苯甲醛肟及其制备方法与流程
苯甲醛肟及其制备方法
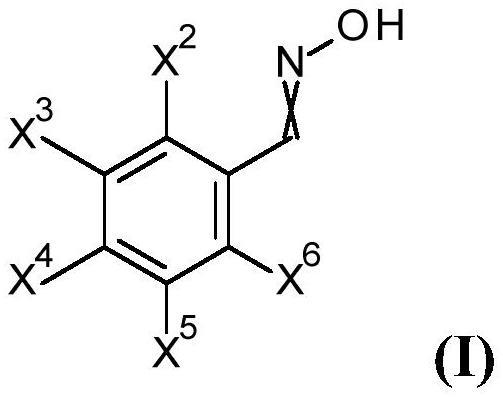
1.本发明涉及一种制备通式(i)的苯甲醛肟的方法及其作为合成农用化学品和药用活性物质的重要前体的用途。
2.苯甲醛肟是农用化学品(如wo 2014/048827a1或wo
3.2019/145245a1)和药用(如j.med.chem.2003,46,284)含苯基异恶唑啉的活性物质的重要前体。
4.文献中多次记载了从相应的苯甲醛开始制备苯甲醛肟,例如在org.lett.2001,3,4209(在有机溶剂中加入碱进行反应)、jp2016166153a(两相体系)、us 20090062342a1(水体系,加入相转移催化剂和无机碱)或molecules 2018,23,2545(水体系,加入有机碱)。wo 2012/130798a1和wo 2014/048827a1记载了从3,5-二氟苯甲醛制备肟。
5.3,5-二氟苯甲醛的制备记载于例如us 4424229a、us 6287646b1和mol.crystals and liquid crystals 2010,528,138中通过格氏形成而使用镁的方法。作为一种可替代的选择,turbo格氏试剂的使用先前已记载于ep 1582523a1中,仲丁基锂使用记载于j.org.chem.2011,76,9391中。此外,ep 1389608a1记载了其通过金属催化的氰化和随后的还原来制备,wo 2019/2224774a1和cn 101565344a记载了相应苄醇的氧化。
6.存在大量的文献关于从卤代苯开始经苯甲醛到相应肟的两步制备苯甲醛肟。然而,还没有在不分离中间产物的情况下进行这两个反应步骤的方法被记载。醛的分离是关键步骤,因为其挥发性和对大气中氧气氧化的敏感性,分离醛会导致形成不需要的副产品和减少产量。因此,避免这种分离是可取的。
7.根据上述现有技术,本发明的目的是找到一种制备通式(i)化合物的方法,用这种方法可以以更高的产量、高纯度以及环境友好的方式获得该化合物,以便可以在工业规模上获得制备活性物质的重要中间体。
8.上述目的通过制备通式(i)化合物的方法实现
[0009][0010]
其中
[0011]
x2为h、c
1-c4烷基、c
1-c4氟代烷基、c
1-c4氟氧烷基(fluoroxyalkyl)、c
1-c4烷氧基、氟、cn,
[0012]
x3为h、c
1-c4烷基、c
1-c4氟代烷基、c
1-c4氟氧烷基、c
1-c4烷氧基、氟、氯、cn,
[0013]
x4为h、c
1-c4烷基、c
1-c4氟代烷基、c
1-c4氟氧烷基、c
1-c4烷氧基、氟、cn,
[0014]
x5为h、c
1-c4烷基、c
1-c4氟代烷基、c
1-c4氟氧烷基、c
1-c4烷氧基、氟、氯、cn,
[0015]
x6为h、c
1-c4烷基、c
1-c4氟代烷基、c
1-c4氟氧烷基、c
1-c4烷氧基、氟、cn,
[0016]
其中通式(ii)的化合物
[0017][0018]
其中基团如上文定义,
[0019]
在第一反应步骤中,在异丙基氯化镁和dmf的存在下反应,形成相应的加合物(iii),在第二反应步骤中,(iii)加入酸和羟胺进一步反应形成通式(iv)的醛
[0020][0021]
其中基团如上文定义,
[0022]
随后形成通式(i)的化合物。
[0023]
通式(i)、(ii)、(iii)和(iv)化合物中的基团优选限定如下:
[0024]
x2是h、甲基、三氟甲基、二氟甲基、二氟甲氧基、三氟甲氧基、氟、甲氧基、cn,
[0025]
x3是h、甲基、三氟甲基、二氟甲基、二氟甲氧基、三氟甲氧基、氟、氯、甲氧基、cn,
[0026]
x4是h、甲基、三氟甲基、二氟甲基、二氟甲氧基、三氟甲氧基、氟、甲氧基、cn,
[0027]
x5是h、甲基、三氟甲基、二氟甲基、二氟甲氧基、三氟甲氧基、氟、氯、甲氧基、cn,
[0028]
x6是h、甲基、三氟甲基、二氟甲基、二氟甲氧基、三氟甲氧基、氟、甲氧基、cn。
[0029]
通式(i)、(ii)、(iii)和(iv)化合物中的基团特别优选限定如下:
[0030]
x2是h,
[0031]
x3是h、甲基、三氟甲基、二氟甲基、氟、氯、甲氧基、cn,
[0032]
x4是氟、h,
[0033]
x5是h、甲基、三氟甲基、二氟甲基、氟、氯、甲氧基、cn,
[0034]
x6是h。
[0035]
通式(i)、(ii)、(iii)和(iv)化合物中的基团非常特别优选限定如下:
[0036]
x2是h,
[0037]
x3是h、氟,
[0038]
x4是h、氟,
[0039]
x5是h、氟,
[0040]
x6是h。
[0041]
通式(i)、(ii)、(iii)和(iv)化合物中的基团最优选限定如下:
[0042]
x2是h,
[0043]
x3是氟,
[0044]
x4是h,
[0045]
x5是氟,
[0046]
x6是h。
[0047]
方法和中间体的描述
[0048]
方案1
[0049][0050]
在第一步中,在持续冷却下向通式(ii)的化合物中加入异丙基氯化镁,异丙基氯化镁可以直接以市售溶液的形式使用(例如在thf中的2摩尔溶液),或者由镁和2-氯丙烷在合适的溶剂(例如thf)中预制而成。然后用dmf(n,n-二甲基甲酰胺)处理反应混合物。得到相应的dmf加合物(iii),在没有后处理和在持续冷却的情况下向(iii)加入含水酸和羟胺(以盐或水溶液的形式),相应的醛(iv)经过连续反应形成通式(i)的化合物。
[0051]
式(i)的化合物可作为几何异构体的混合物存在。
[0052][0053]
e异构体和z异构体之间的比例各不相同,e异构体通常以更大的量存在。
[0054]
反应步骤1
[0055]
第一反应步骤在合适的溶剂中进行。
[0056]
合适的溶剂原则上是所有在特定反应条件下为惰性的有机溶剂,例如脂族、脂环族或芳族烃(例如戊烷、己烷、庚烷、辛烷、壬烷和工业级碳氢化合物、环己烷、甲基环己烷、石油醚、轻石油、苯、甲苯、二甲苯、均三甲苯)以及脂族、脂环族或芳族醚类(例如1,2-二甲氧基乙烷(dme)、二甘醇二甲醚、四氢呋喃(thf)、2-甲基-thf、1,4-二恶烷、甲基叔丁基醚(mtbe)、环戊基甲醚、苯甲醚)或上述溶剂的混合物。优选地,第一反应步骤在甲苯、二甲苯、均三甲苯、苯甲醚、thf、甲基环己烷、2-甲基-thf或甲基叔丁基醚或上述溶剂的混合物中进行。更优选地,使用甲苯、thf或甲苯与thf的混合物。更优选地,溶剂是无水溶剂。
[0057]
此外,在加入异丙基氯化镁的过程中冷却反应混合物。优选地,反应在-10℃和20℃之间的温度进行,更优选地在0℃和15℃的温度之间进行。dmf的添加优选在-10℃和60℃之间的温度进行,更优选在0℃和30℃之间的温度进行。非常特别优选地,向该反应混合物
中加入dmf时的温度与向该反应混合物中加入异丙基氯化镁时的温度相同。此处反应步骤1是通过将通式(ii)化合物以纯物质或在合适溶剂中的溶液的形式计量加入到异丙基氯化镁溶液中进行的。优选地,反应步骤1是通过将异丙基氯化镁计量加入到通式(ii)化合物的溶液中进行的。随后将反应混合物计量加入到dmf溶液中,优选地将dmf计量加入到反应混合物中。
[0058]
异丙基氯化镁以等摩尔量或过量使用,优选5.0至1.05当量,更优选2.0至1.05当量。
[0059]
dmf以等摩尔量或过量使用,优选5.0至1.05当量,更优选2.0至1.05当量。
[0060]
异丙基氯化镁可以以市售溶液的形式使用,如在thf中的2摩尔溶液,或其可以就地自行制备。为此,首先将金属镁进料到合适的有机溶剂中(该有机溶剂在反应条件下是惰性的,特别优选地是二乙醚或四氢呋喃),并加入2-氯丙烷。可以任选地通过本领域技术人员通常已知的方法活化镁,例如通过碘、二溴甲烷或通过添加活性异丙基氯化镁进行活化。随后是反应步骤1,优选地将通式(ii)的化合物计量加入根据本发明所述的异丙基氯化镁溶液中。
[0061]
反应步骤2
[0062]
第二反应步骤在水溶液中进行。
[0063]
此外,在反应混合物的水解过程中/在加入含水酸和以盐或游离碱的水溶液形式的羟胺的过程中,冷却反应混合物。优选地,在0℃和40℃之间的温度范围内进行反应,更优选在0℃和30℃之间的温度进行。
[0064]
来自反应步骤1的反应混合物的水解可以通过加入含水酸或水来实现,优选通过将反应混合物计量加入至含水酸或水实现水解,更优选通过将反应混合物计量加入至水实现。
[0065]
所用的酸优选含水盐酸或含水硫酸,更优选含水硫酸。硫酸可以在反应混合物水解前就存在,优选在优选条件下反应混合物在水中水解发生后计量加入硫酸。含水硫酸优选以在1重量%至80重量%之间,更优选以在10重量%至50重量%之间的浓度使用。优选地,使用0.2至5.0当量的硫酸,更优选使用0.3至2当量的硫酸。
[0066]
羟胺优选以氯盐(nh2oh-hcl)或硫酸盐((nh2oh)2·
h2so4)的形式、纯物质的形式或作为水溶液、或作为游离碱(nh2oh)的水溶液使用,更优选作为硫酸盐的水溶液使用。该溶液优选以5重量%至40重量%,更优选以15重量%至35重量%的浓度使用。优选地,使用0.5至0.7当量的(nh2oh)2·
h2so4,更优选使用0.55至0.6当量的(nh2oh)2·
h2so4。
[0067]
在反应完成时,可以进行通式(i)的化合物的后处理和分离,例如通过相分离、用合适的有机溶剂萃取水相、用水洗涤有机相,并完全或部分去除溶剂。通式(i)的化合物可以以固体物质的形式或作为合适溶剂中的溶液分离。
[0068]
两个步骤的产率为92%至97%,相比之下,现有技术两个步骤中的产率为55至79%。
实施例
[0069]
通过以下实施例更详细地阐述本发明,其对本发明不构成限制。
[0070]
测量方法
[0071]
通过1h/
19
f nmr对产物进行表征。
[0072]
实施例1:(1e)-3,5-二氟苯甲醛肟(i-1)
[0073]
在氩气环境下,将100毫升甲苯中的100.0克(1.0当量)1-溴-3,5-二氟苯装入带有机械搅拌器的1l四颈烧瓶,并在0℃-5℃、1.5小时内逐滴加入279.3毫升<10℃的市售的异丙基氯化镁(275.3克,2m在thf中,1.1当量)。在添加结束时,在《5℃将该混合物再搅拌3小时,然后在《10℃将40.8克(1.1当量)n,n-二甲基甲酰胺在60分钟内计量加入。在此温度再搅拌15-20分钟后,在30分钟内将反应混合物缓慢加入500毫升已预冷至《5℃的水中。在这个过程中,内部温度保持在《10℃。在添加结束时,在《10℃在30分钟内计量加入29.9克(0.3当量)的50%硫酸。随后在《5℃在45分钟内计量加入45.8克(0.55当量)硫酸羟胺在120毫升水中的溶液,在加入结束时将该反应混合物再搅拌3小时,在此期间温度上升至20℃-25℃。在又加入100毫升甲苯后,分离相,用200毫升水洗涤有机相。在100-150mbar和40℃部分除去有机相的溶剂,得到产物在甲苯/thf中的溶液:240.0克(31.5重量%,产率95%)。
[0074]1h nmr(600mhz,cdcl3):δ6.83ppm(m,1h),7.09(m,2h),7.64(br s,1h),8.06(s,1h)ppm。
[0075]
19
f nmr(600mhz,cdcl3):δ-109.0(2f)ppm。
[0076]
实施例2:(1e)-3,5-二氟苯甲醛肟(i-1)
[0077]
在氩气下,将6.8克(1.1当量)镁装入10毫升thf中的500毫升反应器,用2毫升(0.01当量,2m在thf中)市售的异丙基氯化镁将其激活。随后,在30℃在1小时内平行计量加入21.9克(1.1当量)的2-氯丙烷和65毫升thf。该反应混合物在此温度再搅拌1小时,然后冷却到《5℃,在此温度在1小时内将50.0克(1.0当量)1-溴-3,5-二氟溴苯在50毫升甲苯中的溶液计量加入其中。在加入结束时在此温度再搅拌该混合物3小时,然后在《10℃在30分钟内计量加入20.4克(1.1当量)的n,n-二甲基甲酰胺。在此温度再搅拌15-20分钟后,将该反应混合物在30分钟内加入至250毫升冷却至《5℃的水中,内部温度保持在《10℃。在添加结束时,在《10℃在30分钟内计量加入14.9克(0.3当量)的50%硫酸。随后在《5℃在45分钟内计量加入22.9克(0.55当量)硫酸羟胺在50毫升水中的溶液,在加入结束时将该反应混合物再搅拌3小时,在此期间温度上升至20℃-25℃。在又加入50毫升甲苯后,分离相,用100毫升水洗涤有机相。在100-150mbar和40℃部分除去有机相的溶剂,得到产物在甲苯/thf中的溶液:150.0克(24.9重量%,产率94%)。
[0078]
反应(实施例1和2)也可以在以下溶剂中以类似的方式进行:
[0079]
代替甲苯的溶剂产率mtbe99.6甲基环己烷92.92-甲基四氢呋喃73.2二甲苯81.4苯甲醚73.4
[0080]
使用本发明的方法还制备了以下化合物:
[0081]
3,5-二氯苯甲醛肟
[0082]1h nmr(400mhz,cdcl3):δ7.37(t,j=1.9hz,1h),7.45(br s,1h),7.46(d,j=7.5hz,2h),8.03(s,1h)ppm。
[0083]
3-氟-5-甲氧基苯甲醛肟
[0084]1h nmr(400mhz,cdcl3):δ3.79(s,3h),6.85(dt,j=2.3/11.0hz,1h),6.97-7.01(m,2h),8.11(s,1h)ppm。
[0085]
3-氟-5-甲基苯甲醛肟
[0086]1h nmr(400mhz,cdcl3):δ2.37ppm(s,3h),7.09(m,2h),6.89(d,j=9.4hz,1h),7.10-7.14(m,2h),7.46(br s,1h),8.07(s,1h)ppm。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1