粒、载体、分离的任何序列的dna、控制区、分离的任何序列的rna、核酸探针和引物。
[0066]
除非另外特别说明,否则核酸分子可以是单链的、双链的或其混合物。例如,可以存在发夹弯(hairpin turns)或环。除非另外特别说明,否则核酸分子可以包含切口。
[0067]
长核酸.除非另外特别说明,否则“长核酸片段”或“长核酸分子”是长度为至少1kbp的双链核酸,并因此是一种大分子,并且可以跨至整个染色体。它可以来源于任何人造或天然的来源,包括单细胞、细胞群体、液滴、扩增过程等。它可以包括具有另外的结构(诸如结构蛋白组蛋白)的核酸,并从而包括染色质。它可以包括具有与其结合的另外的实体(例如标记体、dna结合蛋白、rna)的核酸。
[0068]
高级核酸结构.“高级核酸结构”或“结构”或“高级结构”是指任何二级、三级或四级dna结构,包括与所述核酸分子结合的任何实体。核酸分子可以是线性的或环状的。核酸可以具有多种结构构型中的任何一种,例如,是单链的,双链的,三链体,复制环或两者的组合,以及具有高级的分子内或分子间二级/三级/四级结构,例如,染色体区域,染色体边界、染色质区域、区室,拓扑关联结构域(tad),染色质环与局部直接调控因子结合,凝缩蛋白关联环、黏连蛋白关联环、引导核酸、argonaut复合物、crispr cas9复合物、核蛋白复合物、绝缘子复合物、增强子-启动子复合物、核糖核酸(rna)、小干扰rna(sirna)、微rna(mirna)、引导rna(grna)、长非编码rna(lncrna)、重复区结合蛋白、端粒修饰蛋白、核酸修复蛋白、调控因子结合蛋白、核酸结合蛋白、蛋白、组蛋白脱乙酰酶(hdac)、染色质重塑蛋白、甲基结合蛋白、转录因子转录复合物、以顺式或者反式方式弯折基因组dna聚合物的弯曲诸如发夹、复制环、三链区等。核酸内的核苷酸可以具有表观基因组状态的任何组合,包括但不限于诸如甲基化或乙酰化状态。核酸可以来源于任何来源,人造的或天然的,包括单细胞、细胞群体、液滴、扩增过程等。在一些实施方案中,这些结构包含化合物和/或核酸和蛋白质的相互作用。在一些实施方案中,这些结构包含线性1d聚合物链之外的核酸的2d和3d构型。这些2d和3d构型可以通过与蛋白质、其他核酸分子或外部边界条件的相互作用来形成。边界条件的非限制性实例包括微米流体室或纳米流体室、基底上或基底中或限定在流体装置内的孔、液滴、核。核酸可以包括具有另外的结构的核酸,另外的结构诸如结构蛋白,包括但不限于诸如任何调控结合位点复合物,增强子/转录因子复合物及其与核酸分子的相互作用、黏连蛋白复合物smc(染色体的结构维持)、atp酶亚单位(smc1和smc3)、非smc调控亚单位(rad21/scc1/mcd1和sa1/sa2/scc3)、sgo1、有丝分裂激酶(polo样激酶1(plk1)和aurora b)、蛋白磷酸酶2a(pp2a)、染色体乘客复合物(cpc)、topo ii去连环(decatenation)、凝缩蛋白、ctcf蛋白、pds5蛋白、wapl蛋白、凝缩蛋白i、凝缩蛋白ii、cap-g、组蛋白及其衍生物复合物,并且因此包括染色质。在一些实施方案中,高级结构可以包含外源核酸基因组整合复合物,特别是包含病毒基因组整合复合物或重组核酸的外源核酸基因组整合复合物。在一
些实施方案中,高级结构可以包括染色体外附加体物理对接复合物,特别是,其中这样的复合物通过结合位点承载染色体。在一些实施方案中,高级核酸结构包含源自宿主染色体的染色体外核酸。以上所有(非限制地)可以是标记的靶,指示基因组组织的某些状态的存在或状态之间的转变(这可能与致病基因组学结果相关)的物理或构象生物标志物。
[0069]
特别地,高级核酸结构可以指细胞核内包含的单独的、集合的各种水平的基因组组织[jerkovic,2021]、[kempfer,2020],或是其子集。这样的基因组组织始于缠绕组蛋白形成核小体的线性一级dna,核小体被组织成簇(clutch),每个簇包含~1-2kb的dna。核小体簇形成尺寸~100kb的染色质纳米结构域(cnd),大多数增强子-启动子(e-p)接触发生在这里。在~1mb的规模,cnd和ccctc结合因子(ctcf)-黏连蛋白依赖性染色质环形成拓扑关联结构域(tad)和环结构域。在高达上百兆碱基的更高规模,染色质分离成基因活性和基因非活性区室(分别为a和b)以及区室特异性接触中枢,形成姐妹染色单体轴。在最高的拓扑水平上,细胞核被组织成染色体区域。
[0070]
杂交.如本文使用的,术语“杂交(hybridization)”、“杂交(hybridizing)”、“杂交(hybridize)”、“退火(annealing)”和“退火(anneal)”在提及互补或基本上互补的核酸的配对时可互换地使用。杂交和杂交强度(即,核酸之间缔合的强度)受诸如以下的因素影响:核酸之间的互补程度、所涉及条件的严格性、形成的杂交体的tm(解链温度)温度和环境条件。“杂交”方法涉及一个核酸与另一个互补核酸(即具有互补核苷酸序列的核酸)的退火。
[0071]
配对可以通过核酸序列与基本上互补或完全互补的序列通过碱基配对接合以形成杂交复合物的任何过程来实现。为了杂交的目的,如果两个核酸序列的至少60%(例如,至少70%、至少80%或至少90%)的单独碱基彼此互补,则两个核酸序列“基本上互补”。
[0072]
在本文件的上下文中,在核酸链和双链核酸分子之间发生杂交的情况下,应理解这样的杂交是在双链核酸分子部分变性或完全变性的条件下完成的,除非另外特别说明。
[0073]
标记体.本文使用的“标记体”是可以与核酸分子结合的物理实体或者直接或间接结合至核酸分子的实体,其可以用于产生可以用探查检测的信号,该信号不同于不具有所述实体的所述核酸会产生的检测信号(或缺乏这种信号)。标记体可以是荧光嵌入染料,当荧光嵌入染料与核酸结合时,可以用于在荧光成像系统中鉴定所述核酸的存在。在另一实例中,标记体可以是与甲基化核苷酸特异性结合,并且当通过纳米孔运送时提供电流阻塞信号,从而报告关于所述分子的甲基化状态的信号的化合物。在另一实例中,荧光探针与核酸的序列特异性地杂交,从而用荧光成像系统提供对所述核酸上存在该序列的证实。在另一实例中,荧光探针与特定蛋白(例如:dna结合蛋白)特异性结合,其中所述蛋白与长核酸分子结合。在一些情况下,标记体不存在本身就是信号。在一些情况下,与标记体相关的信号是来自另一个标记体的信号的减弱、阻断、置换、猝灭或修饰。非限制性实例包括:暗标记体与核酸结合以置换现有的键合荧光体;使暗标记体与所述核酸结合以阻断荧光标记体结合;使与核酸键合的邻近荧光标记体猝灭;直接或间接与结合核酸的荧光标记体键反应以降低其荧光。在一些情况下,在探查核酸分子和标记体时,标记体没有物理附接到所述核酸分子。例如,标记体可以通过可裂解接头附接到核酸分子。在所需的时间,接头被裂解,释放所述标记分子,其然后通过探查被检测。
[0074]
探查.“探查”是用探查系统评估核酸、长核酸分子、高级核酸结构、核酸-蛋白质复合物或其他生物分子的状态的过程。在一些实施方案中,通过测量直接或间接从标记体产
生的信号来探查核酸上至少一个标记体的状态,从而评估核酸的状态。探查可以是二元评估,诸如标记体存在或不存在。探查可以是定量的,诸如分子上有多少标记体。探查可以是沿着线、区域或体积的信号密度或强度。探查可以是沿着分子长度的物理计数或标记体之间的距离。
[0075]
在一些实施方案中,探查用于产生物理图谱的计算机(in-silico)表示。
[0076]
在一些实施方案中,探查用于评估高级核酸结构的物理状态。被探查的结构的物理状态可以包括分子的拓扑结构,诸如环结构、一组分级(hierarchical)环结构的存在,环中存在的超螺旋的数目或来自相同或不同的分子的一个或更多个环缠绕的程度。被探查的结构的物理状态可以包括核酸区域对结合配偶体或者顺式或反式作用因子的可及性。被探查的结构的物理状态可以包括仍然非常接近的部分复制的核酸(诸如okazaki片段)或新合成的核酸的标志物(诸如来自brdu脉冲的结果)的存在。被探查的结构的物理状态可以包括已被实验操作或受遗传异常影响的中期染色体上残留的黏连蛋白的水平(例如,通过消耗黏连蛋白本身或wapl),得到的染色单体显示出实质不同的长度和形状,成为指示某些病理状态的可定量测量的生物标志物(losada等人2005;gandhi等人2006年;shintomi和hirano 2009)。被探查的结构的物理状态可以包括这些染色单体中凝缩蛋白i和ii的量、比例和分布。被探查的结构的物理状态可以包括基因组组织的动态变化,如黏连蛋白释放和姐妹染色单体解析(sister chromatid resolution)的动态变化。
[0077]
在一些实施方案中,被探查的信号可以是荧光的、光致发光的、电磁的、电的、磁的、物理的、化学的、表现出等离子体共振的或通过表面增强的等离子体共振的方式的增强拉曼信号。
[0078]
被探查的信号本质上可以是模拟的或数字的。例如,信号可以是沿着核酸长度的标记体的模拟密度谱,其中测量的信号来源于多于一个标记体。在一些实施方案中,在没有标记体的情况下直接探查核酸的状态,例如通过相位显微术直接探查细胞中的长核酸分子,或通过电流阻塞纳米孔直接探查核酸。探查系统可以单独或组合使用的不同探查方法的非穷尽实例包括荧光成像、明场成像、暗场成像、相对比成像、落射荧光(epi-florescent)成像、全内反射荧光成像、近场/隐失场成像、波导、零模式波导、等离子体信号传递、共聚焦、散射、光片、结构照明、受激发射损耗、超分辨率、随机激活超分辨率、随机结合超分辨率、多光子、电流、电压、功率、电容、电感或反应性信号的纳米孔感测(通过孔的库伦阻塞或跨孔的隧穿效应)、化学感测(例如:通过反应)、物理感测(例如:与感测探针的相互作用)、sem、tem、stm、spm、afm。另外,不同标记体和探查方法的组合也是可行的。例如:对核酸上的嵌入染料进行荧光成像,同时使所述核酸移位通过纳米孔并测量孔电流。
[0079]
探查系统.本文使用的“探查系统”是用于探查样品的自动化或半自动系统。在一些实施方案中,其中样品在流体装置内或流体装置上时被探查,探查系统与流体装置连接并控制流体装置的操作。在一些实施方案中,探查系统包括可以一起由控制器或用户协调的多个单独的系统。例如,用于将样品加载到流体装置中的仪器、用于使所述样品在所述流体装置中流动的仪器、用于对所述流体装置中的所述样品成像的仪器、用于操作用于分析所述成像数据的软件的控制器。在一些实施方案中,探查系统包括所有系统或系统子集的集成。
[0080]
在其中样品被限制在流体装置内或流体装置上的一些实施方案中,探查系统对装
置的操作可以包括:通过在所述实体上施加外力来操纵包装物(package)或长核酸分子的物理位置和构象;将所述包装物或长核酸分子暴露于环境条件或试剂一段时间;光学探查包装物或长核酸分子的静态或动态构型,以便于分析它们的组成,或作为反馈系统的一部分来控制装置的操作;从装置提取所需的包装物或长核酸分子。流体装置和探查系统可以以多种方式连接。非穷尽列表包括:流体端口(开放和密封两者)、电终端、光学窗口、机械垫、加热管或散热器、电感线圈、流体分配、表面扫描探针。探查系统可以对装置执行的潜在功能的非穷尽列表包括:温度监测,施加热,去除热,向端口施加压力或真空,测量真空,测量压力,施加电压,测量电压,施加电流,测量电流,施加电功率,测量电功率,在远场或近场环境中,使装置暴露于聚焦和/或非聚焦的电磁波、收集从装置产生或反射的电磁波光,产生和测量温度、电磁力、表面能或化学浓度差或梯度,将液体分配到装置孔或端口中、或分配到装置表面上,使装置表面或装置表面上的实体与接触探针(例如:afm尖端)接触。
[0081]
在一些实施方案中,对长核酸分子在流体装置的某个区域中的存在的确认和对其在所述装置内的物理位置的控制由探查系统使用反馈控制器系统来控制。对长核酸分子的检测通过检测至少一种被探查的信号来进行。在优选实施方案中,信号是来源于结合到所述长核酸分子的标记体的电磁信号。在一种实施方案中,控制仪器反馈控制系统至少部分地利用长核酸分子内物理图谱谱图(physical map profile)的鉴定或分子内物理图谱谱图的不存在作为输入信息。
[0082]
在一些实施方案中,探查系统包括系统内的本地化计算处理模块、通过直接通信连接的相邻计算处理模块、通过网络连接的外部计算处理模块或其组合。计算处理模块的多种实例包括:pc、微控制器、专用集成微芯片(asic)、现场可编程门阵列(fpga)、cpu、gpu、单片系统(system on chip)、网络服务器、云计算服务或其组合。
[0083]
探查系统可以包括至少一个流体分配尖端,其能够在装置表面上期望的x、y、z坐标处分配流体液滴,并且在一些实施方案中,在流体装置表面上期望的x、y、z坐标处提取流体液滴。流体分配和提取可以以微升、纳升、皮升、飞升或阿升(attoliter)的体积进行。
[0084]
探查系统可以能够同时或按顺序使多于一个光源照射,并且能够同时或按顺序对多于一个颜色成像。如果同时对多于一个颜色进行成像,这可以在不同的相机上、在单个相机但传感器阵列的不同区域上、或者在同一相机的同一传感器上完成。在一些实施方案中,对由控制仪器照射的光的波长进行选择,以便以某种方式与样品、样品标记体或官能化表面相互作用。非限制性实例包括:核酸的光裂解、使可光裂解接头光裂解、操作光镊、激活光激活反应、使光不稳定保护基团脱保护、ir加热(ir thermal heating)。
[0085]
除非另外特别说明,否则在本文件中,通过探查系统对长核酸分子的任何探查包括这样的实施方案:其中长核酸分子的至少一部分与包含嵌入荧光染料的至少一个标记体结合,并且探查系统包括光学荧光成像系统。
[0086]
序列.术语“序列”或“核酸序列”或“寡核苷酸序列”是指一串连续的核苷酸碱基,并且在特定上下文中还指出现在寡核苷酸中的核苷酸碱基相对于彼此的特定位置。
[0087]
测序可以通过当前可用的各种系统进行,诸如但不限于illumina、pacific biosciences、oxford nanopore、life technologies(ion torrent)、bgi的测序系统。
[0088]
限制性内切酶.术语“限制性内切核酸酶”或“限制性内切酶”是指在被称为“限制性位点”或“限制性识别位点”的特异性识别核苷酸序列处或附近切割dna的酶。限制性内切
核酸酶包括能够识别和切割甲基化dna的酶和仅识别非甲基化dna的酶。甲基化dna包括dam甲基化、dcm甲基化和cpg甲基化。限制性内切核酸酶可以识别四个、六个或八个核苷酸长的限制性位点。这些类型的限制性内切核酸酶分别称为4-切割酶(cutter)、6-切割酶和8-切割酶。在一些实施方案中,限制性内切核酸酶可以是以下之一:acll、hindlll、sspl、mluci、pcil、agel、bspmi、bfuai、sexai、mlul、bceai、hpych4iv、hpych4iii、bael、bsaxi、afliii、spel、bsrl、bmrl、bglii、afel、alul、stul、seal、clal、bspdi、pi-scei、nsil、asel、swal、cspci、mfel、nb.bsssi、bsssal、bmgbi、pmll、drain、alel、ecopl5i、pvuii、alwni、btsimuti、ndel、fatl、nlalll、cviaii、msll、fspei、xcml、bstxi、pflmi、bed、ncol、bseyi、faul、xmal、tspmi、smal、nt.cvipii、lpnpi、acil、sadi、bsrbi、hpall、mspl、scrfi、styd4i、bsaji、bsll、btgl、neil、avrii、mnll、nb.bbvci、bbvci、nt.bbvci、sbfl、bpuloi、bsu36i、econi、hpyav、bstni、pspgi、styl、bcgl、pvul、bstui、eagl、rsrii、bsiei、bsiwi、bsmbi、hpy99i、mspall、mspji、sgrai、bfal、bspcni、paer7i、xhol、earl、acul、pstl、bpml、ddel、sfcl、aflii、bpuei、smll、bsobi、aval、mboll、bbsl、xmnl、nb.bsmi、bsml、ecori、hgal、zral、aatll、pflfi、tthl 1ii、pshai、ahdl、drdl、eco53kl、sad、bseri、mlyl、plel、nt.bstnbi、hinfl、ecorv、mbol、sau3ai、dpnii、bfuci、dpnl、bsabi、tfil、bsrdi、nb.bsrdi、bbvl、btsal、nb.btsi、bstapi、sfani、sphl、srfl、nmeaiii、nael、ngomiv、bgll、asisi、btgzi、hinpli、hhal、bsshii、notl、fnu4hi、cac8i、mwol、bmtl、nhel、bspqi、sapl、nt.bspqi、blpl、apeki、tsel、bspl286i、nt.alwi、alwl、bamhi、btsci、fokl、haelll、fsel、sfil、narl、kasl、pluti、sfol、asci、ecil、bsmfi、pspomi、apal、sau96i、nlalv、acc65i、kpnl、bsal、hphl、bsteii、avail、banl、baegi、bsahi、banll、rsal、cviqi、bstzl7i、bcivi、sall、bsmai、bcodi、nt.bsmai、apali、bsgl、acci、hpyl66ii、tsp45i、hpal、pmel、hindi、bsihkai、tspri、apol、nspl、bsrfal、bstyi、haell、cviki-l、ecool09i、ppumi、i-ceul、snabi、i-scel、bsphi、bspei、mmel、taqal、nrul、hpyl88i、hpyl88iii、xbal、bell、hpych4v、fspl、pi-pspi、msci、bsrgi、msel、pad、psil、bstbi、dral、pspxi、bsawi、bsaai或eael。
[0089]
切口酶是仅切割一条dna链的限制性内切酶,但就本发明的目的而言,它们可以以与限制性内切酶基本类似的方式发挥作用,即当伴随着产生单链切口的交替方式(诸如单链特异性核酸酶、通过使dna结合荧光团诸如yoyo-1进行光循环的光裂解、机械剪切、brdu掺入和光刺激、或由一个或更多个种类的切口酶的高密度切割)时,它们可以切断双链dna,无论是形成平端、突出端还是大的突出端。
[0090]
参考物.“基因组参考物”或“参考物”是任何可以与另一基因组数据集进行比较的基因组数据集。可以采用任何数据格式,包括但不限于序列数据、核型分析数据、甲基化数据、基因组功能元件数据诸如顺式调控元件(cre)图谱、一级结构变异图谱数据、高级核酸结构数据、物理作图数据、遗传作图数据、光学作图数据、原始数据、经处理的数据、模拟数据、信号谱(包括电子或荧光产生的信号谱)。基因组参考物可以包括多于一种数据格式。基因组参考物可以代表来自多于一个数据集的一致内容,这些数据集可以来源于不同的数据格式或者可以不来源于不同的数据格式。基因组参考物可以包括生物体或模型的基因组信息的全部、或子集或代表。基因组参考物可以是它所代表的基因组信息的不完整代表。
[0091]
基因组参考物可以源自指示不存在疾病或紊乱状态的基因组,或者可以源自指示疾病或紊乱状态的基因组。此外,基因组参考物(例如,具有长于100bp、长于1kb、长于
100kb、长于10mb、长于1000mb的长度)可以在一个或更多个方面进行表征,非限制性实例包括确定特定特征、特定单体型、特定遗传变异、特定结构变异、特定单核苷酸多态性(snp)及其组合的存在(或不存在),不仅指存在或不存在于基因组参考物的整体中,而且指存在或不存在于基因组参考物的特定区域中,如由邻近的基因组内容物所定义的。此外,基因组参考物的任何合适类型和数目的特征都可以用来基于样品核酸是否显示出与参考物类似的特征,将样品核酸表征为源自(或不源自)指示紊乱或疾病的核酸。
[0092]
在一些情况下,基因组参考物是物理图谱。这可以以任何数量的方式产生,包括但不限于:原始单分子数据、经处理的单分子数据、从序列或模拟产生的物理图谱的计算机表示、通过对多于一个单分子物理图谱进行组装和/或平均化产生的物理图谱的计算机表示,或其组合。例如,基于已知或部分已知的序列,模拟计算机物理图谱可以基于所使用的产生物理图谱的方法来产生。在其中物理图谱包括已知序列处的标记体的实施方案中,可以产生一组离散有序的以碱基对计的区段长度。在一种实施方案中,物理图谱包括基于双螺旋之间的模拟局部氢键解离动力学在碱基对方面,基于核苷酸序列和预测的功能元件数据图谱在化学部分修饰、调控因子关联或结构折叠模式方面,沿着序列长度标记信号密度的连续模拟信号。
[0093]
在一些情况下,基因组参考物是从微阵列(例如:dna微阵列、mmchip、蛋白微阵列、肽微阵列、组织微阵列等)、或核型分析或fish分析获得的数据。在一些情况下,基因组参考物是从接近性3d作图技术或3d物理作图技术获得的数据。
[0094]
在一些情况下,对与基因组参考物的比较进行的表征可以在编程计算机处理器的帮助下完成。在一些情况下,这样的编程计算机处理器可以包括在计算机控制系统中。
[0095]
物理作图.核酸的“物理作图”或“作图”包括从长核酸分子的物理片段提取基因组、表观基因组、功能或结构信息的各种方法,其中所提取的信息可以与分子上的物理坐标关联。作为一般规则,所获得的信息的分辨率低于实际的潜在序列信息,但这两种类型的信息在分子内是空间上相关的(或反相关的),并且因此,前者通常提供关于沿着核酸的物理位置的序列内容的
‘
图谱’。在一些实施方案中,图谱和潜在序列之间的关系是直接的,例如,图谱表示沿着分子长度的ag含量的密度或特定识别序列的频率。在一些实施方案中,图谱与潜在序列之间的关系是间接的,例如,图谱表示与蛋白一起包装成结构的核酸的密度,其继而至少部分地随潜在序列而变化。在一些实施方案中,物理图谱是线性物理图谱,其中提取的信息可以沿着轴的长度分配,例如,沿着长核酸分子的主轴的at/cg比。在优选实施方案中,通过探查沿着长核酸分子的主轴的拉长部分结合的标记体来产生“线性物理图谱”或“1d物理图谱”。为了清楚,以卷曲状态占据3d空间的串可以被表示为直线,并且因此沿着3d卷曲提取的值可以被表示为沿着串的1d表示的分箱值,并且因此构成线性物理图谱。在一些实施方案中,物理图谱是“2d物理图谱”,其中提取的信息可以在包含分子的平面内分配,例如:核型分析。在一些实施方案中,物理图谱是“3d物理图谱”,其中提取的信息可以在分子占据的3d体积中分配。例如,使用超分辨率技术加标签以在(x,y,z)空间中鉴定标签在染色体内的位置,如用oligofisseq[nguyen,2020]或原位基因组测序[payne,2020]展示的。
[0096]
第一种并且是最广泛使用的物理作图形式是核型分析,其中用优先与at或cg区域结合的染色方法处理中期染色体,从而产生与核酸的潜在序列以及核酸的结构和表观基因
组模式相关的“条带”[moore,2001]。然而,由于被成像的核酸的凝缩的性质,这样的方法对于核苷酸序列的分辨率相当低,约5-10mbp。较为近期的对拉长的间期基因组dna使用线性作图的方法通过以下来产生:对在已知限制性位点处消化的核酸进行成像[schwartz,1988,6,147,198](例如:参见图1(a)),对在切口位点处附接的荧光探针进行成像[xiao,2007](例如:参见图01(b)),对核酸分子的甲基化模式的荧光特征进行成像[sharim,2019],对染色质的组蛋白的荧光特征进行成像[riehn,2011],通过传感器对结合到核酸的探针进行电检测[rose,2013,2014/0272954],以及使用纳米孔传感器对核酸上的甲基化特征进行电检测[rand,2017]。
[0097]
另一种线性物理作图方法是沿着拉长的核酸分子的长度测量at/cg相对密度或局部解链温度(例如:参见图1(c))。这样的信号可以用来与其他类似的图谱进行比较,或者与从序列数据计算机产生的图谱进行比较。有很多种产生这种信号的方法。例如,信号可以本质上是荧光的或电的。核酸可以用嵌入染料均匀染色,并且然后部分地解链,导致在at含量丰富的区域中染料的相对损失[tegenfeldt,2009,10,434,512]。另一种方法是使双链核酸暴露于竞争结合核酸的两种不同物类(species)。一种物类是非荧光的并且优先结合at丰富的区域,而另一种物类是荧光的并且没有这种偏倚[nilsson,2014]。又另一种方法是使用差异性地标记at区域和cg区域的两种不同颜色的染料。
[0098]
使用这样的非凝缩的间期核酸聚合物链作图提高了一级序列信息的分辨率,然而图谱去除了任何天然结构折叠或结合支持蛋白信息,并且通常从具有许多潜在异质细胞的汇集的样品的本体溶液(bulk solution)中提取。最近,已经展示了3d物理图谱,其中直接或间接探查在特定位置附接至染色体的标签,以确定它们在3d空间中染色体内的相对位置(对于各种方法的评述,参见[jerkovic,2021])。这些方法可以包括超分辨率显微术方法,诸如sim、smlm和sted、oligopaint fish方法、多重化oligopaint fish方法和oligofisseq方法。另外,还包括原位测序方法,诸如oligofisseq[nguyen,2020]。注意,在本文件中,“3d物理图谱(作图)”不同于本文件其他地方定义的“接近性3d图谱(作图)”。
[0099]
图1(a)、图1(b)、图1(c)展示了用于产生和探查长核酸分子线性物理图谱的多种不同实施方案。在图1(a)中,长核酸分子104的物理图谱通过在特定序列位点(例如:限制性内切酶的识别位点)处裂解分子从而产生裂解事件发生处的缺口105来产生。沿着分子的长度,染料非特异性地附接(例如:使用嵌入染料),使得来自来源亲本分子的子分子可以被探查,以产生沿着亲本分子的物理长度(0106)的信号101。然后,信号可以用来确定个体子分子的长度和顺序{103-x},并从而产生亲本分子的物理图谱。在该方法的大多数实施方案中,亲本分子被梳理到表面上,并且然后被裂解,以便保持子分子的物理接近和相对顺序。然而,这样的实施方案也可以在受限流体装置的拉长通道内以至少部分拉长的状态来实施,使得子分子的顺序可以被探查[ramsey,2015,10,106,848]。在一些实施方案中,不同裂解位点的混合可以被同时使用。
[0100]
在图1(b)中,长核酸分子114的物理图谱通过沿着分子长度稀疏地结合标记体115而产生,其中结合位点与一组特定靶相关(或反相关)。在一些方法中,标记体与序列基序靶直接结合。在一些方法中,产生信号的标记体通过例如以下的过程间接地结合,例如:产生序列特异性切口,然后从切口位点开始掺入核苷酸,所述核苷酸中的一些可以能够产生信号。探查带有标记体的长核酸分子,沿着分子116的物理长度从标记体115产生信号111。信
号之间的距离、长度和顺序的集合{113-x}然后代表分子的物理图谱。在一些实施方案中,进一步的信息还可以通过解释来自各标记位点的信号112的相对幅度来产生。当使用荧光探查时,不同颜色的标记体可以用来代表不同的特异性位点。
[0101]
在图1(c)中,长核酸分子124的物理图谱是通过沿着分子长度密集结合标记体125,使得结合模式与分子的潜在物理序列内容相关(或反相关)而产生的。例如,相对at/cg含量,或相对解链温度,或甲基化cg的相对密度。由于该方法中标记体的密集性质,物理图谱不是长度和顺序的集合,而是沿着分子126的物理长度的强度变化的模拟信号121。
[0102]
产生物理图谱的探查方法通常是荧光成像,然而不同的实施方案也是可行的,包括沿着表面上经梳理的分子的长度的扫描探针,或当分子移位通过时测量通过收缩部的库仑阻塞电流或跨收缩部的隧穿电流的收缩装置。
[0103]
除非另外特别说明,否则物理图谱是指先前提及的任何方法,包括其组合。例如,长核酸分子可以具有用沿着分子长度的荧光标记体从at/tc密度产生的物理图谱,并且然后还具有当分子被运送通过所述收缩装置时用收缩装置沿着分子长度从甲基化谱产生的物理图谱。
[0104]
拉长的dna.大多数使用荧光成像或电信号来提取与潜在基因组、结构或表观基因组内容物相关的信号的线性物理作图方法采用某种形式的方法来至少局部地“拉长”长核酸分子,从而可以可以提高拉长区域中的物理作图的分辨率,并减少歧义。在溶液中处于其天然状态的长核酸分子将形成无归卷曲。因此,已经开发了各种方法来“解卷曲”和拉长分子。
[0105]
通过使长核酸分子的一部分结合在官能化的固体表面上,分子通过使溶液流动而被拉长并最终被拉紧,与基底表面完全接触[bensimon,1997,7,368,234],这种技术通常称为
‘
梳理’dna。替代地,存在其他长聚合物拉长方法,诸如末端锚定在表面上的流体流诱导拉长[gibb,2012],通过层流的水性溶液流体动力聚焦[chan,1999,6,696,022],通过限制纳米通道的线性化[tegenfeldt,2005],在微流体装置中在存在物理障碍特征的情况下由两个成角度的相反的外部施加的力拉动长核酸分子[volkmuth,1992],在流体装置中通过同时暴露于两个相反的外部施加的力而流体动力捕获分子[tanyeri,2011]。
[0106]
大多数情况下,长核酸分子的至少一部分的拉长状态必须由外力维持,然后以其他方式恢复到其自然无规卷曲状态,除非核酸的至少一部分在没有持续外力的情况下通过物理限制保持处于拉长状态[dai,2016]。
[0107]
除非另外特别说明,否则“拉长的”或“部分拉长的”核酸是这样的长核酸片段,其分子主轴的至少一个包含至少1kb的区段可以投射到2d平面,并且不与其自身重叠。为了清楚,对于其中长核酸包含另外的结构的实施方案,例如当核酸包含在染色质中、与组蛋白紧密结合时,主轴是指较大的染色质分子,而不是核酸链本身。因此,本公开内容中的陈述,诸如“沿着分子的长度”,在提及长核酸分子时,是指沿着主轴的长度。
[0108]
接近性3d作图.在本文件中,“接近性3d作图”是指涉及通过使至少两条核酸链(无论是否为同一染色体)直接或间接地交联在一起而捕获至少两条核酸链的接近关系的方案。作为参考,[kempfer,2020]和[szabo,2019]评述了这些不同的技术,其中非穷尽列表包括以下:3c、4c、5c、hi-c、tcc、plac-seq、chia-pet、捕获-c、c-hic、单细胞hic、gam、sprite、chia-drop。
[0109]
条形码.如本文使用的“条形码”是编码信息的短核苷酸序列(例如,至少约4个、6个、8个、10个、12个、14个、16个、18个、20个、25个、30个、35个核苷酸长)。条形码可以是一个连续的序列或者两个或更多个非连续的子序列。条形码可以用于例如鉴定分区或珠或实体中的寡核苷酸附接的分子。在一些实施方案中,与连接到其他珠的寡核苷酸中的条形码相比,珠特异性条形码对于该珠是独特的。在另一实例中,由于独特的“细胞条形码”,来自每个细胞的核酸可以与其他细胞的核酸区分。这样的分区特异性条形码、细胞条形码、或珠条形码可以使用各种方法产生。在一些情况下,分区特异性条形码、细胞条形码、或颗粒条形码使用分开和混合(也称为分开和汇集)合成方案来产生,例如,如[agresti,2014,2016/0060621]中描述的。在一些实施方案中,本文描述的寡核苷酸中可以有多于一种类型的条形码。
[0110]
在一些实施方案中,与条形码关联的信息可以是对单个实体、特定实体、一类实体、一个子集的实体、特异性选择的实体、随机选择的实体、一组实体的鉴定,其中实体可以是分子、高级核酸结构、细胞器、样品、受试者。在一些实施方案中,与条形码关联的信息可以是过程、时间标记(time-stamp)、位置、与另一实体和/或条形码的关系、实验id、样品id、或环境条件。在一些实施方案中,可以使用任何编码技术将多于一个信息内容存储在条形码中。
[0111]
在一些实施方案中,条形码是单链的。在一些实施方案中,条形码是双链的。在一些实施方案中,条形码具有单链组分和双链组分两者。在一些实施方案中,条形码至少部分地包含2d和/或3d结构,例如发夹或dna折纸(origami)结构。
[0112]
在一些实施方案中,条形码中编码的信息使用错误检查和/或错误校正技术来完成,以确保存储在内的信息的正确性。例如,使用汉明码(hamming code)。在条形码中存储了多于一个信息内容的一些情况下,单独的信息段用它们在条形码内相应的核苷酸单独编码。在其他情况下,可以使用编码方案使核苷酸是共有的。在一些情况下,压缩技术可以用来减少所需核苷酸的数目。
[0113]
在一些实施方案中,条形码中编码的信息包括独特地鉴定与之缀合的分子。这些类型的条形码有时被称为“独特分子标识符”或“umi”。在又其他的实例中,可以利用包含对每个分区独特的“分区特异性条形码”和对每个分子独特的“分子条形码”的引物。在进行条形码化之后,分区然后可以被组合,并任选地被扩增,同时基于特定条形码保持“虚拟”分区。因此,例如,在无需保持物理分区的情况下,包含各个条形码的靶核酸的存在或不存在可以被计数或追踪(例如通过测序)。
[0114]
条形码序列的长度决定了可以区分多少个独特条形码。例如,1个核苷酸的条形码可以区分4个或更少的不同样品或分子;4个核苷酸的条形码可以区分256个样品或更少的样品;6个核苷酸的条形码可以区分4096个不同样品或更少的不同样品;以及8个核苷酸的条形码可以索引65,536个不同样品或更少的不同样品。
[0115]
在一些实施方案中,使用选择软件来设计或随机产生条形码序列,所述选择软件用于选择为这样的条形码,所述条形码:没有发夹,或包含均匀的碱基组成(15%-30% a、t、g和c),或没有均聚物(默认允许3个碱基为相同核苷酸),或没有简单重复序列,或没有低复杂度序列,或不与常见载体或衔接子序列相同。此外,条形码可以被设计成即使存在3个错配测序错误而仍是独特的。
[0116]
条形码通常使用固有不精确的过程来合成和/或聚合(例如,扩增)。因此,意在一致的条形码(例如,在单个分区、细胞或珠的所有条形码化核酸间共有的细胞条形码、颗粒条形码或分区特异性条形码)可能包含原型条形码序列的各种n-1缺失或其他突变。因此,在一些实施方案中,被称为“相同”或“基本上相同”的拷贝的条形码可以包括由于例如合成、聚合或纯化错误中的一个或更多个错误而不同的条形码,并且因此可以包含原型条形码序列的各种n-1缺失或其他突变。然而,理论上理想的条形码的这样的微小变化不干扰本文描述的方法、组合物和试剂盒。因此,如本文使用的,在颗粒条形码、细胞条形码、分区特异性条形码或分子条形码的上下文中的术语“独特”涵盖理想条形码序列的各种无意的n-1缺失和突变。在一些情况下,由于条形码合成、聚合和/或扩增的不精确性质引起的问题通过对可能的条形码序列进行与待区分的条形码序列的数量相比的过量采样(例如,至少约2倍、5倍、10倍或更多的可能的条形码序列)或者通过使用错误校正编码技术来克服。条形码技术的使用是本领域熟知的,参见例如[shiroguchi,2012]和[smith,2010]。用于使用条形码技术的另外的方法和组合物包括[agresti,2014,2016/0060621]中描述的那些。
[0117]
在一些实施方案中,条形码的至少一部分还可以用作引物结合位点。在一些实施方案中,引物结合位点用于pcr引物。在一些实施方案中,形成一组独特条形码的所有条形码在所述条形码中包含全局相同的引物结合位点,使得单一引物序列可以用于与所有条形码结合。在一些实施方案中,引物将是引物结合位点的互补序列。在其他实施方案中,引物将是与引物结合位点相同的序列,因为引物将与原始引物结合位点的先前扩增产物结合。在一些实施方案中,可以存在组合。
[0118]
另外,在一些实施方案中,条形码的至少一部分还可以用作引物。
[0119]
可裂解接头.“裂解结构域”或“可裂解接头”表示可以用于可逆地附接至少两个实体的所述至少两个实体之间的连接。在一些实施方案中,所述至少两个实体是大分子。在一些实施方案中,实体中的至少一个是基底,或者被连接到基底。
[0120]
在一些实施方案中,连接实体的裂解结构域是二硫键。可以添加还原剂来破坏二硫键,导致实体的分离。作为另一实例,在有或没有光敏剂诸如金纳米颗粒的情况下,加热也可以导致裂解结构域的降解和实体的分离。在一些实施方案中,激光辐射被用于加热和降解裂解结构域,在一些实施方案中,激光辐射靶向特定位置。在一些实施方案中,裂解结构域是光敏化学键(例如,当暴露于诸如紫外光的光时解离的化学键)。
[0121]
具有光敏化学键的寡核苷酸(例如,可光裂解接头)具有多种优点。它们可以被有效且快速地(例如,在纳秒和毫秒内)裂解。在一些情况下,可以使用光掩模(photo-mask),使得阵列的仅特定区域暴露于裂解刺激(例如,暴露于uv光、暴露于光、暴露于激光诱导的热)。替代地,可以通过数字微镜装置对样品成像或通过检流计镜(galvanometer mirror)和成像透镜的方式引导聚焦光来实现选择性照射。当使用可光裂解接头时,裂解反应由光触发,并且可以是对接头高度选择性的,并因此是双正交的。通常,可光裂解接头的波长吸收位于光谱的近uv范围内。在一些实施方案中,可光裂解接头的吸收最大值为约200nm至约600nm。
[0122]
可以在裂解结构域中使用的光敏化学键的非限制性实例包括[leriche,2012]和[weissleder,2013,2017/0275669]中描述的那些,这两项文献通过引用以其整体并入本文。例如,包含光敏化学键的接头包括3-氨基-3-(2-硝基苯基)丙酸(anp)、苯酰基酯
(phenacyl ester)衍生物、8-喹啉基苯磺酸酯、双香豆素、6-溴-7-烷氧基基香豆素-4-基甲氧基羰基、基于bimane的接头和基于双芳基腙的接头。在一些实施方案中,光敏键是可裂解接头诸如邻硝基苄基(onb)接头的一部分。可以在裂解结构域中使用的光敏化学键的其他实例包括卤化核苷,诸如溴脱氧尿苷(brdu)。brdu胸苷类似物,可以容易地掺入到寡核苷酸中,并且对uvb光(280-320nm范围)敏感。当暴露于uvb光时,发生导致裂解结构域裂解的光裂解反应(例如,在brdu掺入位点紧接5’的核苷处([doddridge,1998]和[cook,1999])。
[0123]
裂解结构域的其他实例包括不稳定化学键,诸如但不限于酯键(linkage)(例如,可用酸、碱或羟胺裂解)、邻二醇键(例如,可经由高碘酸钠裂解)、diels-alder键(例如,可经由热裂解)、砜键(例如,可经由碱裂解)、硅基醚键(例如,可经由酸裂解)、糖苷键(例如,可经由淀粉酶裂解)、肽键(例如,可经由蛋白酶裂解)、无碱基或无嘌呤/无嘧啶(ap)位点(例如,可用碱或ap内切核酸酶裂解)或磷酸二酯键(例如,可经由核酸酶(例如,dna酶)裂解)。
[0124]
在一些实施方案中,裂解结构域包含由能够裂解核酸分子(例如,能够破坏两个或更多个核苷酸之间的磷酸二酯键)的一种或更多种酶识别的序列。键可以经由其它核酸分子靶向酶,诸如限制性内切酶(例如,限制性内切核酸酶)裂解。例如,裂解结构域可以包含限制性内切核酸酶(限制性内切酶)识别序列。限制性内切酶在称为限制性位点的特定识别核苷酸序列处切割双链或单链dna。在一些实施方案中,使用罕见切割限制性内切酶,例如具有长识别位点(长度至少8个碱基对)的酶,来减少在别处裂解的可能性。
[0125]
在一些实施方案中,裂解结构域包含多(u)序列,多(u)序列可以被尿嘧啶dna糖苷酶(udg)和dna糖苷酶-裂解酶内切核酸酶viii(商业上称为user
tm
酶)的混合物裂解。可释放的实体可以在释放后可用于反应。
[0126]
在一些实施方案中,裂解结构域包含切口酶识别位点或序列。切口酶是只裂解dna双链体中的单链的内切核酸酶。因此,裂解结构域可以包含切口酶识别位点,使得该位点的切口使实体之间的物理连接不稳定,并导致它们被分离。
[0127]
在一些实施方案中,裂解结构域包含设计成在存在靶向引导rna的情况下被crispr相关系统识别和切割的核酸序列。在优选实施方案中,设计的靶序列在感兴趣样品基因组内是独特的、极其罕见的或不存在的。
[0128]
在一些实施方案中,裂解结构域包含使得两条链不是100%互补(例如,错配碱基对的数目可以是一个、两个或三个碱基对)的双链核酸。这样的错配例如由muty和t7内切核酸酶i识别,导致核酸分子在错配位置处裂解。
[0129]
在一些实施方案中,裂解结构域包括可以通过使两条链解链以将它们彼此分离而裂解的双链核酸。在一些实施方案中,两条链不是100%互补的,或者含有高比例的at对(》50%),使得所需的解链温度低于基因组的平均解链温度。在一些实施方案中,裂解结构域可以是2个碱基对或更多、或3个碱基对或更多、或4个碱基对或更多、或5个碱基对或更多、或6个碱基对或更多、或8个碱基对或更多、或10个碱基对或更多。在一些实施方案中,结构域解链包括提高温度、或改变ph、改变缓冲液的离子强度、多价离子的螯合或竞争性结合物的添加。
[0130]
结合.如本文使用的“结合(binding)”、“结合(bound)”、“结合(bind)”通常指两个实体(本文中称为“结合配偶体”,例如底物和酶或者抗体和表位)之间的共价或非共价相互
作用。两个或更多个实体之间的任何化学结合是键,包括但不限于:共价键合、σ键合、π键合、离子键合、偶极键合、金属键合、分子间键合、氢键合、范德华键合。由于“结合”是通用术语,以下是结合类型的所有实例:“杂交”、氢结合、小沟结合、大沟结合、点击结合、亲和结合、特异性结合和非特异性结合。其他实例包括:转录因子与核酸结合、蛋白质与核酸结合。
[0131]
特异性和非特异性结合.如本文使用的,术语“特异性结合”和“非特异性结合”必须以文本中使用这些术语的上下文解释。例如,实体可以与核酸分子“特异性结合”,但不具有在某些基因组长度规模上和/或在某些基因组区域内的对所述核酸分子的潜在序列的显著偏好或偏倚。因此,在分子序列的上下文中,实体与所述核酸分子“非特异性结合”。
[0132]
当在物理上不同的分子之间结合的上下文中时,“特异性结合”通常指两个结合配偶体之间在一组特定的条件(例如,生理条件)下使得配偶体彼此结合但不以显著或实质水平结合环境中(例如,生物样品中、组织中)可能存在的其他分子的相互作用。
[0133]
优先结合.术语“优先结合”意指在至少两个不同的结合位点(这些位点可以在同一实体上,或者可以是物理上不同的实体)之间进行比较时,在特定实体和两个位点之间存在非零的结合概率,但是可以存在该特定实体在一个位点上比在另一位点上结合的概率更优先的条件。
[0134]
亲和基团.“亲和基团”是对于与另一个特异性或特定的分子或部分(其“亲和配偶体”)的缔合或结合具有高亲和力或偏好的分子或分子部分。与另一个特异性或特定的分子或部分的缔合或结合可以通过非共价相互作用,诸如氢键合、离子力和范德华相互作用。例如,亲和基团可以是对于蛋白亲和素或链霉亲和素具有高亲和力的生物素。例如,亲和基团也可以指对于生物素具有亲和力的亲和素或链霉亲和素。形成各种亲和色谱方法基础的分子可识别为亲和基团。亲和基团及其结合或缔合的特异性或特定的分子或部分的其他实例包括,但不限于,抗体或抗体片段和它们的相应抗原,诸如地高辛和抗地高辛抗体、凝集素和糖类(例如,糖、单糖、双糖或多糖)、以及受体和受体配体。
[0135]
亲和基团可以能够进行点击化学反应。术语“点击化学”是指由k.barry sharpless引入的化学哲学,该化学哲学描述了定制为通过将包含反应基团的小单元连接在一起而快速且可靠地产生共价键的化学。点击化学不是指特定的反应,而是指一个概念,包括模拟可见于自然界中的反应的反应。在一些实施方案中,点击化学反应是模块化的,范围宽,给出高化学产率,产生无害的副产物,是立体特异性的,表现出大于》84kj/mol的大的热力学驱动力以有利于具有单一反应产物的反应,和/或可以在生理条件下进行。独特的放热反应使反应物“弹性加载(spring loaded)”。在一些实施方案中,点击化学反应表现出高原子经济性,可以在简单的反应条件下进行,使用可容易获得的起始材料和试剂,不使用有毒溶剂或使用良性或容易去除的溶剂(优选地水),和/或通过非色谱方法(结晶或蒸馏)提供简单的产物分离。例如,叠氮化物是环辛炔或任何其他炔烃的配偶体。
[0136]
亲和基团可以形成交联实体中反应基团的一部分。
[0137]
任何一对亲和基团及其结合或缔合的特异性或特定的分子或部分可以具有逆转的角色,例如,使得在第一分子和第二分子之间,在第一种情况下,第一分子被表征为针对第二分子的亲和基团,而在第二种情况下,第二分子被表征为针对第一分子的亲和基团。
[0138]
光不稳定保护基团.“光不稳定保护基团”是与亲和基团相互作用的反应性官能团,使得当光不稳定保护基团暴露于某种光时,结果是所述亲和基团在与它先前受保护的
状态相比时将与其所缔合的结合配偶体结合的可能性增加。在这样的光暴露之前,亲和基团通常被称为被“加笼(caged)”。
[0139]
本领域中已知许多用于加笼的亲和基团的方法以及制备和使用方法,诸如那些保护亲和基团以减少或消除亲和基团对特定靶结合物类具有的亲和力的方法。这些方法可以用于防止成员与不期望的靶结合(否则不期望的靶能够与该成员结合),或者用于其他目的,诸如控制结合的时间和位置。此外,可以利用各种方法来确保亲和基团对靶结合物类的亲和力在脱笼后不降低,至少不实质上降低(与不涉及加笼的情况下具有的亲和力相比)。例如,在通过光刻法制备聚合物阵列的过程内的非限制性实例是用光不稳定保护基团(例如,menpoc、nnpoc、nppoc)保护否则为反应性的官能团。这些反应性官能团然后通过选择性照射被激活以与基底某些区域内的单体偶联,其中光具有能够光解光不稳定保护基团并释放先前保护的或加笼的羟基基团的波长。这种保护笼内的亲和基团的方法当然不限于核酸阵列的光刻法合成,并且该概念的许多变化和改编是本领域熟知的,用于各种方法、化学和应用中的各种分子,诸如核酸、氨基酸、抗体等。
[0140]
诸如生物素的分子的光保护通常通过用可光激活的保护基团对分子进行修饰来实现,其中在分子仍被保护基团加笼时,保护基团位于关键位置处(例如,使特定键失活)以防止不期望的反应。然后无活性的加笼的分子通过适当的辐照(诸如以一个或更多个适当的波长照射,诸如波长短于340nm、350nm、360nm、365nm、375nm、390nm或更大的紫外光)脱笼。
[0141]
许多方法可用于用光不稳定保护基团对聚合物诸如寡核苷酸加笼。例如,加笼保护基团可以放置在核苷酸间磷酸酯、糖上的各个位置或核酸碱基上。某些方法在寡核苷酸的亚磷酰胺合成期间掺入生物素。关于生物素的使用,特别是加笼的受保护生物素的背景,参见美国专利编号[barrett,1989,5,252,743]、[barrett,1989,5,451,683]、[fodor,1989,6,919,211]、和[fodor,1989,6,955,915];美国专利申请公布编号[fodor,1989,2003/0119011]、和[pirrung,1996],为了所有目的将所有这些专利通过引用以其整体并入本文。
[0142]
捕获结构域.“捕获结构域”是直接或通过一系列反应和/或处理步骤与核酸分子、染色质或其它生物分子诸如蛋白质特异性或非特异性结合的实体。
[0143]
在一些实施方案中,捕获结构域是被配置为与一种或更多种分析物相互作用的功能核酸序列,诸如一种或更多种不同类型的核酸(例如,rna分子和dna分子)。在一些实施方案中,功能核酸序列可以包含n-mer序列(例如,随机n-mer序列),这种n-mer序列被选择为与多于一个dna分子相互作用。在一些实施方案中,功能序列可以包含多(t)序列,这种多(t)序列被配置为通过mrna转录物的多(a)尾与信使rna(mrna)分子相互作用。在一些实施方案中,功能核酸序列是蛋白质(例如,转录因子、dna结合蛋白或rna结合蛋白)的结合靶。
[0144]
捕获结构域可以包括核糖核苷酸、脱氧核糖核苷酸和/或肽核酸。它们可以包含能够参与watson-crick型或类似碱基对相互作用的合成核苷酸残基。在一些实施方案中,捕获结构域能够引发逆转录反应以产生与捕获的rna分子互补的cdna。在一些实施方案中,捕获结构域可以引发dna延伸(聚合酶)反应以产生与捕获的dna分子互补的dna。
[0145]
在一些实施方案中,捕获结构域可以作为捕获的dna分子和捕获结构域之间的连接反应的模板。在一些实施方案中,捕获结构域可以连接到捕获的dna分子的一条链。例如,
splintr连接酶以及rna或dna序列(例如,简并rna)可用于将单链dna或rna连接到捕获结构域。在一些实施方案中,具有以rna为模板的连接酶活性的连接酶,例如splintr连接酶、t4 rna连接酶2或kod连接酶,可用于将单链dna或rna连接到捕获结构域。在一些实施方案中,捕获结构域包含夹板寡核苷酸(splint oligonucleotide)。在一些实施方案中,捕获结构域捕获夹板寡核苷酸。在一些实施方案中,捕获结构域包含游离3'末端,其可以例如通过模板依赖性聚合以形成延伸的捕获结构域。在一些实施方案中,捕获结构域包含能够与在与阵列接触的生物样品的细胞中存在的核酸(例如,rna或其他分析物)杂交的核苷酸序列。在一些实施方案中,捕获结构域可以被选择或设计成与靶核酸选择性或特异性结合。例如,捕获结构域可以被选择或设计成通过与mrna多(a)尾杂交的方式来捕获mrna。因此,在一些实施方案中,捕获结构域包含多(t)dna寡核苷酸,例如通过磷酸二酯键连接的一系列连续脱氧胸苷残基,其能够与mrna的多(a)尾杂交。在一些实施方案中,捕获结构域可以包括功能或结构类似于多(t)尾的核苷酸。例如,多(u)寡核苷酸或包含脱氧胸苷类似物的寡核苷酸。在一些实施方案中,捕获结构域包含至少2个、3个、4个、5个、6个、7个、8个、9个、10个、11个、12个、13个、14个、15个、16个、17个、18个、19个或20个核苷酸。在一些实施方案中,捕获结构域包含至少25个、30个、35个、40个、50个核苷酸。例如,fish探针中常用的杂交序列的长度可以是40bp。
[0146]
在一些实施方案中,捕获结构域包含能够与mrna和/或基因组dna结合的序列。例如,捕获结构域可以包含能够与mrna的多(a)尾和/或基因组dna中存在的多(a)均聚序列结合的核酸序列(例如,多(t)序列)。在一些实施方案中,使用末端转移酶将均聚序列添加至mrna分子或基因组dna分子,以产生具有多(a)或多(t)序列的分析物。例如,可以将多(a)序列添加至分析物(例如,基因组dna的片段),从而使分析物能够被多(t)捕获结构域捕获。
[0147]
在一些实施方案中,随机序列,例如随机六聚体或类似序列,可用于形成捕获结构域的全部或一部分。例如,随机序列可以与多(t)(或多(t)类似物)序列一起使用。因此,在捕获结构域包含多(t)(或“多(t)样”)寡核苷酸的情况下,它也可以包含随机寡核苷酸序列(例如,“多(t)随机序列”结构域)。这可以例如位于多(t)序列的5'或3',例如,在捕获结构域的3'末端。多(t)随机序列结构域可以促进对mrna多(a)尾的捕获。在一些实施方案中,捕获结构域可以是完全随机的序列。在一些实施方案中,可以使用简并捕获结构域。在一些实施方案中,捕获结构域可以包含通用序列。在一些实施方案中,捕获结构域可以包含条形码。
[0148]
捕获结构域可以基于其被设计来捕获的特定基因序列或特定基序序列或共有/保守序列(即,序列特异性捕获结构域)。因此,在一些实施方案中,捕获结构域能够选择性地结合期望的核酸亚型或子集,例如特定类型的rna,诸如mrna、rrna、trna、srp rna、tmrna、snrna、snorna、smy rna、scarna、grna、rna酶p、rna酶mrp、terc、sl rna、arna、cis-nat、crrna、incrna、mirna、pirna、sirna、shrna、tasirna、rasirna、7sk、erna、ncrna或其他类型的rna。在非限制性实例中,捕获结构域可以能够选择性地结合期望的核糖核酸子集,例如微生物组rna,如16s rrna。
[0149]
在一些实施方案中,捕获结构域包含“锚”或“锚定序列”,其是设计成确保捕获结构域与预期分析物杂交的核苷酸序列。在一些实施方案中,锚序列包含含有1-mer、2-mer、3-mer或更长的序列的核苷酸的序列。在一些实施方案中,短序列是随机的。例如,包含多
(t)序列的捕获结构域可以被设计成捕获mrna。在这样的实施方案中,锚定序列可以包含随机的3-mer(例如,ggg),其有助于确保多(t)捕获结构域与mrna杂交。在一些实施方案中,锚定序列可以是vn、n或nn。替代地,该序列可以使用特定的核苷酸序列来设计。在一些实施方案中,锚序列位于捕获结构域的3'末端。在一些实施方案中,锚序列位于捕获结构域的5'末端。
[0150]
在一些实施方案中,捕获结构域在接触生物样品之前被封闭。在一些实施方案中,捕获结构域的游离3'末端被封闭或修饰。在一些实施方案中,捕获结构域的游离3'末端可以被杂交的核酸结构(例如,发夹)掩蔽。在一些实施方案中,捕获结构域的游离3'末端可以通过化学修饰来封闭,例如,添加叠氮甲基基团作为化学可逆的封端部分,使得捕获结构域不包含游离3'末端。在生物样品与捕获结构域接触之前封闭或修饰捕获结构域(特别是在捕获结构域的游离3'末端)防止捕获结构域的修饰,例如,防止将多(a)尾添加到捕获结构域的游离3'末端。在一些实施方案中,捕获结构域被光不稳定保护基团加笼,并且可以通过适当暴露于特定波长的光而变得有活性。
[0151]
3'修饰的非限制性实例包括双脱氧c-3'(3'-ddc)、3'反向dt、3'c3间隔区、3'氨基和3'磷酸化。在一些实施方案中,生物样品中的核酸可以被修饰,使得它可以被捕获结构域捕获。例如,可以将衔接子序列(包含能够与捕获结构域结合的互补结合结构域)添加到核酸(例如,片段化的基因组dna)的末端。在一些实施方案中,这是通过连接衔接子序列或延伸核酸来实现的。在一些实施方案中,酶用于在核酸序列的末端掺入另外的核苷酸,例如多(a)尾。在一些实施方案中,捕获结构域可以被可逆地掩蔽或修饰,使得捕获结构域不包含游离3'末端。在一些实施方案中,3'末端被去除、修饰或使其不可接近,使得捕获结构域不受用于修饰生物样品的核酸的过程(例如,连接或延伸)的影响。
[0152]
在一些实施方案中,捕获结构域可以是非核酸结构域。不完全基于核酸的合适捕获结构域的实例包括但不限于模拟本文描述的任何捕获结构域的功能的蛋白质、肽、适配体、抗原、抗体和分子类似物。
[0153]
在一些实施方案中,捕获结构域包含可以与核酸、染色质和/或蛋白质非特异性或特异性结合并因此可以用作交联剂的组分的亲和剂。在一些实施方案中,捕获结构域包含交联部分。在一些实施方案中,交联部分可以通过各种处理或暴露于环境条件或环境条件的变化而激活。
[0154]
在一些实施方案中,捕获结构域包含补骨脂素或其衍生物。在一些实施方案中,捕获结构域包含二氮杂环丙烯或其衍生物。在一些实施方案中,捕获结构域包含甲醛或其衍生物。在一些实施方案中,捕获结构域包含以下中的任何或其衍生物:三氧沙林、甲氧基补骨脂素、羟甲基-4,5',8-三甲基补骨脂素、烷化剂诸如氮芥、顺铂、氯乙基亚硝基脲、丝裂霉素c、双官能醛和双官能醌甲基化物。
[0155]
在一些实施方案中,捕获结构域包括以下中的任一种或其衍生物:二琥珀酰亚胺基戊二酸酯、二琥珀酰亚胺基辛二酸酯、二琥珀酰亚胺基酒石酸酯、己二亚氨酸二甲酯、庚二亚氨酸二甲酯、辛二亚氨酸二甲酯、l,5-二氟-2,4-二硝基苯、n-马来酰亚胺基丙酸酰肼、3-(2-吡啶基二硫代)丙酰基酰肼、双马来酰亚胺基乙烷、二氮杂环丙烯(diazarine)、琥珀酰亚胺基碘乙酸酯、n-马来酰亚胺乙酰-氧基琥珀酰亚胺吡啶基二硫代)丙酸酯、马来酰亚胺基团、硫醇反应性基团、氨基基团诸如伯胺和仲胺、羧基基团、羟基基团、醛基基团、炔基
基团、叠氮基团、羰基、卤代乙酰基(例如,碘乙酰基)基团、亚胺酯基团、n-羟基琥珀酰亚胺酯、巯基基团、吡啶基二硫基团。
[0156]
在一些实施方案中,捕获结构域包含仅在暴露于紫外线或可见光时才变得有反应性的光反应性捕获结构域。两种示例光反应性化学基团包括芳基叠氮类和二氮杂环丙烯类。
[0157]
捕获探针.“捕获探针”或“探针”是指包含捕获结构域和条形码的分子。在一些实施方案中,捕获探针包含可裂解接头或裂解结构域,或可裂解接头或裂解结构域的至少一个残基或至少一部分。例如,捕获探针可以通过可裂解接头连接到实体,并且捕获探针在通过裂解所述接头从所述实体释放之后保留接头的一部分。在一些实施方案中,捕获探针包含引发位点。在一些实施方案中,捕获探针包含一个或更多个荧光团。在一些实施方案中,捕获探针包含fret对的供体或受体,或者可以猝灭附近荧光团的猝灭体(quenching body)。在一些实施方案中,捕获探针包含功能序列,诸如功能结构域。在一些实施方案中,除了捕获结构域之外,捕获探针还包含亲和基团,例如生物素。
[0158]
功能结构域.“功能结构域”包含用于整个分析过程中下游分析步骤的功能核苷酸序列。它们通常可用于测序工作流程中的后续处理。例如,功能序列可以包含测序仪特异性流动池附接序列,和/或测序引物序列,例如用于测序的r1引物序列,和/或用于测序的r2引物序列。功能序列通常可以被选择为与各种不同测序系统(例如,454测序、ion torrent proton或pgm、illumina x10、pacbio、纳米孔、bgi)中的任何一种及其要求相容。可以使用合适的功能序列的这样的商业化测序系统和技术的实例包括(但不限于)roche 454测序、ion torrent proton或pgm测序、illumina x10测序、pacbio smrt测序和oxford纳米孔测序。此外,在一些实施方案中,功能序列可以被选择为与其他测序系统包括非商业化测序系统相容。功能序列及其使用的实例描述于[hindson,2012,2014/0378345]和[hindson,2014 2015/0376609]中,其中每个的全部内容通过引用并入本文。
[0159]
交联剂.如本文使用的术语“交联(crosslink)”、“交联(crosslinking)”或“交联(crosslink)”是指通过与使两个或更多个化合物桥接的“交联剂(crosslinker)”(或“交联剂(crosslinking agent)”或“交联试剂(crosslinking reagent)”)结合而使两个或更多个化合物之间的任何稳定化学缔合成为可能,使得它们可以作为一个单元进一步被处理,其中交联剂包含至少两个亲和基团或捕获结构域,所述至少两个亲和基团或捕获结构域将与它们各自的靶化合物结合。这样的稳定性可以基于共价键合和/或非共价键合,并且可以被化学或光激活逆转。所述两个或更多个化合物的性质可以是相同的、相似的或不同的。类似地,所述两个或更多个亲和基团或捕获结构域的性质可以是相同的、相似的或不同的。
[0160]
梳理.本文定义的“分子梳理(molecular combing)”或“梳理(combing)”是指将大分子特别是核酸分子的至少一部分固定到基底表面、或基底表面上的多孔膜内,使得大分子的至少一部分在基本上平行于所述基底表面的平面中被拉长的过程。拉长部分可以完全固定到基底,或者所述部分的至少一部分具有一定程度的自由。在一些实施方案中,分子的至少一部分在平行于所述基底表面的多孔材料膜内被拉长,或者分子的至少一部分在平行于所述基底表面的多孔材料膜的顶部被拉长,或者分子的至少一部分在两点之间被拉长并悬浮。在一些实施方案中,基底表面是流体装置的至少一部分。
[0161]
在一种实施方案中,单个核酸分子通过一个或两个末端(或接近一个或两个末端
的区域)与修饰的表面(例如,硅烷化玻璃)结合,并且然后被后退空气/水界面(receding air/water interface)基本上均匀地拉伸和对齐。schurra和bensimon(2009)“combing genomic dna for structure and functional studies.”methods mol.biol.464:71-90;另参见美国专利编号[bensimon,1995,7,122,647],这两者通过引用以其整体并入本文。
[0162]
完全拉伸的核酸分子的百分比取决于核酸分子的长度和所用的方法。通常,核酸分子在表面上伸展得越长,就越容易实现完全拉伸。例如,根据[conti,2003],在一些毛细管流动条件下,10kb dna分子的超过40%可以被常规拉伸,而使用相同的条件,4kb分子的仅20%可以被完全拉伸。对于较短的核酸片段,拉伸质量可以通过将盖玻片落到载玻片上来诱导的较强的流动而改进。然而,这种方法可能会将较长的核酸片段剪切成较短的片段,并因此可能不适合拉伸较长的分子。参见例如,[conti,2003]conti等人(2003)current protocols in cytometry john wiley&sons,inc.和[gueroui,2002]gueroui等人(2022年4月30日)“observation by fluorescence microscopy of transcription on single combed dna.”pnas 99(9):6005-6010,二者特此通过引用以其整体并入。另参见[bensimon,1994,5,840,862]、[bensimon,1995,wo 97/18326]、[bensimon,1999,wo 00/73503]、[bensimon,1995,7,122,647],它们特此通过引用以其整体并入。[lebofsky,2003]“single dna molecule analysis:applications of molecular combing.”brief funct.genomic proteomic 1:385-96,特此通过引用以其整体并入。
[0163]
在一些实施方案中,长核酸分子在一端附着到基底,并由各种弱力(例如,电学力、表面张力或光学力)拉伸。在该实施方案中,核酸分子的一端被首先锚定到表面。例如,分子可以通过吸附附着到疏水表面(例如,改性玻璃)。锚定的核酸分子可以被后退弯液面、蒸发或被氮气流拉伸。参见例如,[chan,2006]“a simple dna stretching method for fluorescence imaging of single dna molecules.”nucleic acids research 34(17):e1-e6,通过引用以其整体并入本文。
[0164]
另一种实施方案,通过将长核酸分子溶解在缓冲液的液滴中并沿着基底下行来拉伸核酸分子。在另外的实施方案中,长核酸分子被包埋在琼脂糖或其他凝胶中。包含核酸的琼脂糖然后被融化并沿着基底梳理。
[0165]
在另一种实施方案中,分子在至少一个特定的点附着到基底,允许分子的其余部分有相当大的自由度,使得通过以基本上平行于基底表面的方向对分子施加外力来获得分子的拉长部分。这样的实施方案的实例包括“dna幕(curtain)”[gibb,2012],其中附着点为受控过程,或者附着点可以通过分子与流体特征(例如,如[craighead,2011,专利9,926,552]示出的柱)的相互作用而是随机的。
[0166]
在一些实施方案中,分子梳理可以通过在流体装置中拉长分子产生的流体流来完成,使得在装置中拉长之后,分子以拉长状态呈现在装置表面上,或者在装置表面的多孔膜内。在一些实施方案中,装置的流体通道不完全受限,使得在运送溶液蒸发之后,分子以拉长状态至少部分地固定在装置的表面上。在一些实施方案中,如图2中示出的,分子205在微流体装置(204)的受限拉长通道中被拉长,微流体装置(204)在此处具有提供有助于促进拉长的限制环境和/或物理障碍(203)的通道尺寸(202)。在微流体装置内包围分子的溶液中的胶凝材料然后被胶凝。最后,通过移除顶部(201),同时将分子保持在凝胶膜内,或者通过使用多孔顶部材料,使分子(215)可接近装置表面。
[0167]
微流体装置.如本文使用的术语“微流体装置”或“流体装置”通常是指这样的装置,其被配置为用于流体运送和/或通过流体运送实体,并且具有流体通道,流体可以在流体通道中以不大于约100微米的至少一个最小尺寸流动。最小尺寸可以是任何长度、宽度、高度、半径或横截面轴。微流体装置还可以包括多于一个流体通道。微流体装置的给定流体通道的一个或更多个尺寸可以取决于例如一个通道和/或更多个通道的特定配置以及也包括在装置中的其他特征而变化。
[0168]
本文描述的微流体装置还可以包括任何另外的部件,所述另外的部件可以例如有助于调节流体流动,诸如流体流动调节器(例如,泵、压力源等),有助于防止流体通道堵塞的特征(例如,通道中的漏斗特征;位于通道之间的储库,向流体通道提供流体的储库等)和/或从流体流中去除碎片的特征,诸如例如过滤器。此外,微流体装置可以被配置为流体芯片,其包括向微流体通道的布置供应流体的一个或更多个储库,并且还包括接收通过微流体装置的流体的一个或更多个储库。另外,微流体装置可以由任何合适的材料构成,包括聚合物物类和玻璃,或者由多相不混溶介质包封形成的通道和腔。微流体装置可以包含多个微通道、阀、泵、反应器、混合器和其他用于产生液滴的部件。微流体装置可以包含主动和/或被动传感器、电子和/或磁装置、集成的光学或官能化表面。限定微流体装置通道的物理基底可以是固体或柔性的、可渗透或不可渗透的,或其组合,物理基底可以随位置和/或时间而变化。微流体装置可以包括对至少一个波长的光至少部分透明和/或对至少一个波长的光至少部分不透明的材料。
[0169]
微流体装置可以完全独立,具有对其内包含的所需样品进行操作的所有必需的功能。操作可以是完全被动的,诸如使用毛细管压力来操作流体流[juncker,2002],或者可以包含内部电源,诸如电池。可选地,流体装置可以在外部装置的协助下操作,所述外部装置可以提供功率、电压、电流、磁场、压力、真空、光、热、冷却、感测、成像、数字通信、包封、环境条件等的任何组合。外部装置可以是移动装置,诸如智能电话,或者较大的台式装置。
[0170]
通道内的流体限制可以通过其中流体可以在流体装置内或流体装置上限定的特征内或特征上保持一定时间段的任何方法进行。在大多数实施方案中,流体被通道壁的固体或半固体物理边界所限制。图3示出了具有诸如矩形(302)、三角形(303)、椭圆形(304)和混合几何形状(305)的横截面的通道壁全部限定在流体装置(301)内的实例。在其他实施方案中,流体装置内的流体限制可以通过固体物理特征与表面能特征的组合[casavant,2013]或不混溶流体[li,2020]而被至少部分地限制。被至少部分地限制在物理边界内的流体的实例包括物理限定在流体装置(306)的表面上的各种通道,诸如凹槽(307、308)和矩形(309、310),所有这些通道都填充有足以使表面张力允许液体被物理保持在通道内而不溢出的最小量的液体。在其他实施方案中,通道(311)可以由流体装置的拐角(312)中的凹槽限定,或者通道(314)可以由流体装置的两个物理分离的边界(313和315)限定,或者通道(321)可以由流体装置的拐角(320)限定。在其它实施方案中,通道(317)由流体装置(316)表面上的亲水部分(318)限定,其中亲水部分由流体装置表面上的疏水部分(319)界定。在所有情况下,这些实施方案是非限制性实例。
[0171]
应理解,本文描述的一些原理和设计特征可以缩放到更大的装置和系统,包括采用通道横截面达到毫米或甚至厘米规模的通道和特征的装置和系统。因此,当将一些装置和系统描述为“微流体”时,在某些实施方案中意图该描述同等地适用于一些更大规模的装
置。另外,应理解,本文描述的一些原理和设计特征可以缩放到更小的装置和系统,包括采用通道横截面为数百纳米、或甚至数十纳米、或甚至单纳米规模的通道和特征的装置和系统。因此,当将一些装置和系统描述为“微流体”时,在某些实施方案中意图该描述同等地适用于一些更小规模的装置。作为实例,装置可以具有直径为数毫米的输入孔,以容纳由移液器加载的液体,输入孔与长度为数厘米、数百微米宽、数百nm深的通道流体连接,流体通道则与直径为0.1nm-10nm的纳米孔收缩装置流体连接。
[0172]
根据本发明的某些方面,各种材料和方法可以用于形成物件(article)或部件,诸如本文描述的物件或部件,例如通道,诸如微流体通道、室等。例如,各种物件或部件可以由固体材料形成,其中通道可以通过微加工、膜沉积工艺形成,诸如旋涂和化学气相沉积、激光制造、光刻技术、键合(bonding)技术、沉积技术、层压技术、模制技术、包括湿化学或等离子体工艺的蚀刻方法、多相不混溶介质包封等。对于图案化,可以采用各种方法,包括但不限于:光刻法、电子束蚀刻法、纳米压印蚀刻法、afm蚀刻法、stm蚀刻法、聚焦离子束蚀刻法、冲压、压花、模制和蘸笔蚀刻法(dip pen lithography)。对于键合,可以采用各种方法,包括但不限于:热键合、胶键合、表面活化键合、熔融键合、阳极键合、等离子体活化的键合、激光键合和超声键合。
[0173]
包装物.“包装物(package)”是能够在实体的限定边界内容纳内容物的任何实体。在一些实施方案中,边界由物理屏障诸如脂质双层或表面活性剂限定。在一些实施方案中,没有屏障,诸如由混合两种不混溶的流体形成的液滴。包装物的非穷尽列表包括:细胞、细胞核、囊泡、外泌体、线粒体、细胞器、细菌、病毒、泡(bubble)、人工膜包装物、油包水液滴、水包油液滴、水-油-水液滴、油-水-油液滴。在所有情况下,包装物可以通过各种方式裂解(或破裂)以释放内容物。
[0174]
凝胶.“凝胶”被定义为包含已交联(“胶凝”)的“胶凝剂”的基本上稀释或多孔的体系。凝胶的非限制性实例包括琼脂糖、聚丙烯酰胺、水凝胶[cal
ó
,2015]、dna凝胶[2020]。在本文件的上下文中,凝胶和半凝胶是等同的,其中半凝胶是具有不完全交联和/或低浓度的胶凝剂的凝胶。在一些实施方案中,凝胶可以包含光可降解特性。在一些实施方案中,胶凝剂的交联可以包括光激活。
[0175]
环境条件.“环境条件”可以包括围绕生物分子的物理、物质、化学的任何特性,这些特性可以影响所述生物分子的物理状态、热力学状态、化学状态或对其他试剂的反应性。对生物分子的影响可以由于环境条件的存在或者环境条件的改变引起。环境条件可以包括温度、压力、湿度水平、ph、离子浓度、流速或方向。环境条件可以是光波长的通量、偏振、强度。环境条件可以包括溶液组成,例如溶液中特定试剂的浓度,或溶液中某些试剂的比例,或用于特定缓冲液的盐组成。环境条件可以包括作用在生物分子上的外力,例如溶液或空气流速。环境条件可以包括导热特性、导电特性、光学不透明度或透明特性。环境条件可以是电场或磁场。
[0176]
fret对.当两个荧光团(“fret对”)非常接近,并且当一个荧光团(供体d)的发射光谱与另一个荧光团(受体a)的激发光谱重叠时,以及当d处于激发态时,发生荧光共振能量转移(“fret”)。能量转移速率kt由下式给出:
[0177]
kt=1/τd(zf/z)6
[0178]
其中τd是d的荧光寿命,z是d和a之间的距离,并且zf是半径。当z=zf时,
发射速率和能量转移速率相等,并且50%的激发供体通过能量转移被失活。zf的值是根据d和a的光谱参数计算的,并考虑了它们的相对取向。fret的效用源自能量转移效率对d和a之间距离的六次方的强烈依赖性。当d和a之间的距离接近zf时,发生最高效的能量转移,对于大多数有机荧光染料对,zf在2纳米和7纳米之间(wu&brand(1994)anal.biochem.218:1-13)。由于这种强烈的距离依赖性,fret通常被称为“光谱尺”,其能够测量纳米范围内的距离(stryer(1978)ann.rev.biochem.47:819-846)。
[0179]
外力.“外力”是任何施加在物体上使得该力可以从静止状态扰动物体的力。非限制性实例包括流体流动施加的流体动力学拉力[larson,1999](其可以通过压差、重力、毛细管作用、电渗透来模拟)、电场、电动力学力、电泳力、脉冲电泳力、磁力、介电力、离心加速度或其组合。另外,外力可以间接施加,例如,如果珠结合到实体,并且然后珠经受外力,诸如磁场或光学放大器(optical teaser)。
[0180]
树枝状大分子.术语“树枝状大分子”是指超支化(hyper-branched)聚合物,或包含超支化聚合物的实体。在某些实施方案中,树枝状大分子,诸如pamam树枝状大分子,允许对球形聚合物尺寸的精确控制。
[0181]
树枝状大分子包括树枝状大分子核心。在某些实施方案中,树枝状大分子核心可以是炔丙胺、乙二胺、三乙醇胺、季戊四醇、叠氮-丙基(烷基)胺、羟乙基(烷基)胺、四苯基甲烷、均苯三甲酰氯、二氨基己烷、二氨基丁烷、胱胺或丙二胺。在特定方面,树枝状大分子的核心是乙二胺。在一些方面,树枝状大分子核心至少部分包含核酸聚合物。树枝状大分子还包括重复单元或臂。在某些方面,重复单元是炔丙胺、乙二胺、三乙醇胺、季戊四醇、丙胺、丙亚胺、叠氮-丙基(烷基)胺、羟乙基(烷基)胺、四苯基甲烷、均苯三甲酰氯、二氨基己烷、二氨基丁烷、胱胺、丙二胺和赖氨酸。在特定方面,重复单元是酰胺胺(amidoamine)。树枝状大分子可以是核酸树枝状大分子。树枝状大分子可以包含树枝状大分子的组合。在某些方面,树枝状大分子具有约1nm至约1000nm的直径。
[0182]
在流体狭缝通道装置中对dna结构进行荧光分析
[0183]
以下一组公开内容包括用于在用流体狭缝通道装置内用至少一种荧光标记体研究包含至少一种高级核酸结构的长核酸分子的装置和方法。在优选实施方案中,至少一种标记体包含嵌入荧光染料。通过在流体装置的流体狭缝通道中对高级核酸结构进行荧光成像,可以将所述结构的状态和动态控制在用于探查的期望的光学平面内,同时允许用户在用探查系统探查之前和探查期间对所述结构的环境条件和试剂暴露条件的控制的更大灵活性。图4示出了一种示例性微流体装置,其中待研究的高级核酸结构垂直限制在流体狭缝通道内。
[0184]
狭缝的深度可以取决于多种因素,包括光学探查系统的聚焦深度、被研究的高级核酸结构的物理性质,或者用于物理控制样品的熵障(entropic barrier)和凹陷的物理限制。在一些实施方案中,深度可以小于5微米、或小于4微米、或小于3微米、或小于2微米、或小于1微米、或小于0.8微米、或小于0.7微米、或小于0.6微米、或小于0.5微米、或小于0.4微米、或小于0.3微米、或小于0.2微米、或小于0.1微米、或小于0.09微米、或小于0.08微米、或小于0.07微米、或小于0.06微米、或小于0.05微米或小于0.03微米。在一些实施方案中,核酸结构来源于在微流体装置中裂解的包装物,并且然后在狭缝内收集其内容物。在一些实施方案中,在将样品引入微流体装置之前将荧光标记和染料结合至结构,而在其他实施方
案中,标记和染料在装置中结合至结构。
[0185]
图4展示了一种特定的实施方案,其中具有高级核酸结构的长核酸分子(401)被限制在狭缝流体通道(406)内。在该特定实施方案中,高级结构由黏连蛋白复合物(408和409)形成的环(402和403)组成。另外,荧光标记体(405)位于或接近正向和反向ctcf位点。狭缝流体通道被设计成足够浅,使得长核酸内的感兴趣的结构的一部分可以在探查所述部分期间保持在拉长状态。用这样的装置和方法探查高级核酸结构是非常有利的,因为感兴趣的结构的至少一部分可以保持在探查系统的焦深内,允许观察大范围的关联和相互作用。在优选实施方案中,大部分结构可以在探查期间保持在探查系统的焦深内。另外,流体环境允许实时捕捉动态。例如,可以对长核酸分子通过黏连蛋白复合物挤出(407)的过程进行观察和定量。挤出速度可以通过测量环尺寸的变化,或者通过在分子上包括荧光物理图谱,或者通过在长核酸分子上添加可以作为物理参考点的标记体来确定。在一些实施方案中,可以响应于试剂的引入或去除来监测结构的物理构象的动态变化。在一些实施方案中,可以响应于环境条件的变化或保持来监测结构的物理构象的动态变化。标记体可以与长核酸分子位点特异性或非特异性结合,以便为追踪提供地标。在诸如图4中示出的一些实施方案中,位点特异性ctcf正向或反向标记体可用于监测ctcf/黏连蛋白复合物相互作用。
[0186]
图5展示了一种特定的实施方案,其中具有高级核酸结构的长核酸分子(501)被限制在狭缝流体通道(505)内。在该特定实施方案中,高级结构由拓扑关联结构域(tad)(502和503)组成。狭缝流体通道被设计成足够浅,使得tad之间的互连核酸分子(506)的至少一部分可以保持在拉长状态,允许通过探查系统探查该区域内的物理图谱。以这样的装置和方法探查高级核酸结构是非常有利的,因为tad之间的互连区域可以通过与参考物的比较通过物理图谱来鉴定,允许推断tad的基因组组成。例如,通过将核酸的拉长部分(506)的物理图谱与参考物进行比较而鉴定所述部分的位置,允许确定每个tad(502和503)中包含什么基因、启动子和增强子的什么部分。在一些实施方案中,狭缝深度被选择为足够深(50-2000nm),使得tad能够被物理布置成类似于野生细胞环境中的体积的体积。在这样的构型中,tad内的核酸将预期具有高度的非拉长部分,使得tad内或tad之间的核酸连接部分的物理作图具有挑战性。然而,可以通过荧光探查进行关于dna质量、密度和物理构象的观察以及关于tad的接近关系的观察。另外,可以进行对随环境条件或环境条件变化(包括试剂交换)而变化的tad的探查。在一些实施方案中,狭缝深度被选择成使得大部分tad被压缩在等于或小于探查系统的焦深的2d平面内,如图5中示出的。在该实施方案中,长核酸将在2d平面内与其自身重叠,然而可以分析荧光成像的长核酸分子的图像识别软件,以追踪一幅或更多幅图像内的分子聚合物链。另外,通过用标记体标记分子以产生物理图谱,可以推断接近关系。例如,分子的不同tad部分之间的接近关系507。
[0187]
在其中高级核酸结构的一部分没有完全包含在探查系统的焦深内的一些实施方案中,可以在已知步进高度(单位为nm)的不同深度捕获多于一幅图像,并且然后通过图像分析将所述图像拼接在一起,以产生所述结构的计算机3d渲染(in-silico 3d rendering)。
[0188]
在一些实施方案中,当在装置中时,整个或部分或高级核酸结构可以暴露于试剂和环境条件的期望组合。这些试剂可以包括能够原位修饰结构的酶。在一些实施方案中,各种微流体特征、流体流和力用于在装置中对核酸进行物理操作和拉长。在一些实施方案中,
可以在核酸的至少处于拉长状态的一部分上产生物理图谱。该物理图谱可以用作地标来鉴定被研究的高级核酸结构在基因组内的可能起源。在一些实施方案中,物理图谱随着结构动态改变物理构型而产生。在一些实施方案中,各种微流体特征可以包括凹陷(pits)、立柱(posts)、柱(pillars)、槽(troughs)、通道(channels)、凸起(bumps)、拓扑变化(topological variations)和/或拐角(corners)。在一些实施方案中,与核酸结构结合的荧光探针可以是特异性的或非特异性的。在一些实施方案中,荧光探针附接至转录复合物的组分。附接可以是通过特异性结合,或可以是通过组分的修饰和/或突变以包括荧光体。在一些实施方案中,用于标识不同实体的荧光团可以是荧光团的不同组合。在一些实施方案中,不同实体上的不同荧光团可以形成fret对。
[0189]
在多孔凝胶材料中捕获长dna以用于3d分析
[0190]
在另一组实施方案中,包含高级核酸结构的长核酸分子被固定在流体装置的流体通道内的多孔凝胶材料内,使得可以保持物理接近关系以进行探查。利用由探查系统控制的流体装置来将核酸固定在凝胶内是非常有利的,因为这样的装置和方法允许对所述结构被固定的时间点进行观察和选择。另外,被固定在由探查系统控制的流体装置中的凝胶中允许对将样品物理定位到流体装置内或流体装置上的期望位置的控制,并且允许对将试剂和环境条件暴露于样品的空间靶向和定时控制,允许样品处理和探查的自动化,并且允许长核酸分子来源于各种输入样品类型。
[0191]
在一些实施方案中,其中长核酸分子被固定在凝胶中的流体通道是流体装置内的受限流体通道。在一些实施方案中,其中长核酸分子被固定在凝胶中的流体通道是流体装置上的非受限流体通道。
[0192]
在一些实施方案中,被固定在凝胶中的时间点可以参考某个事件、或一系列事件或事件的组合来选择。在一些实施方案中,参考点可以直接处于事件时或约处于事件时。在一些实施方案中,参考点可以在事件之前或之后。在一些实施方案中,可以存在关联的持续时间,例如,事件之前5分钟,或事件之后5分钟。
[0193]
在一些实施方案中,被固定在凝胶中的时间点可以参考细胞周期的某个时间点来选择,例如当细胞处于间期、或前期、或前中期、或中期、或后期或末期时。在一些实施方案中,被固定在凝胶中的时间点可以参考细胞的某个检查点来选择。在一些实施方案中,凝胶中的固定可以参考细胞生命中的某个时间点来选择,例如在细胞凋亡期间。在一些实施方案中,被固定在凝胶中的时间点可以参考当细胞经历特定形态变化时的某个时间点来选择,例如起泡(blebbing)、质壁分离(plasmolysis)、核碎裂、核固缩、胞吞作用、吞噬作用、病毒颗粒分泌期间的出芽/裂解或dna片段化。在一些实施方案中,被固定在凝胶中的时间点可以参考当细胞中的核酸结构经历物理组成、形态、密度、构象或拓扑变化时的时间点来选择。在一些实施方案中,被固定在凝胶中的时间点可以参考当细胞经历特定酶促活性时的时间点来选择,例如转录、复制复合物的形成或解构、环挤出、基因调控、转录复合物的形成或解构、染色体凝缩或压缩的形成或解构、姐妹染色单体解析。在一些实施方案中,被固定在凝胶中的时间点可以参考当包装物或长核酸分子暴露于特定试剂、试剂组合或试剂浓度时的时间点来选择。在一些实施方案中,被固定在凝胶中的时间点可以参考当包装物或长核酸分子暴露于特定环境条件或环境条件变化时的时间点来选择。在一些实施方案中,被固定在凝胶中的时间点可以参考当包装物或长核酸分子暴露于特定蛋白质、酶或外源核
酸诸如病毒基因组整合或染色体外病毒附加体通过ebna1结合位点与宿主染色体物理对接时的时间点来选择。
[0194]
图6展示了基于对细胞的观察将长核酸分子固定在凝胶中的实施方案装置和方法。在时间点(i),包含至少一个长核酸分子(此处染色体:601)的细胞(602)流动(609)通过第一流体通道(610),该第一流体通道包含在缓冲液中的胶凝剂。在该实施方案中,第一流体通道(610)与第二流体通道(605)交会(606),并且围绕该交会部的所有出口的是流体过滤特征(607、608、604),在过滤特征之间具有流体开口,其中过滤特征之间的流体开口小到足以阻挡所述完整细胞或长核酸分子通过,除非在所述完整细胞或长核酸分子上施加足够大的外力。在稍后的时间点(ii),细胞将进入交会区域(606)并被捕获而不能前进通过过滤特征,然而过滤特征将允许在交会部中进行溶液和试剂交换。然后通过用光学探查系统的探查来观察细胞,直到达到期望的固定时间。在一些实施方案中,在观察期间可以保持或改变试剂和环境条件。在一些实施方案中,细胞或细胞内的分子和实体可以在进入交会区域之前或进入交会区域之后用染料染色或标记。染料的性质可以是荧光的或非荧光的。当达到期望的时间点时,胶凝剂然后被胶凝,在第一通道(611)中产生多孔凝胶材料。接下来(iii),通过使试剂(622)流动(621)通过第二通道流入交会区域(606),可以裂解细胞,去除非染色体物质,并且剩余的染色体物质保持适当地固定在凝胶中。然后可以在固定在凝胶中的同时进一步处理长核酸材料,包括试剂暴露、保持或改变环境条件,和进行探查。
[0195]
在一些实施方案中,流体通道的顶部是多孔的或可移除的。因此,对于试剂交换,第二通道不是必要的。对于其中顶部是多孔的实施方案,试剂交换可以通过顶部通过流动或扩散来完成。对于其中顶部是可移除的实施方案,例如键合到氧化硅流体装置的pdms顶部,顶部可以在胶凝剂胶凝之后被移除,以允许通过扩散直接与凝胶中的物质进行试剂交换。
[0196]
在一些实施方案中,在流体装置中不存在流体过滤特征(607、608、604)。包装物或长核酸分子与胶凝剂一起流入第一通道,并且当包装物或长核酸定位于交会部(606)时流动停止,并且然后胶凝剂被胶凝,允许试剂通过第二通道通过多孔凝胶交换。
[0197]
在一些实施方案中,通过分析探查数据,从长核酸分子群体中选择至少一个长核酸分子,以固定在多孔凝胶中。在一些实施方案中,长核酸分子群体被固定在凝胶中,并且使用选择标准从群体中取出至少一个单个分子,或者从群体中取出至少一个单个分子的至少一部分。
[0198]
在一些实施方案中,选择标准包括对物理图谱的分析。在一些实施方案中,将物理图谱与参考物进行比较。在一些实施方案中,物理图谱与选择的至少一个长核酸分子相关。在一些实施方案中,物理图谱与来自未选择的分子群体的至少一个长核酸分子相关。在一些实施方案中,选择标准包括长核酸分子上标记体的存在或不存在。
[0199]
在一些实施方案中,包含至少一个长核酸分子的至少一个包装物选自包装物的群体。在一些实施方案中,至少部分地通过对探查数据的分析来为包装物选择标准提供信息。在一些实施方案中,包装物选择标准包括至少一个标记体的存在或不存在。在一些实施方案中,包装物选择标准包括包装物形态、或包装物类型、或来源组织类型、或疾病类型、或与疾病的关联或细胞周期的状态。
[0200]
在一些实施方案中,多孔凝胶材料内的长核酸分子当在凝胶中时经历任何接近性
3d作图过程(参见定义)的至少一部分。这样的过程可以包括试剂交换、环境条件、交联反应、消化、限制性裂解、酶促反应或连接。
[0201]
在一些实施方案中,多孔凝胶材料内的长核酸分子当在凝胶中时经历任何3d物理作图过程(参见定义)的至少一部分。这样的过程可以包括试剂交换、标签的杂交、标记体的结合、引物的杂交、条形码的杂交、酶促过程(诸如连接、消化、产生切口、限制性裂解或聚合)、环境条件。另外,当长核酸分子固定在凝胶内时,可以进行光学探查或各种试剂的循环与3d物理作图过程的全部或部分。
[0202]
在一些实施方案中,在固定在凝胶材料中之前或之后,通过蛋白质消化或蛋白质变性(例如用蛋白酶k)或更靶向的驰豫(诸如用topo ii、wapl处理),使长核酸分子至少部分地去除蛋白质。在一些实施方案中,染色体的至少一部分在非湍流条件下暴露于蛋白质消化或变性,使得允许染色体通过聚合物自我回避(polymer self-avoidance)以使熵最小化在流体装置的流体通道或室内膨胀(swell)到更大的物理尺寸,其后膨胀的染色体(the expanded chromosome)然后被固定在多孔凝胶中。在优选实施方案中,在膨胀过程期间保持或修改环境条件,以近似平均地保持染色体内接近关系的比例。在一些实施方案中,环境条件的这种修改通过反馈系统来控制,其至少部分地由在膨胀过程期间光学探查染色体产生的数据来提供信息。图7(a)展示了该实施方案,其中(i)长核酸分子(此处是处于中期的染色体703)在流体装置的流体通道(701)中,并暴露于蛋白质消化酶(702)。在蛋白质消化之后,(ii)由于聚合物自我回避,染色体的尺寸从原始体积膨胀(704)。在膨胀后,(iii)在通道中包围染色体的胶凝剂被胶凝,允许试剂交换(706)以参与与所述膨胀染色体的反应,同时所述染色体保持固定在多孔凝胶(705)中。在一些实施方案中,在蛋白质消化或变性之前引入胶凝剂。在一些实施方案中,在蛋白质消化或变性之后引入胶凝剂。
[0203]
图7(b)展示了图7(a)的放大描述,其中(i)长核酸分子(此处是包含染色体核酸(711)的染色质,其通过核小体(712)保持致密构型)在流体装置的流体通道(701)中,并暴露于消化核小体的蛋白质消化酶(702)。在蛋白质消化之后,(ii)从核小体中释放染色体核酸,并膨胀原始体积的倍数,使得两点之间存在从消化前(713)到消化后(714)增加的距离关系。在膨胀后,(iii)包围核酸(711)的胶凝剂以胶凝固定,允许试剂交换(706)参与与所述膨胀染色体的反应,同时所述染色体保持固定在多孔凝胶(705)中。
[0204]
在固定在多孔凝胶中的同时探查膨胀的染色体对于其中同时探查多于一个荧光信号,并且每个信号对应于染色体上的特定位置的3d物理作图应用特别有利。首先,通过使染色体膨胀,可以进行的独立同时测量的数目增加,因为对于任何给定的光学探查系统,存在对在一定体积内可以进行的这种测量的密度的固有限制。其次,本来通常太近而无法光学分辨的两个荧光信号之间的距离测量在膨胀后可以变得可分辨。通过测量染色体的膨胀程度,可以将测量的距离长度校正到它们在膨胀之前的原始值。在一些应用中,至少部分地通过使用膨胀过程期间或之后的染色体的光学探查数据来估计膨胀之前染色体内两个位置之间的绝对距离。在一些应用中,只需要膨胀的染色体内的位置的相对定位。
[0205]
在一些实施方案中,将核酸分子从装置中取出,并且在外部进行3d作图反应。在一些实施方案中,将核酸分子暴露于至少一种类型的限制性内切酶,以在多于一个位置裂解分子。在一些实施方案中,在裂解之后,在至少一对裂解的末端上连接、结合、附接或掺入条形码。在一些实施方案中,条形码对于来源于单细胞的核酸是独特的。在一些实施方案中,
流体装置能够捕获至少两个不同细胞并保持至少两个不同细胞的物理分离。
[0206]
通过将染色体包埋在可光降解的基质中产生空间分辨的或亚染色体采样的基因组文库
[0207]
提供了用于通过以下从整个群体中选择性分离一部分核酸的方法:将该群体包埋在可光裂解或可光降解的基质(901),优选地可光裂解或可光降解的水凝胶中,然后随后基于水凝胶的空间限制的光降解(905)仅释放该群体的子集。在从接近核酸的区域(906)中消除水凝胶后,可以进行酶促、化学和机械操作,包括取出(907)到独特的容器(908)中。非限制性应用是提取存在于完整细胞核、裂解的细胞核、化学固定或部分化学固定的细胞核、膨胀的细胞核、包埋在基质中的膨胀的细胞核中的dna。另外的非限制性应用包括分离沉积在平坦表面、结构化表面、流体装置或三维装置上的部分凝缩或完全凝缩的染色体,替代地提取沉积在整个细胞、固定的细胞、裂解的细胞或基质中膨胀的细胞的rna,如在膨胀显微术的实践中的。该方法可以更广泛地用于固定的细胞核或完整细胞的空间剖析。
[0208]
在一些实施方案中,从染色体群体中分离染色体的至少一部分的方法,其中(a)将染色体群体固定在可光降解的凝胶中,(b)用光学探查系统探查所述固定的染色体,并且基于至少部分地由所述探查提供信息的选择标准选择染色体的至少一部分,以及(c)通过对固定染色体的所述部分的凝胶的靶向光降解从群体中分离所述部分,并且然后(d)将选择部分从剩余的固定的群体中洗脱出来并收集它。在一些实施方案中,选定的染色体的至少一部分从其来源染色体光裂解。在一些实施方案中,选定的染色体的至少一部分具有至少一个结合的标记体。在一些实施方案中,将凝胶中的所述固定的染色体暴露于至少一种试剂。在一些实施方案中,将凝胶中的所述固定的染色体暴露于至少一种环境条件。在一些实施方案中,所述凝胶是水凝胶。在一些实施方案中,所述凝胶是可光降解凝胶。在一些实施方案中,选定的染色体的至少一部分包括拉长的长核酸分子。在一些实施方案中,选定的染色体的至少一部分包括物理图谱。在一些实施方案中,所述物理图谱与参考物的比较至少部分地为选择标准提供信息。在一些实施方案中,物理图谱是线性物理图谱。在一些实施方案中,物理图谱是2d物理图谱。在一些实施方案中,物理图谱是3d物理图谱。在一些实施方案中,在被固定在凝胶中之前,在基底上、在流体装置中或在流体装置上梳理染色体。在一些实施方案中,在被固定在凝胶中之前,在流体装置内的流体通道中或在流体装置上至少部分地拉长染色体。
[0209]
优选实施方案获取凝缩的染色体并将其沉积在平坦或结构化的玻璃或聚合物基底上,并操作它们以选择性分离存在于一个或更多个特定染色体位置的dna。该实施方案利用细胞遗传学分析的广泛知识来制备染色体的展片(spread),对它们进行染色,并分析它们的显带模式以检测高级结构变异。该实施方案建立在此基础上,通过允许细胞遗传学家、其他科学家或自动化仪器探查包含至少一个标记体的染色体,与参考物进行比较,以便选择个体分子、个体分子的一部分、个体分子的多于一个部分或多于一个分子的多于一个部分,以通过选择性分离感兴趣区域(可能通过从感兴趣区域产生dna测序文库)来进一步分析。
[0210]
该方法依赖于通过将可见光或紫外辐射聚焦到光敏基质上以高分辨率的方式特异性分解基质的能力。在优选实施方案中,基质是下文描述的可光降解水凝胶。该方法还依赖于探查系统与下文描述的可光降解系统的耦合,所述探查系统优选地由包含明场光学器
件的显微镜组成,最优选地由荧光探查系统组成,所述荧光探查系统由与荧光光学器件组合的明场光学器件组成。
[0211]
该方法可以以不同的自动化程度来进行。以下实施方案是不利的,因为它不能很好地扩展到多于一个样品,对操作者是不方便的,并且可能具有对分离的核酸的不完全传质(mass transfer),但是提出它是为了说明自动化对于基本技术不是必需的:手动显微镜的操作者可以使用不会使基质裂解的光来观察样品,并且可以相对于预校准的位置手动定位显微镜载物台。然后,操作者打开光源,将可见光或紫外辐射引导向处于预校准位置的基质部分。随后,操作者用手动移液器并对辐照部位进行流体操作,以手动抽吸已经从基质中释放的核酸。然后,操作者将内容物转移到微量离心管或等效的管中,并继续使用商业可得的测序文库试剂盒,根据制造商的方案从样品中制备dna测序文库。
[0212]
在更优选的实施方案中,自动化帮助用户更精确地靶向感兴趣的区域,例如通过以任意形状整合精确定时的光脉冲来将聚焦的光靶向到样品以启动基质光降解。系统可以从紧邻处采集一系列样品,例如,用户可以通过在样品的图像上追踪图案来将感兴趣区域的中心标识为限定半径的圆、多边形或自由形式的形状。仪器可以在一次通过中采集整个区域,或者可以手动或以自动方式将该区域细分为多于一个子区域,并对每个子区域进行顺序提取,以提高空间采样分辨率。
[0213]
在一些实施方案中,仪器可以通过以系统的方式对基质进行顺序光降解来收集多个样品,例如通过获取薄样品并对其进行二维光栅扫描,或者通过将厚切片分成三维体积。某些采样几何形状有利于使剩余的完整基质的稳定性最大化并使采样位置之间的串扰最小化,但是当样品被细分为多种几何形状并且每个部分以多种顺序采样时,可以享受整体技术的益处。
[0214]
在一些实施方案中,整个单个染色体(902)可以选自染色体群体(904和902),诸如来自单细胞的染色体展片。可以基于探查来选择染色体,以鉴定一个或更多个标记体,诸如giemsa条带,或者替代地可以对它们进行荧光探查,以鉴定荧光标记体诸如fish探针的存在。例如,可以基于染色体由于插入而具有异常大的臂(903)来选择染色体。在其他实施方案中,操作者或自动化仪器可以从多个细胞中重复鉴定和选择相同的染色体,例如,从人类患者分离的细胞中观察到的7号染色体的多个拷贝可以一起分离和提取并汇集,或者逐个分离以用于单独的下游分析。在其他实施方案中,染色体的一部分可以被分离,不限于一个或更多个端粒区域、整个姐妹染色单体、着丝粒区域、整个臂、部分臂或以上任何组合。
[0215]
在一些实施方案中,该方法可以在核型分析之后使用,以便对通过核型分析观察到的潜在结构变异进行后续诊断,或者基于关于样品或患者的先前信息分析怀疑含有结构变异的特定位点。
[0216]
在其他实施方案中,dna序列可以通过序列特异性手段鉴定,并用于指导dna的分离。例如,对已经整合到宿主基因组中的病毒dna特异性的fish探针可以用来鉴定单个染色体中病毒dna已经整合的区域。然后,由荧光探查系统获得的荧光信号将用于该位置周围的靶向水凝胶光降解并收集样品以使用短读段测序进一步表征整合位点。
[0217]
在其他实施方案中,样品的选定部分的下游分析方法可以包括通过将样品移动到另一个流体装置或表面或者通过将样品移动到同一装置的相邻部分来产生物理图谱。
[0218]
在一些实施方案中,该方法可以与包含多于一个样品和一个或更多个探查区域或
操作区域的流体装置协同使用。通过仅使部分分析物选择性释放(可能地基于分析物的成像),该方法可以允许仅分析物的某些拷贝前进通过装置并进入探查区域或操作区域。
[0219]
在其他实施方案中,可以固定培养物或完整组织中的细胞,并将可光降解基质放置在细胞上。然后可以通过去除基质对细胞进行选择性采样。这可以以不知情(blind)方式发生,例如通过在样品上放置nx m网格的采样位置,或者可以以更协调的方式发生,例如通过沿着背腹轴间隔对脑组织切片采样。可以沿着单个或多于一个神经元的投射(projection)的长度对它们进行采样,并在沿着投射的不同点分离mrna,以用于通过rnaseq分析。
[0220]
在其他实施方案中,细胞核可以用固定剂(优选地福尔马林)和围绕细胞核聚合的可光降解水凝胶固定。通过光降解、染色质降解和洗脱以及样品抽吸的连续步骤,可以以一次一个体素(voxel)剖析细胞核,并从每个体素构建dna文库。测序数据的比较将允许构建基因组内容物图谱。优选地,高分辨率基因组空间标记的hi-c方法在水凝胶聚合之前完成,并且体素数据和hi-c数据的组合用于创建高分辨率3d图谱。
[0221]
染色体抗收缩方法
[0222]
可光降解水凝胶分离和释放的基因组分辨率取决于核酸的密度,在2d释放的情况下以每平方微米的碱基对为单位,或者在3d释放的情况下以每立方微米的碱基对为单位。如下文描述的降低密度的方法将允许更精细的基因组分辨率。
[0223]
长核酸或核酸:蛋白质复合物的膨胀或收缩程度可以通过成熟的方法控制,大量染色体细胞遗传学分析的实验室方案证明了这一点[lawce2017]。染色体在细胞周期期间经历不同的压缩阶段,从弥散的间期异染色质到前中期期间紧密结合的姐妹染色单体。小分子colcemid的添加能够通过阻断细胞周期进程并使染色体处于凝缩状态并且姐妹染色单体是附接的,对观察到的染色体的尺寸产生显著影响。染色体的压缩程度,以及因此在g显带和其他类型的细胞遗传学分析期间观察到的显带模式,对制备的染色体的临床效用有很大影响。对调节压缩程度的需要已经导致了各种各样的调节压缩的方案,压缩程度通常被测量为检测到的每个单倍体组的染色体条带总数。
[0224]
通过两类互补的实验室方法可以实现对染色体尺寸的更精细控制[lawce 2017]。首先,替代的细胞周期同步化方法更温和,诸如添加甲氨蝶呤以中断嘌呤途径,然后添加胸苷以将细胞从阻断中释放出来。然后,同步化的细胞可以用低得多的浓度的colcemid阻滞(arrest),这比单独的colcemid产生更加膨胀的染色体,或者可以简单地在观察到染色体适当压缩时收获细胞。第二类方法用多种化学试剂,诸如秋水仙碱、velban、溴化乙啶、放线菌素d、吖啶橙、hoechst 33258、dapi、偏端霉素a、5-氮杂胞苷、9-氨基吖啶和brdu,来处理染色体。盐的特征和浓度也被用于调节染色体形态,包括kcl、ohnuki低渗溶液、稀释的hanks bss和柠檬酸钠。另外的方法通过蛋白水解消化的方式使染色质变松。这可以伴随着通过添加拓扑酶诸如拓扑异构酶ii来解开交叉的dna链的方法。
[0225]
[drouin 1991]以每单倍体组1000个条带的水平展示了示例性的高分辨率染色体显带,其中在用胸苷小心地进行细胞周期释放(cell cycle release)后,制备染色体展片并使其显带,产生具有5-10mb/微米之间的膨胀线性密度(取决于序列)的拉长的染色体。
[0226]
染色质破坏和dna片段化
[0227]
即使在从水凝胶中部分释放凝缩的染色体之后,将dna保持在一起的共价键、dna
之间的拓扑相互作用和组成染色质的蛋白质/核酸复合物的非共价连接仍将阻止dna自由扩散。这些相互作用需要在可以释放核酸之前被破坏,并且它们被破坏的方式会影响产物对下游处理的适用性。
[0228]
在应用是创建聚焦短读段测序文库的一种实施方案中,在水凝胶光降解之前,dna用嵌入染料yoyo-1染色。在水凝胶光降解的每一步之后,用488nm的光辐照同一区域,以通过自由基损伤进一步刺激dna裂解。根据用于光降解水凝胶的光的波长,yoyo-1可能已经开始降解dna,并且随后的辐照是不必要的。
[0229]
在最优选的实施方案中,在每个降解过程之后,胰蛋白酶和nebnext dsdna片段化酶(new england biolabs)的混合物流过暴露的核酸和/或染色质,并在37℃孵育10min。
[0230]
在其他实施方案中,蛋白酶和内切核酸酶被添加到溶液中,同时拴系至微珠,这阻止它们进入水凝胶的仍然聚合的区域。
[0231]
在其他实施方案中,核酸和/或染色质在被水凝胶包封之前被蛋白酶和内切核酸酶部分或全部消化。这可以在低ph进行,以增强核酸对基底的亲和力并防止dna洗脱,并且随后可以在样品完全包埋在水凝胶中后升高ph。
[0232]
用于基因组文库构建或其他下游分析方法的dna片段化可以通过各种方式发生,不限于机械破坏、流体动力剪切、雾化、由声波或聚焦声波破坏、限制性内切核酸酶消化、切口酶和单链核酸酶消化、在有或没有单链核酸酶的情况下由嵌入荧光团的荧光光循环产生的单链切口积累的光裂解,以及转座酶裂解和插入。dna从染色体的释放和片段化是借助于蛋白水解酶(不限于蛋白酶k或胰蛋白酶)消化包被dna的染色质蛋白来辅助的。
[0233]
光降解系统
[0234]
一种光学装置实施方案,其中通过将特定波长和通量(fluence)的光传递到精确定义的区域来光降解可光降解的水凝胶基质,该区域的坐标对于来自探查系统的探查事件的记录是已知的。
[0235]
此处在本文件中,“高分辨率”是指选择性释放和回收在仍然附接至支持物的核酸的部分1微米以内的核酸的部分的能力。可以重复该过程以顺序释放相邻部分,使得物质被系统地剖析和分离。此处,1微米的规格意味着》80%的核酸被保留在装置的一个区域中,但是1微米之外的《20%的核酸已经被去除。在其他实施方案中,分辨率为2微米、4微米、8微米、16微米、32微米、500nm、250nm、125nm、63nm或32nm。
[0236]
在光学上,有多种方式可以将光引导至水凝胶,以便以高分辨率的方式对其进行光降解。高分辨率需要高na照射,诸如常规通过良好校正的成像物镜来实现。在一种非限制性实施方案中,nikon 60x 1.4na vc既用于用红光成像,红光不会使onb衍生的水凝胶基质明显地光降解,并且也用于用uv或紫光辐照。照射通路照射数字微镜装置,并且然后使其对样品成像,使得装置的中心部分与主检测相机可见的图像部分尺寸相同。例如,这将包括对0.47英寸对角线dlp471tp装置(texas instruments)的中心2048x 2048个像素成像,这对应于1/60x的放大倍数。为了实现该装置的全光学分辨率成像,用大于物镜na/放大倍数(1.4/60=0.023)的数值孔径对其进行照射,并且光瞳(pupil)近似成像到物镜后光瞳。通过设置数字微镜装置中每个像素的开或关的状态,并且然后施加一定剂量的光降解光,可以选择性照射任意图案的水凝胶。
[0237]
在替代实施方案中,准直激光束可用于对样品进行光降解,并且通过对共聚焦显
2013]形成对照。这种水凝胶不需要施加在分析物上,并且可以在添加分析物之前预聚合,但是不能实现水凝胶的3-d切片。
[0246]
在另一种实施方案中,通过将乙二醇壳聚糖与两端用onb衍生化的peg单体聚合形成peg-4-(3-(1-(n-羟基琥珀酰亚胺基碳酸酯)乙基)-4-硝基苯氧基)丁酸酯来形成可光降解的水凝胶[wu 2019]。
[0247]
在替代实施方案中,光化学被用于水凝胶的构建和破坏两者,优选地通过使用具有不同波长敏感度的光化学基团。使用构建波长的辐照来选择性沉积水凝胶,而使用光降解波长的辐照来选择性去除水凝胶。在替代实施方案中,水凝胶包含偶氮苯基团,偶氮苯基团响应于不同波长的光而可逆地变形。在一种实施方案中,水凝胶包含n,n-二甲基氨基乙基甲基丙烯酸酯(dmaema)和4-甲基-[7-(甲基丙烯酰基)氧基-乙基-氧基]香豆素(cma),用405nm光聚合,并且用254nm光光降解[he 2011]。
[0248]
在替代实施方案中,在包含至少一个标记体的核酸样品已经被探查装置探查并且一部分已经被选择用于进一步分析之后,选定的分子或分子的一部分,或者一个或更多个分子的多于一个部分可以通过将水凝胶前体施加到整个样品并且仅在感兴趣样品不在的位置使其光聚合来选择性地操作。在最优选的实施方案中,水凝胶前体是peg da,其在存在irgacure 2959的情况下通过用320nm和390nm之间的光辐照而固化。
[0249]
封闭和开放的装置及对洗脱和收集的影响
[0250]
可光降解水凝胶可以放置在开放或封闭的流体装置中。开放系统的实例是显微镜盖玻片,其暴露于显微镜物镜,并且如果适用的话,下侧的光学耦合流体将样品沉积在上侧,并且然后暴露于空气中。空气可以包括温度、湿度、压力或组成分子的分压受控的环境调节空气。例如,装置的其他特征可以存在于装置的顶表面之上。盖玻片和样品可以是孔底。
[0251]
可光降解的水凝胶可以替代地放置在如早前定义的封闭的微流体装置内。在这种情况下,水凝胶的深度和平面范围可以受限于流动通道的深度、流动通道的形状、表面化学或表面图案化、或者一种或更多种交替流体流的流动。
[0252]
每种方法都有优点和缺点。在其中整个流动通道充满水凝胶的封闭装置的情况下,任何冲洗掉解聚的水凝胶或洗脱样品的尝试将首先要求在相当大的程度上清除入口和出口端口附近的水凝胶,以便连续的流体路径可用于将流体从入口引导到出口。一些实施方案首先使水凝胶的薄层聚合,然后在水凝胶上放置顶板以在顶板和水凝胶之间形成具有足够间隙的流动通道(flowlane),使得流体可以在入口和出口端口之间移动,而完全不需要切割通道(channel)。相反,优选的实施方案通过在聚合之前将水凝胶前体选择性递送到装置的特定部分的方式来限制水凝胶聚合的程度。在流体通路能够将试剂移入和移出水凝胶区域时,封闭微流体系统能够以最小流体体积实现对水凝胶区域的非常快速和彻底的洗涤,这转化为试剂(诸如蛋白酶、内切核酸酶或聚合酶)的最小花费。开放系统仍然可以实现相同程度的洗涤,但可能需要递送更大量的试剂。
[0253]
从开放装置提取样品可以通过将流体沉积在水凝胶上,任选地混合流体,然后抽吸流体来进行。优选实施方案利用机器人移液臂和可更换的移液器吸头,并将样品沉积在384孔板中。
[0254]
从封闭装置提取样品可以通过使样品流过流动通道到流体装置的另一部分,或者
流出流体装置并通过毛细管进入另一流体装置,或者流出流体装置以沉积在收集系统(最优选地位于384孔板上的自动级分收集器)中来进行。或者,可以将样品置于乳剂中的流体液滴中(事先与条形码试剂混合或未与条形码试剂混合)。
[0255]
fret接近性标记体对
[0256]
在另一组实施方案中,至少两个荧光标记体分别结合到高级核酸结构的不同区域,使得在至少两个标记体的集合内,存在一对当彼此足够接近时可以形成fret对的标记体。与依赖于交联并因此将接近信息限制在单个时间点的探查长核酸分子片段之间的接近关系的方法不同,所公开的方法可以随着样品的演变动态地形成fret对,允许在一段持续时间内监测实时接近关系动态。当被探查系统荧光探查时,通过探查由fret对产生的fret信号(其中fret对各自结合到结构内的两个不同位置),可以监测这两个位置彼此足够接近以产生fret信号的次数和每次的持续时间。另外,可以使用fret信号的幅度、角度和偏振来推断关于fret对彼此之间的距离和取向的信息。
[0257]
在一些实施方案中,在细胞周期的至少一部分内,例如当细胞处于间期、或前期、或前中期、或中期、或后期或末期时,探查fret信号的性质。在一些实施方案中,在细胞生命的至少一部分内,例如在细胞凋亡期间,探查fret信号的性质。在一些实施方案中,在细胞形态变化(例如起泡、质壁分离、核碎裂、核固缩或dna片段化)的至少一部分期间探查fret信号的性质。在一些实施方案中,在细胞中核酸结构的物理变化(包括物理组成、形态、密度、构象或拓扑的变化)的至少一部分期间探查fret信号的性质。在一些实施方案中,在细胞检查点的至少一部分期间探查fret信号的性质。在一些实施方案中,在其中细胞经历特定酶促活性(例如转录、环挤出、基因调控、转录复合物的形成或解构)的至少一部分时间段期间探查fret信号的性质。
[0258]
在一些实施方案中,在当包装物或长核酸分子暴露于特定试剂、试剂组合或试剂浓度时的至少一部分时间段期间探查fret信号的性质。在一些实施方案中,在当包装物或长核酸分子暴露于环境条件时的至少一部分时间段期间探查fret信号的性质。在一些实施方案中,在当包装物或长核酸分子暴露于特定蛋白质或酶时的至少一部分时间段期间探查fret信号的性质。
[0259]
在一些实施方案中,高级核酸结构可以结合一个受体和多于一个可能的供体。在一些实施方案中,高级核酸结构可以结合一个供体和多于一个可能的受体。在一些实施方案中,高级核酸结构可以结合多于一个可能的供体和多于一个可能的受体。
[0260]
图10(a)展示了一种实施方案,其中具有由环(1006)和黏连蛋白复合物(1004)组成的高级核酸结构的长核酸分子(1007)用两个标记体标记,当彼此足够接近时,这两个标记体一起可以形成fret对。在该特定实施方案中,供体标记体(1001)结合在ctcf位点(1003)处或附近,而受体标记体(1002)与黏连蛋白复合物结合。对于这样的实施方案,在探查fret对期间,当fret对在环挤出(1006)过程期间变得接近时,可以检测到fret信号。
[0261]
图10(b)展示了一种实施方案,其中具有由环和转录复合物(1013)组成的高级核酸结构的长核酸分子(1016)用两个标记体标记,当彼此足够接近时,这两个标记体一起可以形成fret对。在该特定实施方案中,供体标记体(1011)结合在启动子位点处或附近,而受体标记体(1014)结合在增强子位点处或附近。对于这样的实施方案,在探查fret对期间,当fret对在转录过程期间变得接近时,可以检测到fret信号。
[0262]
在一些实施方案中,对至少一个个体fret受体标记体的fret信号性质的统计数据由光学探查系统记录。在一些实施方案中,多于2个个体受体,或多于10个个体受体,或多于100个个体受体。
[0263]
在一些实施方案中,从多于10个受体、或多于100个受体、或多于1,000个受体或多于10,000个受体的群体中收集由受体群体产生的fret信号的性质的统计数据。
[0264]
在一些实施方案中,fret对标记体在高级核酸结构被限制在流体装置内或流体装置上时与所述结构结合。在一些实施方案中,结合到高级核酸结构的fret对在所述结构被限制在流体装置内时被荧光探查。
[0265]
可裂解接近性条形码化交联剂(cpbc)
[0266]
在这组实施方案方法中,我们描述了此处定义为“可裂解接近性条形码化交联剂”(cpbc)的实体。cpbc是包含至少两个捕获探针的实体,其中至少一个捕获探针是可释放捕获探针,其可通过使将所述至少一个可重新封闭的捕获探针连接到cpbc的一个或更多个可裂解接头裂解而与cpbc物理分离。通过经由捕获结构域与它们各自的靶生物分子结合使cpbc与一种或更多种生物分子交联,这些生物分子的接近关系可以在可释放捕获探针已经从cpbc释放之后通过条形码保持和追踪。
[0267]
在优选实施方案中,cpbc所包含的捕获探针各自具有共同形成“关系集”的条形码,使得对关系集内的一个条形码的身份的了解允许确定同一关系集内的其他条形码的身份。在一些实施方案中,形成关系集的所有条形码都是相同的。在一些实施方案中,关系集中的至少两种条形码包含彼此的互补序列。在一些实施方案中,关系集中的至少一种条形码可以通过数学函数或查找表从同一关系集中的另一种条形码确定。在优选实施方案中,每个cpbc具有独特条形码关系集,使得如果给出来源于多于一个cpbc的条形码的集合,则条形码的集合可以被分选为来源关系集。例如,如果给出来源于cpbc-1、cpbc-2和cpbc-3的条形码集合,则该集合可以被分选为三个不同的关系集:来源于cpbc-1的条形码关系集、来源于cpbc-2的条形码关系集以及来源于cpbc-3的条形码关系集。
[0268]
cpbc提供了在一段持续时间内或在特定时间点鉴定和追踪液体环境内各种生物分子或生物分子的部分的接近关系的非常有利的方法,因为生物分子或生物分子的部分的接近关系和时间关系两者可以从结合的捕获探针确定。在一些实施方案中,可以在期望的时间点将cpbc引入到包含至少一些靶生物分子的环境中。在一些实施方案中,引入cpbc以靶向流体装置内或流体装置上的生物分子。在一些实施方案中,至少一个捕获探针的至少一个捕获结构域可以在期望的时间被激活或失活,以与其靶生物分子交联。在一些实施方案中,激活或失活机制是特定波长的光、或温度或ph。
[0269]
在一些实施方案中,可以在不同的时间点将多于一个cpbc引入到包含靶分子的环境中。在优选实施方案中,条形码内包含的至少一部分信息可以与引入时间或引入顺序关联。
[0270]
cpbc可以被设计成使得捕获探针所占据的物理体积可以针对特定应用来选择。以这种方式,cpbc可以被设计成研究跨越大范围物理距离的生物分子或生物分子的部分之间的接近关系。在一些实施方案中,可以期望的是选择特定尺寸的cpbc来追踪不同尺寸的高级核酸结构的接近关系。
[0271]
cpbc特别有利于其中期望保持被追踪的生物分子(或多于一个生物分子)的长程
顺序或完整性的应用。与需要在捕获后消化核酸分子以保持相邻分子或分子部分的接近关系的现有常用的接近性检测方法(参见“接近性3d作图”的定义)不同,cpbc通过条形码保持接近关系的信息,所述条形码可以通过可裂解接头从cpbc释放。以此方式,不仅可以通过条形码保持接近关系,还可以保持核酸分子的长程序列内容物。当被探查的样品是单细胞,并且保持长核酸分子的长程完整性对于鉴定结构变异至关重要时,这是特别重要的。此外,通过裂解可裂解接头,可以允许分子重新定位成不同的物理构型,允许来自新cpbc或来自原始cpbc的剩余捕获探针的捕获探针的另一轮结合。
[0272]
在一些实施方案中,通过直接对条形码测序或间接对条形码的扩增产物测序来鉴定条形码的身份。在一些实施方案中,条形码的身份是通过结合标记体来鉴定的,标记体被设计成与特定条形码、条形码集或条形码子集特异性结合。
[0273]
在优选实施方案中,将至少一个cpbc引入环境中,其中与每个捕获探针关联的捕获结构域结合其靶生物分子的条件是优选的。在至少一个可释放捕获探针通过探针的捕获结构域与其靶生物分子结合后,然后通过裂解可裂解接头从剩余的cpbc释放至少一个可释放捕获探针。
[0274]
图11(a)确定了cpbc的一种可行的实施方案。此处,cpbc包含由可裂解接头(1102)连接的两个捕获探针(1101和1103)。在该特定实施方案中,第一捕获探针(1101)包含捕获结构域(1104)和条形码(1105)。另外,第二捕获探针(1103)包含条形码(1106)和捕获结构域(1107)。此处,条形码(1105和1106)属于关系集。
[0275]
图11(b)展示了图11(a)中描述的cpbc用于捕获长核酸分子的部分的接近信息的一种可行的实施方案。此处,存在交联到同一长核酸分子(1111)的两个不同的cpbc(1112和1113),每个cpbc具有独特的条形码关系集,第一cpbc(1112)具有关系集a,并且第二cpbc(1113)具有关系集b。此处在该实例中,与这两个cpbc关联的所有捕获结构域被设计成非特异性结合核酸或染色质,使得在时间点(i)长核酸分子(1111)在长核酸分子(1111)的两个部分彼此足够接近使得它们可以被cpbc交联桥接的区域被这两个cpbc(1112和1113)交联。在稍后的时间点(ii),cpbc内的可裂解接头被裂解,来源于同一cpbc的捕获探针从彼此释放。沿着长核酸分子(1111)的长度,现在有4个结合的捕获探针(1121、1122、1123、1124),其中分子的部分的先前接近关系的信息由捕获探针中包含的条形码保持。因此,来自第二cpbc的先前接近关系1126的信息由条形码关系集b保持,并且来自第一cpbc的先前接近关系1127的信息由条形码关系集a保持。
[0276]
图12展示了图11(a)中描述的cpbc用于在两个不同时间点捕获长核酸分子的部分的接近信息的另一种可行的实施方案。首先(i)长核酸分子1201的部分的接近关系是通过在两个非常接近的位置用两个单独的cpbc(第一cpbc(1203)和第二cpbc(1202),各自分别具有独特条形码关系集:b和a)使分子交联来捕获的。如在先前的实施方案中,此处cpbc包含与核酸或染色质非特异性结合的捕获结构域。然后使两个cpbc各自的可裂解结构域裂解,从而去除交联,并允许长核酸分子(1201)在物理上重新配置,使得在稍后的时间点(ii)引入具有条形码关系集c的另外的第三cpbc(1225)。该第三cpbc交联到同一长核酸分子(1201),同时先前的接近关系由结合的捕获探针(来自第二cpbc(1202)的1221和1222,以及来自第一cpbc(1203)的1223和1224)保持。随后,(iii)在来自第三cpbc(1225)的可裂解接头裂解之后,长核酸分子(1201)具有6个与其结合的捕获探针,这些探针保持了所述分子的
区段的先前接近和时间关系。此处,来自第二cpbc的接近关系1237由关系集a的结合捕获探针(1231和1234)保持,并且来自第一cpbc的接近关系1239由关系集b的结合捕获探针(1232和1236)保持,而来自第三cpbc的时间较晚的接近关系1238由关系集c的结合捕获探针(1233和1235)保持。
[0277]
图13(a)描述了cpbc(1305)的另一种实施方案。在该实施方案中,可裂解接头由杂交在一起的两个核酸分子(1304和1302)组成,其中分离两个捕获探针的裂解机制可以包括多种方法,如先前在“可裂解接头”的定义中描述的,特别是当可裂解接头包含核酸时。在该特定实施方案中,两个核酸分子(1304和1302)杂交在一起,但不共有磷酸二酯主链,并且通过它们之间的杂交键的解链而实现裂解。在优选实施方案中,两条链不是100%互补的,并且具有》50%的at碱基对含量,使得它们的有效解链温度低于它们各自的捕获探针可以交联的任何靶长核酸分子的平均解链温度。在一些实施方案中,捕获探针的条形码包含待解链的核酸的至少一部分。在一些实施方案中,捕获探针的条形码不包含待解链的核酸的任何部分。在该特定实施方案中,在使可裂解接头解链(裂解)后,(ii)两个捕获探针(1311和1312)从彼此释放,其中第一捕获探针1311包含捕获结构域(1301)和条形码(1304),并且第二捕获探针1312包含捕获结构域(1303)和条形码(1302)。
[0278]
图13(b)展示了图13(a)中描述的cpbc用于捕获长核酸分子的部分的接近信息的一种可行的实施方案。此处,存在两个不同的cpbc,第一cpbc(1322)和第二cpbc(1323),各自个具有独特的条形码关系集,第一cpbc(1322)具有关系集a,并且第二cpbc(1323)具有关系集b。此处在该实例中,与这两个cpbc关联的所有捕获结构域被设计成非特异性结合核酸或染色质,使得在时间点(i)长核酸分子(1321)在长核酸分子(1321)的两个区段彼此足够接近使得它们可以通过cpbc交联的区域通过这两个cpbc(1322和1323)交联。在稍后的时间点(ii),cpbc内的可裂解接头被裂解(解链),来源于同一cpbc的捕获探针从彼此释放。沿着长核酸分子(1321)的长度,现在有4个结合的捕获探针(1331、1332、1333、1334),其中分子的部分的先前接近关系的信息由捕获探针中包含的条形码保持。因此,来自第二cpbc的先前接近关系1335的信息由条形码关系集b保持,并且来自第一cpbc的先前接近关系1336的信息由条形码关系集a保持。
[0279]
在该方法的另一组实施方案中,cpbc可以包含核酸树枝状大分子结构、dna纳米技术结构或折纸结构。在一些实施方案中,这样的结构可以包括环、连接体(junctions)或结(knots)。对于这样的实施方案,cpbc可以包含多于两个捕获探针。在一些实施方案中,cpbc可以包含3个、或4个、或5个、或6个、或7个、或8个、或9个、或10个、或11个或12个捕获探针。在一些实施方案中,cpbc可以包含多于2个捕获探针、多于3个捕获探针、多于5个捕获探针、多于10个捕获探针、多于15个捕获探针、多于20个捕获探针、多于50个捕获探针、多于100个捕获探针、多于500个捕获探针。
[0280]
图14展示了一种实施方案,其中cpbc包含4个捕获探针,每个捕获探针与相应的核酸分子和捕获结构域关联,其中4个核酸分子形成一种霍利迪连接体。在该特定实施方案中,cpbc首先(i)使长核酸分子的4个区段(1409、1410、1407、1412)交联,所述区段可以来源于一个或多于一个长核酸分子。在该特定实施方案中,捕获探针包含捕获结构域和包含条形码和可裂解接头的一部分两者的核酸分子。第一捕获探针1421包含捕获结构域1405和核酸聚合物1401。第二捕获探针1422包含捕获结构域1406和核酸聚合物1402。第三捕获探针
1423包含捕获结构域1407和核酸聚合物1403。第四捕获探针1424包含捕获结构域1408和核酸聚合物1404。类似于先前图13(a)的实例,捕获探针彼此杂交以在杂交区域形成可裂解接头,而不共有磷酸二酯主链。此处,第一捕获探针(1421)的一部分核酸聚合物与第四捕获探针的一部分杂交,并且第二部分核酸聚合物与第二捕获探针的一部分杂交。在该特定实施方案中,展示了通过两个可裂解接头连接到cpbc的可释放捕获探针(1421)的实例。对其他捕获探针重复该顺序,如图14时间点(ii)中示出的,于是使得所有捕获探针释放。
[0281]
在图14的该特定实施方案中,包含四个捕获探针的cpbc能够使落入由cpbc占据的体积或接近性捕获区域(1413)内的靶分子(或多于一个靶分子)交联。因此,可以通过选择具有不同支链长度、支链数量和物理构型的dna折纸结构来修改待捕获的期望的接近距离。
[0282]
对于一些实施方案,通过包含至少一个单链核酸聚合物的分子经由所述至少一个单链核酸聚合物的至少一部分与另一个核酸聚合物杂交的自组装,至少部分组装cpbc。对于一些实施方案,可以包括另外的核酸分子聚合物物质作为填充物,以修饰接触探针或支链的长度或柔性,使得cpbc的接近性捕获区域(1413)可以根据需要进行修改和调节。在一些实施方案中,填充物不包含捕获探针的条形码或可裂解接头的任何部分。
[0283]
在该方法的另一组实施方案中,cpbc可以包含中心“珠”,其可以被官能化,使得至少两个捕获探针可以连接到珠,其中至少一个捕获探针是通过可裂解接头连接到珠的可释放捕获探针。在一些实施方案中,珠可以是树枝状大分子、或聚苯乙烯珠、或纳米颗粒、或纳米点或量子点。在一些实施方案中,珠包含荧光特性。例如,珠可以包括荧光珠。在一些实施方案中,珠包含光致发光特性。例如,珠可以包括当用uv光刺激时能够发射特定波长的光的固态量子点。在一些实施方案中,珠包含磁特性。例如,珠可以包括磁珠。
[0284]
图15描述了一种实施方案cpbc,其包含直径1503的珠(1502),其上有4个捕获探针(1513、1523、1533、1543),它们通过各自的可裂解接头(1514、1524、1534、1544)与珠结合。在该特定实施方案中,存在关系集的两个子集:q1和q2。捕获探针1513和1533共有来自同一子集q1的条形码,而捕获探针1523和1543共有来自同一子集q2的条形码。在该特定的实例中,属于子集q1和q2的所有条形码属于同一个亲本关系集q,它将该集合中的所有条形码与图15中示出的cpbc关联。然而,子集q1和q2还与捕获结构域的类型关联,此处子集q1与靶向第一生物分子靶类型的捕获结构域(1511和1531)关联,并且子集q2与靶向第二生物分子靶类型的捕获结构域(1521和1541)关联。这是有利的,因为它允许追踪落入接近性捕获区域(1501)内的特定生物分子类型(或生物分子的部分),使得它们可以被cpbc交联。
[0285]
在cpbc内可以使用各种不同的关系子集。在一些实施方案中,子集可以与珠内的物理位置关联。例如,上半球或下半球,或者与珠核心的距离。对于可渗透的珠,诸如非致密的树枝状大分子,可以有几层壳,每层壳具有不同的直径和独特的关系子集。子集可以与捕获结构域的靶类型关联。子集可以与捕获探针的物理长度、构象或刚性关联。子集可以与可以激活捕获结构域使得其可以与其靶生物分子交联的特定的激活类型关联。例如,特定波长的光、热、ph或催化剂。子集可以与封闭捕获结构域的特定类型的笼关联(例如:与光不稳定保护基团关联),直到所述捕获结构域被解除封闭。例如:在时间点1,子集q1的cpbc的脱笼的捕获探针与附近所有靶分子交联。然后,在稍后的时间点2,子集q2的所述cpbc的加笼的捕获探针随后通过暴露于一种波长的光而脱笼,并且然后自由地与附近所有靶分子交联。
[0286]
在一些实施方案中,所有捕获结构域靶向同一靶类型。在一些实施方案中,每个捕获结构域靶向不同的靶类型。在一些实施方案中,存在捕获同一靶类型的至少两个捕获结构域。在一些实施方案中,cpbc可以仅与一种类型的靶类型、或至少一种靶类型、或至少两种不同的靶类型、或至少三种不同的靶类型或至少五种不同的靶类型交联。
[0287]
在一些实施方案中,靶类型可以是核酸、长核酸、染色质、长核酸分子的富含at的区域、长核酸的富含cg的区域、特定基因、特定增强子、特定启动子、调控区域、非功能区域、端粒区域、着丝粒区域、特定序列、蛋白质、特定蛋白质、酶、特定酶、dna结合蛋白、特定dna结合蛋白、有机分子、atp、特定高级核酸结构。
[0288]
图16展示了包含两个不同关系子集q1和q2的cpbc的实施方案,其中q2和与特定蛋白质的结合关联,并且q1和与核酸聚合物的非特异性结合关联。在该特定实施方案中,特定直径(1605)的珠(1610)具有4个捕获探针,其中两个(1604和1606)与关系子集q2关联,并通过它们各自的捕获结构域与cpbc的接近区域(1608)内的特定蛋白质靶类型(1603和1607)交联。其他两个捕获探针(1611和1602)与关系子集q1关联,并通过它们各自的捕获结构域与长核酸分子(1609和1601)交联,长核酸分子也在cpbc的接近区域内。
[0289]
在稍后的时间点(ii),与cpbc关联的各个捕获探针随后通过可裂解接头的裂解从珠释放。释放后,长核酸分子(1601和1609)保持与它们各自的捕获探针(1602和1611)结合,并且特定蛋白质(1603和1607)保持与它们各自的捕获探针(1604和1606)结合。
[0290]
在一些实施方案中,cpbc包含至少两个不同的捕获探针,其中两个捕获探针具有不同类型的可裂解接头。在一些实施方案中,与捕获探针关联的可裂解接头类型可以通过属于独特关系子集的条形码来鉴定。当希望在不同时间点释放捕获分子的特定亚群时,这可以是非常有利的。例如,来自关系子集q1的条形码与具有与长核酸分子结合的捕获结构域的捕获探针关联,并通过1型可裂解接头连接到珠。类似地,来自关系子集q2的条形码与具有与蛋白质类型结合的捕获结构域的捕获探针关联,并通过2型可裂解接头连接到所述珠。对于这样的实施方案,许多可能的工作流程都是可行的。在一种示例工作流程中,在与接近cpbc的捕获的分子(或分子的部分)交联后,与长核酸交联的捕获探针首先通过与1型可裂解接头关联的裂解机制从珠释放。然后,在稍后的时间点,并且优选地在不同的环境中,与蛋白质交联的捕获探针然后通过与2型可裂解接头关联的裂解机制从珠释放。这是非常有利的,因为分析捕获分子和鉴定与其结合的捕获探针关联的条形码所需的下游过程可以是不同的。在该特定实例中,包含磁特性的cpbc珠将是非常有利的,因为在1型接头被裂解后,可以容易地从溶液中提取珠。
[0291]
在一些实施方案中,cpbc的物理体积或平均直径可以被选择以捕获不同尺寸的接近体积或接近区域中的靶分子。在一些实施方案中,cpbc可以具有大于5nm、或大于10nm、或大于15nm、或大于20nm、或大于30nm、或大于50nm、或大于75nm、或大于100nm、或大于150nm、或大于200nm、或大于500nm、或大于750nm、或大于1000nm的平均直径。特别地,cpbc的尺寸可以被选择以靶向各种特定类型的高级核酸结构。另外,在一些应用中,不同尺寸的cpbc可以同时或按顺序地用于特定样品,以便探测与不同cpbc的不同接近关系。
[0292]
在生物分子及其结合的捕获探针通过使可裂解接头裂解而从cpbc释放之后,可以进行多种潜在的交联后分析,以既鉴定与捕获探针关联的条形码,又分析生物分子本身。对于其中生物分子是长核酸分子的实施方案,可以对分子进行测序,或将其包封在液滴中,或
者从所述分子产生物理图谱,或兼而有之。在一些实施方案中,当分子在微流体装置中至少部分拉长时,通过探查结合到分子的荧光标记体来产生物理图谱。在一些实施方案中,当分子通过梳理方法被至少部分固定在微流体装置的表面上时,通过探查结合到分子的荧光标记体来产生物理图谱。对长核酸分子进行梳理是特别有利的方法,因为结合的捕获探针随后容易用于允许与所述探针反应的各种溶液和试剂交换。例如,包含探针条形码的核酸聚合物可以与序列特异性荧光标签结合,或原位扩增,或原位测序。在一些实施方案中,条形码可以通过合成测序、通过杂交测序或通过连接测序。特别地,这样的对探针的探查可以在参考探针所结合的长核酸分子的潜在物理图谱记录探针的物理位置之后进行。
[0293]
图17描述了一种实施方案,其中长核酸分子(1701)在基底表面(1702)上梳理,并沿着分子的长度用荧光标记体(1703)标记,使得可以通过用光学探查系统进行荧光探查来产生物理图谱。另外,分子和与关系集a(1731和1734)关联的捕获探针结合,关系集a保留沿着分子的这两个位置之间的先前接近关系(1737)的信息,并且分子和与关系集b(1732和1736)关联的捕获探针结合,关系集b保留沿着分子的这两个位置之间的先前接近关系(1739)的信息。
[0294]
在一些实施方案中,基底表面(1702)(具有结合捕获探针的长核酸分子在其上被梳理)是流体装置。
[0295]
在一些实施方案中,用至少一种类型的限制性内切酶消化与捕获探针结合的长核酸分子可以是有利的。在将长核酸分子消化成较小的分子后,将会有包含原始长核酸分子的至少一部分的较小分子的子集,以及包含核酸条形码的结合的捕获探针,它们在一起形成一对。在对核酸聚合物对进行适当的预处理使得另外的核苷酸可以添加到末端之后,另外的核酸条形码可以添加到它们的末端,并用于将它们标识为“对(pair)”。方法可以包括sprit和chia-drop[jerkovic,2021]。
[0296]
在一些实施方案中,将至少一种cpbc引入样品可以是有利的,其中样品被限制在流体通道内或流体装置的外表面上。在一些实施方案中,样品可以固定在外表面上,或者固定在流体通道壁上。在一些实施方案中,样品可以固定在已经胶凝的材料中。在一些实施方案中,样品的2d或3d空间中的至少一个特定区域可以通过光激活占据所述空间的任何cpbc而被靶向以用于与cpbc交联。在一些实施方案中,cpbc的荧光特性用于在微流体装置内和样品内通过荧光探查来追踪所述cpbc物理位置。
[0297]
在一些实施方案中,每个至少一个捕获探针包含生物素,使得在从cpbc释放后,然后捕获探针本身可以用链霉亲和素或包含链霉亲和素的实体捕获。
[0298]
在一些实施方案中,结合到至少一个可释放捕获探针的长核酸分子可以在从cpbc释放所述捕获探针之前或之后进行酶促处理。对长核酸分子的酶促处理可以包括产生切口、消化、核苷酸(包括修饰的、标记的、终止的或可逆终止的核苷酸)的聚合酶掺入,以及连接。
[0299]
在一些实施方案中,并非所有由cpbc捕获的捕获生物分子都在短时间期间内被捕获。例如,生物分子可以在数秒钟、数分钟、数小时或数天的时间段内被捕获。在一些实施方案中,生物分子通过扩散、或流体流动或外部施加的力进入cpbc的接近区域。在一些实施方案中,生物分子可以在不同的时间点进入cpbc的接近区域。在一些实施方案中,cpbc在微流体装置的流体通道内捕获生物分子。
实施例
[0300]
实施例1:捕获凝胶中的膨胀染色体的流体装置
[0301]
作为用于在流体装置中使染色体膨胀,并且然后将所述染色体固定在凝胶中以用于进一步荧光探查的初始概念验证,描述了模型装置、探查系统和使用方法。
[0302]
首先设计并组装了一种流体装置,该流体装置包括与pdms膜键合的borofloat玻璃基底中的蚀刻通道。将0.120mm厚的双面抛光硼硅酸盐玻璃晶片(pyrex 7740)均匀地涂覆铬粘合层(20nm)和铜籽晶层(seed layer)(200nm),并且然后在基底上涂覆光致抗蚀剂(az9260)膜,通过光掩模曝光,并根据制造商的说明显影,以使流体装置形成类似于图6中示出的图案。在该示例装置中,第一通道(610)和第二通道(605)两者均为500微米宽,形成500微米乘500微米的交会区域(606)。过滤特征(607、604)包括直径为2微米的柱,最近邻柱之间的间距为0.5微米(注:在本实施例中,没有如图6中示出的过滤特征608)。然后在铜的暴露区域上将镍电镀至2微米的厚度,之后通过溶剂去除光致抗蚀剂并通过反应离子蚀刻(rie)去除cr/cu电镀层以暴露玻璃基底。然后在电感耦合等离子体蚀刻机中使用c4f8和o2的等离子体气体混合物将被电镀镍掩蔽的暴露玻璃基底蚀刻至5微米的深度,以限定流体通道。
[0303]
然后在加热的水、氨和过氧化氢的混合物中彻底洗涤玻璃基底,以去除任何残留的有机物质,并促进颗粒从表面的去除。最后,将1mm厚的pdms膜和图案化的玻璃暴露于表面活化等离子体,并且然后在室温键合在一起以封装通道。入口和出口端口通过与封闭通道对齐打孔穿过pdms来提供。然后将luer锁连接插入入口和出口pdms端口,以促进流体连接和泵送。
[0304]
血液样品取自患者,并使用商业可得的微流体分选仪(parsortix,angle plc)按照制造商的方案富集潜在的ctc并洗脱。培养洗脱的细胞并使细胞周期阻滞。使用alexa 488标记的抗细胞角蛋白抗体、alexa594标记的抗cd45抗体和dapi的混合物对细胞进行染色,并收获。
[0305]
将流体装置安装在支架上,允许与流体管道对接,以便在装置内进行溶液交换,并允许光学显微镜进行探查。使用配备有60
×
/1.00水浸物镜和scmos相机的倒置显微镜进行荧光成像。装置内的温度由与装置背面保持接触的加热器控制。
[0306]
在接收细胞之前,用1%十二烷基硫酸钠、缓冲溶液(0.5tbe、3%b-巯基乙醇(bme)和0.5%triton x-100)和1mg/ml的bsa冲洗装置10分钟。
[0307]
将染色的细胞通过入口端口引入流体装置,并使用压力驱动流使其沿着第一通道(610)流动。通过在用明场显微术检测到细胞时停止流动,在交会区域(606)中单独检查细胞。然后针对所有三种荧光团对细胞成像,并基于满足所有三个标准来选择ctc候选物以便固定在凝胶中:(a)存在细胞核(b)细胞角蛋白阳性和(c)cdc45阴性。如果细胞不符合标准,则恢复流动,并将另一个细胞流入交会区域进行检查。
[0308]
接下来,在发现ctc候选细胞后,使碱性裂解溶液沿着第二通道(605)流动并通过交会区域(606),其裂解细胞并释放中期染色体。在释放后,染色体由柱保持在交会区域中,而裂解的细胞残渣被过滤掉。接下来,通过以1-10微米/秒的速率使蛋白酶k和rna酶流动通过第二通道以进行消化,以及使yoyo-1流动通过第二通道以对染色体染色(所有试剂来自thermo fisher),来制备染色体以将其固定在凝胶中。所有消化的分子、非染色体核酸物质
和多余的试剂通过柱过滤,将蛋白质被剥离的染色体保持在交会区域中。然后由探查系统探查染色的染色体,以监测它们在37摄氏度30分钟的时间段内的膨胀,在此期间,染色体膨胀到其原始长度的约10倍的长度。另外,在此期间,在探查系统的协调和控制下,无试剂缓冲溶液偶尔以振荡方式流过第二通道,以使染色体在交会区域内温和地分散。
[0309]
接下来,将1.5%(w/w)低胶凝琼脂糖(ix型琼脂糖,sigma-aldrich)的溶液流过第二通道,置换先前的溶液,并将染色体浸入交会区域中的凝胶溶液中,并且然后将装置置于4℃30分钟以确保琼脂糖胶凝。将染色体固定在胶凝的琼脂糖中,然后移除pdms盖,将限制在通道内、距离表面小于5微米的多孔凝胶中的固定的染色体暴露,沿着2d平面膨胀。
[0310]
在这种状态下,染色体是各种不同的原位标记和成像方案容易可及的。
[0311]
实施例2:从观察到具有异常显带模式的单个人类染色体的一部分创建dna文库
[0312]
血液样品取自人类患者,用甲氨蝶呤使细胞周期同步化,用胸苷释放,用colcemid阻滞,在低渗溶液中膨胀,并滴到具有聚丙烯壁和170um盖玻片底部的孔中(801),以产生前中期染色体展片(802)。对展片进行g显带,并置于倒置式nikon te2000显微镜中。显微镜配备了定制为用于对样品进行数字镜装置成像的uv光引擎(young optics nvr plus dlp uv engine)。
[0313]
通过将聚(乙二醇)-二(光不稳定丙烯酸酯)(peg-dpa)与作为氧化还原引发剂和催化剂的过硫酸铵(aps)和四甲基乙二胺(temed)混合,在样品(803)上使可光降解的水凝胶聚合。立即将溶液移液到玻璃上,并允许在室温固化30分钟。随后用20ul的25mm mes ph 5.8洗涤流体孔5次。
[0314]
使用638nm led作为光源,获得显带染色体的明场图像,并将标记模式与g显带模式表进行比较。在17号染色体的单个姐妹染色单体上的位置17p5处鉴定出染色体插入,并做出决定将其切下以供进一步分析。
[0315]
操作者在控制计算机上使用自定义编写的软件追踪围绕染色体图像的框区(box)。当框区完成后,计算机缩放框区并将框区坐标转换成二进制掩模图案,并用该图案对dlp芯片进行编程,然后打开385nm led并辐照染色体区域(804)。这使水凝胶的紧密接近染色体的区域光降解(805)。
[0316]
控制计算机指导机器人移液器吸头用20ul nebuffer r2.1进行2次洗涤,然后添加在nebuffer r2.1中的ecori和胰蛋白酶,并在37℃孵育10min。这使染色质的蛋白质部分消化(807),并将暴露的dna(806)切成小段(808)。然后抽吸样品并转移(809)至微量离心管,并用20ul另外的nebuffer r2.1洗涤装置,将其与第一抽吸物汇集。然后使用ultra ii dna文库制备试剂盒(neb)将样品用于构建dna测序文库。
[0317]
实施例3:可裂解接近性条形码化交联剂
[0318]
作为初始概念验证,我们在此描述了使用可裂解接近性条形码化交联剂(cpbc)的示例方法。在本实施例中,cpbc由两个捕获探针组成,每个捕获探针由附接到长度为15个碱基对的双链核酸条形码的补骨脂素基团组成的捕获结构域组成,其中两个捕获探针具有相同的条形码。然后通过包含双链核酸的可裂解接头将两个捕获探针连接到每一个,该双链核酸包含pam序列,后面是20个碱基对长度的识别区段,当用作crisr-cas9系统的一部分时,该识别区段可以被专门设计的引导rna识别以激活裂解。所有cpbc被制备成确保cpbc具有区别于彼此的独特条形码,并且cpbc在其可裂解接头区域共有相同的识别区段。
[0319]
将包含悬浮于溶液中的人类中期血细胞的样品暴露于cpbc和uv辐射30分钟,以启动补骨脂素和细胞染色体内包含的核酸的交联。交联后,细胞被溶解,并且蛋白质被消化。cpbc通过crisp-cas9系统对识别区段的识别而被裂解,将捕获探针彼此分离。然后在低熔点琼脂糖塞中纯化具有结合的捕获探针的长核酸分子[zhang,2012]。将样品电洗脱到含有每10个核苷酸对1个染料的比例的yoyo-1的低盐变性缓冲液(0.1
×
tbe、20mm nacl、2%β-巯基乙醇)中,并在18℃孵育过夜。以最少操作将样品用甲酰胺以1:1稀释,并加热至31℃,持续10分钟[tegenfeldt,2009,10,434,512],然后在冰上猝灭以产生沿着长核酸分子长度的yoyo-1的at/cg密度物理图谱。
[0320]
然后,通过将dna溶液分配到保持在45度角的硅烷化玻璃上,将长核酸分子梳理在表面上,允许溶液的拖尾弯月面(trailing meniscus)将长核酸分子末端附着到疏水性玻璃表面。然后用荧光成像系统探查长核酸分子,以产生分子物理图谱的计算机表示,然后将其与预先计算的参考物理图谱进行比较,所述参考物理图谱源自通过[2005]的方法分析解链状态的人类基因组组装grch37的序列。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1