APOL1的抑制剂及其使用方法与流程
apol1的抑制剂及其使用方法
1.本技术要求2020年8月26日提交的美国临时专利申请号63/070,705的优先权益,所述申请的内容以引用方式整体并入本文。
2.本公开提供了可抑制载脂蛋白l1(apol1)的化合物和使用这些化合物治疗apol1介导性疾病的方法,所述疾病例如胰腺癌、局灶性节段性肾小球硬化(fsgs)和/或非糖尿病性肾病(ndkd)。在一些实施方案中,fsgs和/或ndkd与2种常见的apol1遗传变异体(g1:s342g:i384m,以及g2:n388del:y389del)中的至少一种相关。在一些实施方案中,胰腺癌与apol1水平升高(诸如例如胰腺癌组织中apol1水平升高)相关。
3.fsgs是一种罕见的肾脏疾病,据估计全球发病率为0.2至1.1/100,000/年。fsgs是造成蛋白尿和肾功能进行性下降的足细胞(肾小球内脏上皮细胞)疾病。ndkd是涉及并非由糖尿病引起的足细胞或肾小球血管床的损害的肾脏疾病。ndkd是特征在于高血压和肾功能进行性下降的疾病。人类遗传学支持g1和g2 apol1变体在诱导肾脏疾病中的因果作用。具有2个apol1风险等位基因的个体患终末期肾脏疾病(eskd)的风险增加,包括原发性(特发性)fsgs、人免疫缺陷病毒(hiv)相关fsgs、ndkd、动脉肾硬化、狼疮性肾炎、微量白蛋白尿和慢性肾脏疾病。参见p.dummer等人,semin nephrol.35(3):222-236(2015)。
4.fsgs和ndkd可以根据潜在的病因学分为不同的亚组。fsgs的一个同质亚组的特征在于载脂蛋白l1(apol1)基因中称为g1和g2的独立常见序列变体的存在,其被称为“apol1风险等位基因”。g1编码一对相关的非同义氨基酸变化(s342g和i384m),g2编码蛋白质的c末端附近的2个氨基酸缺失(n388del:y389del),而g0为祖先(低风险)等位基因。在具有apol1遗传风险变体的患者中也发现了ndkd的独特表型。在apol1介导性fsgs和ndkd中,与没有或仅有1个apol1遗传风险变异的患者相比,具有两个风险等位基因的患者出现更高水平的蛋白尿和更快的肾功能丧失。或者,在amkd中,具有一个风险等位基因的患者也可能出现较高水平的蛋白尿和加速的肾功能丧失。参见g.vajgel等人,j.rheumatol.,2019年11月,jrheum.190684。
5.apol1是仅在人类、大猩猩和狒狒中表达的44kda蛋白质。apol1基因在人类的多个器官中表达,包括肝脏和肾脏。apol1主要由肝脏产生,并且含有允许分泌到血流中的信号肽,在血流中其与高密度脂蛋白亚群结合循环。apol1负责保护免受侵袭性寄生虫布氏布氏锥虫(trypanosoma brucei brucei/t.b.brucei)的侵害。apol1被布氏锥虫内吞并转运到溶酶体,在溶酶体中它插入溶酶体膜并形成导致寄生虫肿胀和死亡的孔。
6.虽然所有3种apol1变体(g0、g1和g2)都具有裂解布氏锥虫的能力,但apol1 g1和g2变体提供了针对已进化出抑制apol1 g0的血清抗性相关蛋白(sra)的寄生虫物种的额外保护;apol1 g1和g2变异体提供了针对引起昏睡病的锥虫的额外保护。g1和g2变异体逃避sra的抑制;g1提供了针对冈比亚锥虫(t.b.gambiense)的额外保护,而g2提供了对罗得西亚锥虫(t.b.rhodesiense)的额外保护。
7.在肾中,apol1在足细胞、内皮细胞(包括肾小球内皮细胞)和一些肾小管细胞中表达。apol1 g1或g2(但非g0)在转基因小鼠中的足细胞特异性表达诱导结构和功能变化,包括蛋白尿、肾功能降低、足细胞异常和肾小球硬化。与这些数据一致,apol1的g1和g2变体在
诱导fsgs和加速其在人类中的进展中起因果作用。具有apol1风险等位基因(即,apol1 g1或apol1g2等位基因的纯合子或复合杂合子)的个体发生fsgs的风险增加,并且如果他们发生fsgs,则他们将面临肾功能迅速下降的风险。因此,抑制apol1可能对携带apol1风险等位基因的个体产生积极影响。
8.尽管apol1的正常血浆浓度相对较高并且在人类中可能变化至少20倍,但循环的apol1与肾脏疾病没有因果关系。然而,肾脏中的apol1被认为是肾脏疾病发展的原因,包括fsgs和ndkd。在某些情况下,促炎细胞因子如干扰素或肿瘤坏死因子-α可使apol1蛋白合成增加大约200倍。另外,若干研究已表明,apol1蛋白可在细胞膜中形成ph门控na
+
/k
+
孔,导致细胞内k
+
的净流出,最终导致局部和全身炎症反应的活化、细胞肿胀和死亡。
9.与欧洲血统的人相比,近期撒哈拉以南非洲血统的人患eskd的风险明显更高。在美国,eskd导致女性寿命损失的年数几乎与乳腺癌一样多,并且男性寿命损失的年数比结肠直肠癌更多。
10.fsgs和ndkd是由足细胞损伤引起的,所述足细胞是肾小球滤过屏障的一部分,因此导致蛋白尿。蛋白尿患者发生终末期肾脏疾病(eskd)和发生蛋白尿相关并发症如感染或血栓栓塞事件的风险更高。对于fsgs或ndkd,没有标准化治疗方案,也没有批准的药物。目前,fsgs和ndkd采用对症治疗(包括使用肾素血管紧张素系统阻断剂控制血压)进行管理,并且fsgs和重度蛋白尿患者可能会接受大剂量的类固醇。目前对ndkd的治疗选项是基于血压控制和肾素血管紧张素系统的阻断。
11.皮质类固醇单独使用或与其他免疫抑制剂组合使用可诱导少数患者病情缓解(少数患者出现如蛋白尿缓解),并伴有多种副作用。然而,即使在最初对皮质类固醇和/或免疫抑制剂治疗有反应的患者中,缓解也常常是不可逆转的。因此,患者,特别是具有2apol1风险等位基因的近代撒哈拉以南非洲血统的个体,经历了导致终末期肾病(esrd)的快速疾病进展。因此,对于用于fsgs和ndkd的治疗的医疗需求没有得到满足。说明性地,鉴于apol1在诱导和加速肾脏疾病的进展中起致病作用的证据,apol1的抑制应当对患有apol1介导性肾脏疾病的患者,特别是那些携带两种apol1风险等位基因(即,对于g1或g2等位基因是纯合的或复合杂合的)的患者具有积极影响。
12.此外,apol1是在多种癌症中异常表达的基因(lin等人,cell death and disease(2021),12:760)。最近,发现与邻近组织相比,apol1在人胰腺癌组织中异常升高,并且与胰腺癌患者的不良预后相关。在体内和体外实验中,apol1敲低显著抑制癌细胞增殖并促进胰腺癌细胞凋亡。
13.本公开的一个方面提供了至少一种选自以下的化合物:式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
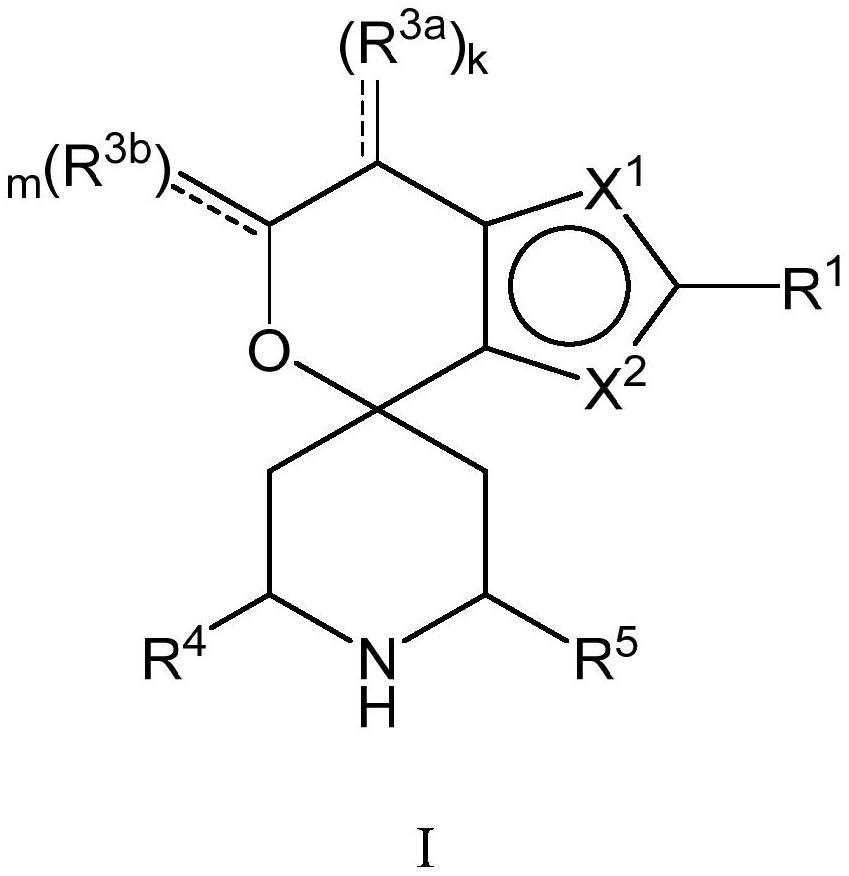
’0的化合物)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐,其可用于治疗由apol1介导性疾病,例如fsgs和ndkd。例如,至少一种化合物是由式i表示的化合物:
[0014][0015]
其中x1、x2、r1、r
3a
、r
3b
、r4、r5、k和m是如本文公开的实施方案中所定义。
[0016]
在一些实施方案中,本公开的至少一种化合物是由以下结构式表示的化合物:
[0017][0018]
式i
[0019]
其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐,其中:
[0020]
x1选自s和-cr
2a
并且x2选自s和-cr
2b
,其中:
[0021]
x1和x2之一是s;
[0022]
当x1为s时,则x2为-cr
2b
;并且
[0023]
当x2是s时,则x1是-cr
2a
;
[0024]
r1选自氢、卤素、-oh、氰基、c
1-c6烷基、c
1-c6烷氧基、c
3-c6环烷基和苯基,其中:
[0025]
r1的所述c
1-c6烷基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2和c
1-c4烷氧基;
[0026]
r1的所述c
1-c6烷氧基任选地被1至3个独立地选自卤素的基团取代;
[0027]
r1的所述c
3-c6环烷基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷基、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)和-c(=o)n(c
1-c4烷基)2;并且
[0028]
r1的所述苯基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷基、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)和-c(=o)n(c
1-c4烷基)2;
[0029]r2a
选自氢、卤素、氰基、-oh、=o和c
1-c6烷基,其中:
[0030]r2a
的所述c
1-c6烷基任选地被1至3个独立地选自卤素、氰基、-oh和c
1-c4烷氧基的基团取代;
[0031]r2b
选自氢、卤素、氰基、-oh、=o和c
1-c6烷基;
[0032]r3a
选自卤素、氰基、-oh、c
1-c6烷基和=o;其中:
[0033]r3a
的所述c
1-c6烷基任选地被1至3个独立地选自卤素、氰基和-oh的基团取代;
[0034]r3b
选自c
1-c2烷基和=o;其中:
[0035]r3b
的所述c
1-c2烷基任选地被1至3个独立地选自卤素、氰基和-oh的基团取代;
[0036]
当r
3a
选自卤素、氰基、oh、c
1-c6烷基时或当r
3b
选自c
1-c2烷基时,在每次出现时为单键;或者当r
3a
为=o时或当r
3b
为=o时,每次出现时为双键;
[0037]
r4选自c
1-c6烷基、-c(=o)o(c
1-c4烷基)、c
2-c6炔基和其中:
[0038]
r4的所述c
1-c6烷基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)、-c(=o)n(c
1-c4烷基)2、c
3-c6环烷基、5至10元杂环基、苯基和5至10元杂芳基;
[0039]
环a选自c
3-c
12
碳环基、3至12元杂环基、c6和c
10
芳基和5至10元杂芳基,其中环a任选地被1、2、3、4或5个ra基团取代;其中:
[0040]
ra在每次出现时独立地选自卤素、氰基、c
1-c6烷基、c
2-c6烯基、c
1-c6烷氧基、c
1-c6卤代烷基、c
1-c6卤代烯基、c
1-c6卤代烷氧基、-c(=o)nrhri、-nrhri、-nrhc(=o)rk、-nrhc(=o)ork、-nrhc(=o)nr
irj
、-nrhs(=o)
prk
、-ork、-oc(=o)rk、-oc(=o)ork、-oc(=o)nrhri、-[o(ch2)q]ro(c
1-c6烷基)、-s(=o)
prk
、-s(=o)
p
nrhri、-c(=o)ork、c
3-c
12
碳环基、3至12元杂环基、c6和c
10
芳基以及5至10元杂芳基;其中:
[0041]
ra的所述c
1-c6烷基、所述c
1-c6烷氧基和所述c
2-c6烯基各自任选地被1至3个独立地选自以下的基团取代:c6至c
10
芳基(任选地被1至3个rm基团取代:)、5至10元杂环基(任选地被1至3个rm基团取代)、5至10元杂芳基(任选地被1至3个rm基团取代)、氰基、-c(=o)rk、-c(=o)ork、-c(=o)nrhri、-nrhri、-nrhc(=o)rk、-nrhc(=o)ork、-nrhc(=o)nr
irj
、-nrhs(=o)
prk
、-ork、-oc(=o)rk、-oc(=o)ork、-oc(=o)nrhri、-s(=o)
prk
、-s(=o)
p
nrhri和c
3-c6碳环基(任选地被1至3个rm基团取代);
[0042]
ra的所述c
3-c
12
碳环基、所述3至12元杂环基、所述c6和c
10
芳基和所述5至10元杂芳基各自任选地被1至3个独立地选自以下的基团取代:卤素、氰基、c
1-c4烷基、-nrhri和-ork;其中:
[0043]
rh、ri和rj在每次出现时各自独立地选自氢、c
1-c4烷基、c6c
10
芳基和c
3-c6环烷基;其中:
[0044]
rh、ri和rj中任一者的所述c
1-c4烷基任选地被1至3个独立地选自卤素、氰基和-oh的基团取代;
[0045]rk
在每次出现时各自独立地选自氢、c
1-c4烷基、5至10元杂环基和c
3-c6碳环基;其
中:
[0046]rk
中任一者的所述c
1-c4烷基任选地被1至3个独立地选自卤素、氰基和-oh的基团取代;
[0047]rm
在每次出现时独立地选自卤素、氰基、氧代、c
1-c6烷基、c
1-c6烷氧基、-s(=o)
prk
和-ork;其中:
[0048]rm
的所述c
1-c6烷基任选地被1至3个独立地选自卤素、氰基和-oh的基团取代;
[0049]
r5选自c
1-c6烷基、-c(=o)o(c
1-c4烷基)、c
3-c
12
碳环基、3至12元杂环基、c6和c
10
芳基以及5至10元杂芳基;其中:
[0050]
r5的所述c
1-c6烷基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)和-c(=o)n(c
1-c4烷基)2;
[0051]
r5的所述c
3-c
12
碳环基、所述3至12元杂环基、所述c6和c
10
芳基以及所述5至10元杂芳基各自任选地被1至3个独立地选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)(任选地被-oh取代)、-n(c
1-c4烷基)2、c
1-c5烷基(任选地被-oh取代)、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)、-nhc(=o)(c
1-c4烷基)、-c(=o)(c
1-c4烷氧基)和-c(=o)n(c
1-c4烷基)2;
[0052]
k是选自0、1和2的整数;其中
[0053]
当r
3a
选自卤素、氰基、-oh和c
1-c6烷基时,k是1或2;并且
[0054]
当r
3a
为=o时,k为1;
[0055]
m是选自0、1和2的整数,其中:
[0056]
当r
3b
选自c
1-c2烷基时,m是1或2;并且
[0057]
当r
3b
为=o时,m为1;
[0058]
p是选自1和2的整数;并且
[0059]
q和r各自是选自1、2、3和4的整数。
[0060]
在一些实施方案中,r4选自c
1-c6烷基和
[0061]
在一些实施方案中,r5选自c
1-c6烷基、-c(=o)o(c
1-c4烷基)、c
3-c
12
碳环基、3至12元杂环基、c6和c
10
芳基以及5至10元杂芳基,其中:
[0062]
r5的所述c
1-c6烷基任选地被1至3个选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)以及-c(=o)n(c
1-c4烷基)2;
[0063]
r5的所述c
3-c
12
碳环基、所述3至12元杂环基、所述c6和c
10
芳基和所述5至10元杂芳基各自任选地被1至3个选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷基、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)和-c(=o)n(c
1-c4烷基)2。
[0064]
在一些实施方案中,本公开的至少一种化合物(例如,至少一种式i化合物)是由以
下结构式表示的化合物:
[0065][0066]
式i0[0067]
其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐,其中:
[0068]
x1和x2各自选自s和-cr2,其中:
[0069]
x1和x2之一是s;
[0070]
当x1为s时,则x2为-cr
2b
;并且
[0071]
当x2是s时,则x1是-cr
2a
;
[0072]
r1选自卤素、氰基、c
1-c6烷基、c
1-c6烷氧基、c
3-c6环烷基和苯基;其中:
[0073]
r1的所述c
1-c6烷基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2和c
1-c4烷氧基;
[0074]
r1的所述c
1-c6烷氧基任选地被1至3个独立地选自卤素的基团取代;
[0075]
r1的所述c
3-c6环烷基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷基、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)和-c(=o)n(c
1-c4烷基)2;并且
[0076]
r1的所述苯基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷基、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)和-c(=o)n(c
1-c4烷基)2;
[0077]r2a
选自氢、卤素、氰基、-oh、=o和c
1-c6烷基;其中:
[0078]r2a
的所述c
1-c6烷基任选地被1至3个独立地选自卤素、氰基、-oh和c
1-c4烷氧基的基团取代;
[0079]r2b
选自氢、卤素、氰基、-oh、=o和c
1-c6烷基;
[0080]r3a
选自卤素、氰基、-oh、c
1-c6烷基和=o;其中:
[0081]r3a
的所述c
1-c6烷基任选地被1至3个独立地选自卤素、氰基和-oh的基团取代;
[0082]r3b
选自c
1-c2烷基和=o;其中:
[0083]r3b
的所述c
1-c2烷基任选地被1至3个独立地选自卤素、氰基和-oh的基团取代;
[0084]
当r
3a
选自卤素、氰基、-oh、c
1-c6烷基时或当r
3b
选自c
1-c2烷基时,在每次出现时为单键;或者当r
3a
为=o时或当r
3b
为=o时,每次出现时为双键;
[0085]
r4选自c
1-c6烷基和其中:
[0086]
r4的所述c
1-c6烷基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)、-c(=o)n(c
1-c4烷基)2、c
3-c6环烷基、5至10元杂环基、苯基和5至10元杂芳基;
[0087]
环a选自c
3-c
12
碳环基、3至12元杂环基、c6和c
10
芳基和5至10元杂芳基,其中环a任选地被1、2、3、4或5个ra基团取代;其中:
[0088]
ra在每次出现时独立地选自卤素、氰基、c
1-c6烷基、c
2-c6烯基、c
1-c6烷氧基、c
1-c6卤代烷基、c
1-c6卤代烯基、c
1-c6卤代烷氧基、-c(=o)nrhri、-nrhri、-nrhc(=o)rk、-nrhc(=o)ork、-nrhc(=o)nr
irj
、-nrhs(=o)
prk、-ork、-oc(=o)rk、-oc(=o)ork、-oc(=o)nrhri、-[o(ch2)q]ro(c
1-c6烷基)、-s(=o)
prk
、-s(=o)
p
nrhri、c
3-c
12
碳环基、3至12元杂环基、c6和c
10
芳基以及5至10元杂芳基;其中:
[0089]
ra的所述c
1-c6烷基和所述c
2-c6烯基各自任选地被1至3个独立地选自以下的基团取代:氰基、-c(=o)rk、-c(=o)ork、-c(=o)nrhri、-nrhri、-nrhc(=o)rk、-nrhc(=o)ork、-nrhc(=o)nr
irj
、-nrhs(=o)
prk、-ork、-oc(=o)rk、-oc(=o)ork、-oc(=o)nrhri、-s(=o)
prk
、-s(=o)
p
nrhri和c
3-c6环烷基;
[0090]
ra的所述c
3-c
12
碳环基、所述3至12元杂环基、所述c6和c
10
芳基和所述5至10元杂芳基各自任选地被1至3个选自以下的基团取代:卤素、氰基、c
1-c4烷基、-nrhri和-ork;其中:
[0091]
rh、ri和rj在每次出现时各自独立地选自氢、c
1-c4烷基和c
3-c6环烷基;其中:
[0092]
rh、ri和rj中任一者的所述c
1-c4烷基任选地被1至3个选自卤素、氰基和-oh的基团取代;
[0093]rk
在每次出现时各自独立地选自氢、c
1-c4烷基和c
3-c6环烷基;其中:
[0094]rk
中任一者的c
1-c4烷基任选地被1至3个选自卤素、氰基和-oh的基团取代;
[0095]
r5选自c
1-c6烷基、-c(=o)o(c
1-c4烷基)、c
3-c
12
碳环基、3至12元杂环基、c6和c
10
芳基以及5至10元杂芳基;其中:
[0096]
r5的所述c
1-c6烷基任选地被1至3个选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)以及-c(=o)n(c
1-c4烷基)2;
[0097]
r5的所述c
3-c
12
碳环基、所述3至12元杂环基、所述c6和c
10
芳基和所述5至10元杂芳基各自任选地被1至3个选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷基、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)和-c(=o)n(c
1-c4烷基)2;
[0098]
当r
3a
选自卤素、氰基、-oh、c
1-c6烷基时,k是选自0、1和2的整数;或者当r
3a
为=o时,k为选自0和1的整数;
[0099]
当r
3b
选自c
1-c2烷基时,m为选自0、1和2的整数;并且当r
3b
为=o时,m为选自0和1的整数;
[0100]
p是选自1和2的整数;并且
[0101]
q和r各自是选自1、2、3和4的整数。
[0102]
在本公开的一个方面,式i化合物选自化合物1至391(例如化合物1至220),使得至少一种实体选自化合物1至391(例如化合物1至220)、这些化合物中任一者的药学上可接受的盐、前述任一者的溶剂化物和前述任一者的氘化衍生物。
[0103]
在一些实施方案中,本公开提供了包含至少一种选自以下的实体的药物组合物:式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如选自式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。在一些实施方案中,药物组合物可以包含至少一种选自以下的化合物:化合物1至391(例如化合物1至220)、这些化合物中任一者的药学上可接受的盐、前述任一者的溶剂化物和前述任一者的氘化衍生物。这些组合物可还包含至少一种额外活性药物成分和/或至少一种载体。
[0104]
本公开的另一个方面提供了治疗apol1介导性疾病(例如,apol1介导性肾脏疾病)的方法,其包括向有需要的受试者施用至少一种选自以下的实体或包含至少一种实体的药物组合物:式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如,式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。在一些实施方案中,所述方法包括施用至少一种选自以下的实体:化合物1至391(例如化合物1至220)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。
[0105]
本公开的另一个方面提供了治疗apol1介导性癌症(例如像胰腺癌)的方法,其包括向有需要的受试者施用至少一种选自以下的实体或包含至少一种实体的药物组合物:式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如,式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。在一些实施方案中,所述方法包括施用至少一种选自以下的实体:化合物1至391(例如化合物1至220)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。
[0106]
本公开的另一方面提供了治疗fsgs和/或ndkd的方法,其包括向有需要的受试者施用至少一种选自以下的实体或包含至少一种实体的药物组合物:式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、
iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如,式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。在一些实施方案中,所述方法包括施用至少一种选自以下的实体:化合物1至391(例如化合物1至220)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。
[0107]
在一些实施方案中,治疗方法包括向有需要的受试者施用至少一种另外的活性剂,其与至少一种选自以下的实体处于同一药物组合物中,或作为单独的组合物:式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如,式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。在一些实施方案中,所述方法包括施用至少一种选自化合物1至391(例如化合物1至220)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐的实体与至少一种另外的活性剂,它们处于同一药物组合物中或处于单独的组合物中。
[0108]
还提供了抑制apol1的方法,其包括向有需要的受试者施用至少一种选自以下的实体或包含至少一种实体的药物组合物:式i、iia、iib、iiia、iiib、iva、ivb、v、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如,式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。在一些实施方案中,抑制apol1的方法包括施用至少一种选自以下的实体或包含至少一种实体的药物组合物:化合物1至391(例如化合物1至220)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。
附图说明
[0109]
图1描绘了化合物181磷酸盐水合物在25
±
2℃和40% rh下的xrpd衍射图。
[0110]
图2描绘了化合物181磷酸盐水合物在25
±
2℃和5% rh(黑色迹线)或90%(灰色迹线)下的xrpd衍射图。
[0111]
图3描绘了化合物181磷酸盐水合物的tga热分析图。
[0112]
图4描绘了化合物181磷酸盐水合物的dsc曲线。
[0113]
图5描绘了化合物181磷酸盐水合物的固态
13
c nmr光谱。
[0114]
图6描绘了化合物181磷酸盐水合物在43% rh下的固态
19
f nmr光谱。
[0115]
图7描述了相对湿度对化合物181磷酸盐水合物的固态
19
f nmr光谱的影响。
[0116]
图8描绘了化合物181磷酸盐水合物在43% rh下的固态
31
p nmr光谱。
[0117]
图9描述了相对湿度对化合物181磷酸盐水合物的固态
31
p nmr光谱的影响。
[0118]
图10描绘了化合物181游离形式一水合物的xrpd衍射图。
[0119]
图11描绘了化合物181游离形式一水合物的tga热分析图。
[0120]
图12描绘了化合物181游离形式一水合物的dsc曲线。
[0121]
图13描绘了化合物181游离形式一水合物的固态
13
c nmr光谱。
[0122]
图14描绘了脱水化合物181游离形式一水合物的固态
13
c nmr光谱。
[0123]
图15描绘了化合物181游离形式一水合物的固态
19
f nmr光谱。
[0124]
图16描绘了脱水化合物181游离形式一水合物的固态
19
f nmr光谱。
[0125]
图17描绘了化合物181磷酸盐甲醇溶剂化物的xrpd衍射图。
[0126]
图18描绘了化合物181磷酸盐甲醇溶剂化物的固态
13
c nmr光谱。
[0127]
图19描绘了化合物181磷酸盐甲醇溶剂化物的固态
19
f nmr光谱。
[0128]
图20描绘了化合物181磷酸盐甲醇溶剂化物的固态
31
p nmr光谱。
[0129]
图21描绘了化合物181磷酸盐mek溶剂化物的xrpd衍射图。
[0130]
图22描绘了化合物181磷酸盐mek溶剂化物的固态
13
c nmr光谱。
[0131]
图23描绘了化合物181磷酸盐mek溶剂化物的固态
19
f nmr光谱。
[0132]
图24描绘了化合物174磷酸盐半水合物的xrpd衍射图。
[0133]
图25描绘了化合物174磷酸盐半水合物的tga热分析图。
[0134]
图26描绘了化合物174磷酸盐半水合物的dsc曲线。
[0135]
图27描绘了化合物174磷酸盐半水合物的固态
13
c nmr光谱。
[0136]
图28描绘了脱水化合物174磷酸半水合物的固态
13
c nmr光谱。
[0137]
图29a描绘了化合物174磷酸盐半水合物的固态
31
p nmr光谱。
[0138]
图29b描绘了脱水化合物174磷酸盐半水合物的固态
31
p nmr光谱。
[0139]
图30描绘了化合物174半水合物的xrpd衍射图。
[0140]
图31描绘了化合物174半水合物的tga热分析图。
[0141]
图32描绘了化合物174半水合物的dsc曲线。
[0142]
图33描绘了化合物174半水合物的固态
13
c nmr光谱。
[0143]
图34描绘了脱水化合物174半水合物的固态
13
c nmr光谱。
具体实施方式
[0144]
定义
[0145]
术语“选自(selected from)”和“选自(chosen from)”在本文中可互换使用。
[0146]
如本文所用的术语“apol1”意指载脂蛋白l1蛋白,并且术语“apol1”意指载脂蛋白l1基因。
[0147]
术语“apol1介导性疾病”是指与异常apol1(例如,某些apol1遗传变异体;apol1水平升高)相关的疾病或病状。在一些实施方案中,apol1介导性疾病是apol1介导性肾脏疾病。在一些实施方案中,apol1介导性疾病与具有两个apol1风险等位基因的患者相关,例如,g1或g2等位基因纯合或复合杂合的患者。在一些实施方案中,apol1介导性疾病与具有一个apol1风险等位基因的患者相关。
[0148]
术语“apol1介导性肾脏疾病”是指损害肾功能并可归因于apol1的疾病或病患。在一些实施方案中,apol1介导性肾脏疾病与具有两个apol1风险等位基因的患者相关,例如,
g1或g2等位基因纯合或复合杂合的患者。在一些实施方案中,apol1介导性肾脏疾病选自eskd、ndkd、fsgs、hiv相关性肾病、动脉肾硬化、狼疮性肾炎、微量白蛋白尿和慢性肾脏疾病。在一些实施方案中,apol1介导性肾脏疾病是慢性肾脏疾病或蛋白尿。
[0149]
如本文所用,术语“fsgs”是指局灶性节段性肾小球硬化,这是导致蛋白尿和肾功能进行性下降的足细胞(肾小球内脏上皮细胞)疾病,并与2种常见apol1遗传变异体有关(g1:s342g:i384m和g2:n388del:y389del)。
[0150]
如本文所用,术语“ndkd”是指非糖尿病性肾脏疾病,其特征在于严重的高血压和肾功能的进行性下降,并与2种常见的apol1遗传变异体(g1:s342g:i384m和g2:n388del:y389del)相关。
[0151]
术语“eskd”和“esrd”在本文中可互换使用,指终末期肾脏疾病或终末期肾病。eskd/esrd为肾脏疾病的最后阶段,即肾衰竭,并且意味着肾已经停止足够好地工作,使得患者在不进行透析或肾移植的情况下无法存活。在一些实施方案中,eskd/esrd与两个apol1风险等位基因相关联。
[0152]
在提及本公开的化合物时,术语“化合物”是指除了分子的组成原子之间可能存在同位素变化之外具有相同化学结构的分子的集合,除非另有说明为立体异构体的集合(例如,外消旋体的集合、顺式/反式立体异构体的集合、或(e)和(z)立体异构体的集合)。因此,本领域技术人员应清楚,由含有指示的氘原子的特定化学结构表示的化合物还将含有较少量的在该结构中的一个或多个指定氘位置处具有氢原子的同位素体。本公开的化合物中此类同位素体的相对量将取决于许多因素,包括用于制备化合物的试剂的同位素纯度和用于制备化合物的各种合成步骤中同位素的并入效率。然而,如上文所阐述,全部此类同位素体的相对量将小于化合物的49.9%。在其他实施方案中,此类同位素体全部的相对量将为化合物的小于47.5%、小于40%、小于32.5%、小于25%、小于17.5%、小于10%、小于5%、小于3%、小于1%或小于0.5%。
[0153]
如本文所用,“任选地被取代的”与表述“取代或未取代的”可互换使用。通常,术语“取代的”,无论前面是否有术语“任选地”,都是指用特定取代基的基团置换给定结构中的氢基团。除非另外指示,否则“任选地被取代”的基团可在该基团的每个可取代位置处具有取代基,并且当任何给定结构中的多于一个位置可以被选自指定基团的多于一个取代基取代时,在每个位置处的取代基可以相同或不同。本公开所设想的取代基的组合为导致形成稳定或化学上可行的化合物的取代基的组合。
[0154]
术语“同位素体”是指其中化学结构与参考化合物仅在其同位素组成方面不同的物种。另外,除非另有说明,否则本文描述的结构还意在包括仅在一个或多个同位素富集原子的存在方面不同的化合物。例如,除了用氘或氚替换氢或用
13
c或
14
c替换碳之外,具有本发明结构的化合物都在本公开的范围内。
[0155]
除非另有说明,否则本文绘示的结构还意在包括结构的所有异构形式,例如外消旋混合物、顺式/反式异构体、几何(或构象)异构体,如(z)和(e)双键异构体,以及(z)和(e)构象异构体。因此,本发明化合物的几何和构象混合物在本公开的范围内。除非另有说明,否则本公开的化合物的所有互变异构形式都在本公开的范围内。
[0156]
如本文所用,术语“互变异构体”是指化合物的两种或更多种异构体中的一种,这些异构体以平衡状态一起存在,并易于通过原子例如氢原子或基团在分子内的迁移而互
换。
[0157]
如本文所用的“立体异构体”是指对映异构体和非对映异构体。
[0158]
如本文所用,“氘化衍生物”是指具有与参考化合物相同的化学结构但一个或多个氢原子被替换为氘原子(“d”或“2
h”)的化合物。应认识到,取决于合成中使用的化学材料的来源,合成的化合物中会出现天然同位素丰度的一些变化。尽管存在这种变化,但与本文描述的氘化衍生物的稳定同位素取代程度相比,天然丰富的稳定氢同位素的浓度小而无关紧要。因此,除非另有说明,否则当提及本公开的化合物的“氘化衍生物”时,至少一个氢被远高于其天然同位素丰度(通常为约0.015%)的氘取代。在一些实施方案中,本公开的氘化衍生物对于每个氘原子具有至少3500(在每个指定氘处52.5%氘掺入)、至少4500(67.5%氘掺入)、至少5000(75%氘掺入)、至少5500(82.5%氘掺入)、至少6000(90%氘掺入)、至少6333.3(95%氘掺入)、至少6466.7(97%氘掺入)或至少6600(99%氘掺入)的同位素富集因子。
[0159]
如本文所用,术语“同位素富集因子”意指指定同位素的同位素丰度与天然丰度之间的比率。
[0160]
如本文所用,术语“烷基”或“脂族”意指完全饱和的直链(即,线性或无支链)或支链的、取代或未取代的烃链。除非另有规定,否则烷基基团含有1至20个烷基碳原子。在一些实施方案中,烷基基团含有1至10个脂族碳原子。在一些实施方案中,烷基基团含有1至8个脂族碳原子。在一些实施方案中,烷基基团包含1至6个烷基碳原子,并且在一些实施方案中,烷基基团包含1至4个烷基碳原子。在其他实施方案中,烷基基团包含1至3个烷基碳原子,并且在其他实施方案中,烷基基团包含1至2个烷基碳原子。在一些实施方案中,烷基基团是取代的。在一些实施方案中,烷基基团是未取代的。在一些实施方案中,烷基是线性或直链或无支链的。在一些实施方案中,烷基基团是支链的。
[0161]
如本文所用,术语“环烷基”或“环状烷基”是指完全饱和的单环c
3-8
烃或螺环、稠合或桥连的双环或三环c
8-14
烃,其中所述双环环系统中的任何单个环具有3至7个成员。在一些实施方案中,环烷基基团是取代的。在一些实施方案中,环烷基基团是未取代的。在一些实施方案中,环烷基是c3至c
12
环烷基。在一些实施方案中,环烷基是c3至c8环烷基。在一些实施方案中,环烷基是c3至c6环烷基。单环环烷基的非限制性实例包括环丙基、环丁基、环戊基和环己基。
[0162]
如本文所用,术语“碳环基”或“脂环族”包括术语“环烷基”或“环状烷基”,并且是指单环c
3-8
烃或螺环、稠合或桥连的双环或三环c
8-14
烃,其是完全饱和的或部分饱和的,因为它含有一个或多个不饱和单元,但不是芳族的,其中所述双环系统中的任何单个环具有3至7个成员。双环碳环基包括与苯基稠合的单环碳环的组合。在一些实施方案中,碳环基基团是取代的。在一些实施方案中,碳环基基团是未取代的。在一些实施方案中,碳环基是c3至c
12
碳环基。在一些实施方案中,碳环基是c3至c
10
碳环基。在一些实施方案中,碳环基是c3至c8碳环基。
[0163]
如本文所用,术语“杂烷基”或“杂脂族”是指如上定义的烷基或脂族基团,其中一个或两个碳原子独立地被一个或多个氧、硫、氮、磷或硅置换。
[0164]
如本文所用,术语“烯基”意指含有一个或多个双键的直链(即直链或无支链)、支链、取代或未取代的烃链。在一些实施方案中,烯基基团是取代的。在一些实施方案中,烯基
基团是未取代的。在一些实施方案中,烯基基团是直链的。在一些实施方案中,烯基基团是支链的。
[0165]
如本文所用,术语“杂环”、“杂环基”、“杂环脂族”或“杂环”是指非芳族(即,完全饱和或部分饱和,因为它含有一个或多个不饱和单元,但不是芳族)的单环或螺环、稠合或桥连的双环或三环环系统,其中一个或多个环成员是独立选择的杂原子。双环杂环基包括下列单环的组合:与单环杂环基稠合的单环杂芳基;与另一个单环杂环基稠合的单环杂环基;与苯基稠合的单环杂环基;与单环碳环基/环烷基稠合的单环杂环基;以及与单环碳环基/环烷基稠合的单环杂芳基。
[0166]
在一些实施方案中,杂环包含被一个或多个氧代基团(例如,例如,c=o基团、s=o基团或so2基团)取代的环原子。
[0167]
在一些实施方案中,“杂环”、“杂环基”、“杂环脂族”或“杂环的”基团具有3至14个环成员,其中一个或多个环成员是独立地选自氧、硫、氮和磷的杂原子。在一些实施方案中,双环或三环环系统中的每个环含有3至7个环组成原子。在一些实施方案中,杂环具有至少一个不饱和碳-碳键。在一些实施方案中,杂环具有至少一个不饱和碳-氮键。在一些实施方案中,杂环具有一个独立地选自氧、硫、氮和磷的杂原子。在一些实施方案中,杂环具有一个为氮原子的杂原子。在一些实施方案中,杂环具有一个为氧原子的杂原子。在一些实施方案中,杂环具有两个各自独立地选自氮和氧的杂原子。在一些实施方案中,杂环具有三个各自独立地选自氮和氧的杂原子。在一些实施方案中,杂环是取代的。在一些实施方案中,杂环是未取代的。在一些实施方案中,杂环基是3至12元杂环基。在一些实施方案中,杂环基是3至10元杂环基。在一些实施方案中,杂环基是3至8元杂环基。在一些实施方案中,杂环基是5至10元杂环基。在一些实施方案中,杂环基是5至8元杂环基。在一些实施方案中,杂环基是5或6元杂环基。单环杂环基的非限制性实例包括哌啶基、哌嗪基、四氢吡喃基、氮杂环丁烷基、四氢噻吩基1,1-二氧化物等。
[0168]
术语“杂原子”意指氧、硫、氮、磷或硅中的一或多者(包括氮、硫、磷或硅的任何氧化形式;任何碱性氮的季铵化形式;或杂环的可取代氮,例如n(如3,4-二氢-2h-吡咯基中)、nh(如吡咯烷基中)或nr
+
(如n-取代的吡咯烷基中))。
[0169]
如本文所用的术语“不饱和”意指部分具有一个或多个不饱和单元或不饱和程度。不饱和是其中化合物中并非所有可用的价键都被取代基占完并因此化合物含有双键或三键的状态。
[0170]
如本文所用,术语“烷氧基”或“硫代烷基”是指如前所定义的烷基基团,其中烷基基团的一个碳分别被氧(“烷氧基”)或硫(“硫代烷基”)原子置换,前提条件是氧和硫原子连接在两个碳原子之间。烷氧基基团的非限制性实例包括甲氧基、乙氧基、甲基甲氧基等。“环状烷氧基”是指含有至少一个烷氧基基团但不为芳族的单环、螺环、双环、桥接双环、三环或桥接三环烃。环状烷氧基基团的非限制性实例包括四氢吡喃基、四氢呋喃基、氧杂环丁烷基、8-氧杂双环[3.2.1]辛烷基和氧杂环庚烷基。在一些实施方案中,“烷氧基”和/或“硫代烷基”基团是取代的。在一些实施方案中,“烷氧基”和/或“硫代烷基”基团是未取代的。
[0171]
如本文所用,术语“卤代烷基”、“卤代烯基”和“卤代烷氧基”分别是指被一个或多个卤素原子取代的直链或支链烷基、烯基或烷氧基。卤代烷基基团的非限制性实例包括-chf2、-ch2f、-cf3、-cf
2-,和全卤代烷基,诸如-cf2cf3。卤代烷氧基基团的非限制性实例包
括-ochf2、-och2f、-ocf3和-ocf
2-。
[0172]
术语“卤素”包括f、cl、br和i,即分别为氟、氯、溴和碘。
[0173]
术语“氨基烷基”意指被氨基基团所取代或含有氨基基团的烷基基团。
[0174]
如本文所用,“氨基”是指为伯胺、仲胺或叔胺的基团。
[0175]
如本文所用,“羰基”基团是指c=o。
[0176]
如本文所用,“氰基”或“腈”基团是指-c≡n。
[0177]
如本文所用,“羟基”基团是指-oh。
[0178]
如本文所用,“硫醇”基团是指-sh。
[0179]
如本文所用,“tert”和“t
‑”
各自是指叔。
[0180]
如本文所用,“芳族基团”或“芳族环”是指含有共轭的平面环系的化学基团,所述环系具有由[4n+2]个p轨道电子组成的离域π电子轨道,其中n为0至6的范围的整数。芳族基团的非限制性实例包括芳基和杂芳基基团。
[0181]
单独使用或作为较大部分(如在“芳基烷基”、“芳基烷氧基”或“芳氧基烷基”中)的一部分使用的术语“芳基”是指具有总共五至十四个环成员的单环或螺环、稠合或桥连双环或三环环系统,其中所述系统中的每个环是仅含碳原子的芳族环,并且其中双环或三环环系统中的每个环含有3至7个环成员。芳基的非限制性实例包括苯环(c6)和萘环(c
10
)。在一些实施方案中,芳基基团是取代的。在一些实施方案中,芳基基团是未取代的。
[0182]
单独使用或作为较大部分的一部分(如在“杂芳基烷基”或“杂芳基烷氧基”中)使用的术语“杂芳基”是指具有总共五至十四个环成员的单环或螺环、稠合或桥接的双环和三环环系统,其中所述系统中的至少一个环是芳族的,所述系统中的至少一个环含有一个或多个杂原子,并且其中双环或三环环系统中的每个环含有3至7个环成员。双环杂芳基包括下列单环的组合:与另一个单环杂芳基稠合的单环杂芳基;和与苯基稠合的单环杂芳基。在一些实施方案中,杂芳基基团是取代的。在一些实施方案中,杂芳基基团具有一个或多个选自氮、氧和硫的杂原子。在一些实施方案中,杂芳基基团具有一个杂原子。在一些实施方案中,杂芳基基团具有两个杂原子。在一些实施方案中,杂芳基基团为具有五个环组成原子的单环环系。在一些实施方案中,杂芳基基团为具有六个环组成原子的单环环系。在一些实施方案中,杂芳基基团是未取代的。在一些实施方案中,杂芳基是3至12元杂芳基。在一些实施方案中,杂芳基是3至10元杂芳基。在一些实施方案中,杂芳基是3至8元杂芳基。在一些实施方案中,杂芳基是5至10元杂芳基。在一些实施方案中,杂芳基是5至8元杂芳基。在一些实施方案中,杂芳基是5或6元杂芳基。单环杂芳基的非限制性实例是吡啶基、嘧啶基、噻吩基、噻唑基、异噁唑基等。
[0183]
在一些实施方案中,杂芳基包含被一个或多个氧代基团(例如,例如,c=o基团、s=o基团或so2基团)取代的环原子。说明性地,杂芳基基团的非限制性实例是苯并[d]噁唑-2(3h)-酮基团。
[0184]
含氮基团如胺基团的有用保护基的非限制性实例包括例如氨基甲酸叔丁酯(boc)、苄基(bn)、四氢吡喃基(thp)、氨基甲酸9-芴基甲酯(fmoc)、氨基甲酸苄酯(cbz)、乙酰胺、三氟乙酰胺、三苯基甲胺、亚苄基胺和对甲苯磺酰胺。添加(通常称为“保护”的过程)和去除(通常称为“去保护”的过程)此类胺保护基团的方法为本领域熟知的,并且可以在例如p.j.kocienski,protecting groups,thieme,1994中获得,其以引用的方式整体并入本
文,并且在greene和wuts,protective groups in organic synthesis,第3版(john wiley&sons,new york,1999)和第4版(john wiley&sons,new jersey,2014)中获得。
[0185]
可用于本公开的方法中的合适溶剂的非限制性实例包括但不限于水、甲醇(meoh)、乙醇(etoh)、二氯甲烷或“亚甲基氯”(ch2cl2)、甲苯、乙腈(mecn)、二甲基甲酰胺(dmf)、二甲基亚砜(dmso)、乙酸甲酯(meoac)、乙酸乙酯(etoac)、庚烷、乙酸异丙酯(ipac)、乙酸叔丁酯(t-buoac)、异丙醇(ipa)、四氢呋喃(thf)、2-甲基四氢呋喃(2-me thf)、甲基乙基酮(mek)、叔-丁醇、乙醚(et2o)、甲基叔丁基醚(mtbe)、1,4-二噁烷和n-甲基吡咯烷酮(nmp)。
[0186]
可用于本公开的方法中的合适的碱的非限制性实例包括但不限于1,8-二氮杂双环[5.4.0]十一碳-7-烯(dbu)、叔丁醇钾(kotbu)、碳酸钾(k2co3)、n-甲基吗啉(nmm)、三乙胺(et3n;tea)、二异丙基乙胺(i-pr2etn;dipea)、吡啶、氢氧化钾(koh)、氢氧化钠(naoh)、氢氧化锂(lioh)和甲醇钠(naome;naoch3)。
[0187]
本公开包括所公开的化合物的药学上可接受的盐。化合物的盐在酸与化合物的碱性基团如氨基官能团或者碱与化合物的酸性基团如羧基官能团之间形成。
[0188]
如本文所用,术语“药学上可接受的”是指在合理的医学判断范围内适合与人类和其他哺乳动物的组织接触而没有过度的毒性、刺激、过敏反应等并且与合理的收益/风险比相称的组分。“药学上可接受的盐”意指在施用于接受者时能够直接或间接提供本公开的化合物的任何无毒盐。合适的药学上可接受的盐有例如s.m.berge等人,j.pharmaceutical sciences,1977,66,1至19中所公开的那些。
[0189]
常用于形成药学上可接受的盐的酸包括无机酸如硫化氢(hydrogen bisulfide)、氢氯酸、氢溴酸、氢碘酸、硫酸和磷酸,以及有机酸如对甲苯磺酸、水杨酸、酒石酸、酸式酒石酸(bitartaric acid)、抗坏血酸、马来酸、苯磺酸(besylic acid)、富马酸、葡萄糖酸、葡糖醛酸、甲酸、谷氨酸、甲磺酸、乙磺酸、苯磺酸(benzenesulfonic acid)、乳酸、草酸、对-溴苯基磺酸、碳酸、琥珀酸、柠檬酸、苯甲酸和乙酸,以及相关的无机和有机酸。这样的药学上可接受的盐因此包括硫酸盐、焦硫酸盐、硫酸氢盐、亚硫酸盐、亚硫酸氢盐、磷酸盐、磷酸一氢盐、磷酸二氢盐、偏磷酸盐、焦磷酸盐、氯化物、溴化物、碘化物、醋酸盐、丙酸盐、癸酸盐、辛酸盐、丙烯酸盐、甲酸盐、异丁酸盐、癸酸盐、庚酸盐、丙炔酸盐、草酸盐、丙二酸盐、琥珀酸盐、辛二酸盐、癸二酸盐、富马酸盐、马来酸盐、丁炔-1,4-二酸盐、己炔-l,6-二酸盐、苯甲酸盐、氯苯甲酸盐、甲基苯甲酸盐、二硝基苯甲酸盐、羟基苯甲酸盐、甲氧基苯甲酸盐、邻苯二甲酸盐、对苯二甲酸盐、磺酸盐、二甲苯磺酸盐、苯乙酸盐、苯丙酸盐、苯丁酸盐、柠檬酸盐、乳酸盐、β-羟基丁酸盐、乙醇酸盐、马来酸盐、酒石酸盐、甲磺酸盐、丙磺酸盐、萘-1-磺酸盐、萘-2-磺酸盐、扁桃酸盐及其他盐。在一些实施方案中,药学上可接受的酸加成盐包括与矿物酸如氢氯酸和氢溴酸形成的盐以及与有机酸如马来酸形成的盐。
[0190]
衍生自适宜的碱的药学上可接受的盐包括碱金属盐、碱土金属盐、铵盐和n
+
(c
1-4
烷基)4盐。本公开还设想本文公开的化合物的任何碱性含氮基团的季铵化。碱金属和碱土金属盐的合适非限制性实例包括钠、锂、钾、钙和镁。药学上可接受的盐的其他非限制性实例包括铵、季铵以及使用抗衡离子(诸如卤离子、氢氧根、羧酸根、硫酸根、磷酸根、硝酸根、低碳数烷基磺酸根和芳基磺酸根)形成的胺阳离子。药学上可接受的盐的其他合适的非限制性实例包括苯磺酸盐和葡糖胺盐。
[0191]
术语“患者”和“受试者”在本文可互换使用并且指包括人的动物。
[0192]
术语“有效剂量”和“有效量”在本文中可互换使用并且是指产生其施用的期望效果(例如,改善fsgs和/或ndkd的症状、减轻fsgs和/ndkd的严重程度或fsgs和/或ndkd的症状、和/或减少fsgs和/或ndkd的进展或fsgs和/或ndkd的症状)的化合物的量。有效剂量的确切量将取决于治疗目的,并且将由本领域技术人员使用已知技术确定(参见例如lloyd(1999)the art,science and technology of pharmaceutical compounding)。
[0193]
如本文所用,术语“治疗”及其同源词是指减慢或停止疾病进展。如本文所用,“治疗”及其同源词包括但不限于以下:完全或部分缓解、降低肾衰竭(例如,esrd)以及疾病相关并发症(例如水肿、易感染或血栓栓塞事件)的风险。可以根据本领域已知或随后开发的方法和技术容易地评估这些症状中任一者的改善或其严重程度的减轻。
[0194]
当与组合物或剂型的成分的剂量、量或重量百分比结合使用时,术语“约”和“大约”包括本领域普通技术人员认为可提供与自指定剂量、量或重量百分比所获得的药理学作用等效的药理学作用的指定剂量、量或重量百分比的值或所述剂量、量或重量百分比的范围。
[0195]
至少一种选自以下的实体可以每天一次、每天两次或每天三次施用,例如用于治疗fsgs:式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如,式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。在一些实施方案中,式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如,式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)选自化合物1至391(例如,选自化合物1至220)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。在一些实施方案中,每天施用一次至少一种选自以下的实体:式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如选自式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。在一些实施方案中,每天施用一次至少一种选自化合物1至391(例如,化合物1至220)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐的实体。在一些实施方案中,每天施用两次至少一种选自以下的实体:式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、
iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如选自式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。在一些实施方案中,每天施用两次至少一种选自化合物1至391(例如,选自化合物1至220)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐的实体。在一些实施方案中,每天施用三次至少一种选自以下的实体:式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如选自式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。在一些实施方案中,每天施用三次至少一种选自化合物1至391(例如,选自化合物1至220)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐的实体。
[0196]
在一些实施方案中,每天一次、每天两次或每天三次施用2mg至1500mg或5mg至1000mg的至少一种选自以下的实体:式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如选自式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。在一些实施方案中,每天一次、每天两次或每天三次施用2mg至1500mg或5mg至1000mg的至少一种选自化合物1至391(例如,选自化合物1至220)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐的实体。
[0197]
本领域普通技术人员应认识到,在公开化合物的量时,相关的化合物的药学上可接受的盐形式的量为相当于化合物的游离碱的浓度的量。本文公开的化合物、药学上可接受的盐、溶剂化物和氘化衍生物的量基于参考化合物的游离碱形式。例如,“1000mg至少一种选自式i化合物及其药学上可接受的盐的化合物”包括1000mg式i化合物和相当于1000mg式i化合物的式i化合物的药学上可接受的盐的浓度。
[0198]
如本文所用,术语“环境条件”意指室温、露天条件和不受控制的湿度条件。
[0199]
如本文所用,术语“结晶形式”和“形式”可互换地指在晶格中具有特定分子堆积排列的晶体结构(或多晶型物)。结晶形式可以通过一种或多种表征技术,包括例如x射线粉末衍射(xrpd)、单晶x射线衍射、固态核磁共振(ssnmr)、差示扫描量热法(dsc)、红外辐射(ir)和/或热重分析(tga)来彼此鉴别和区分。因此,如本文所用,术语“化合物[x]的形式a”或“化合物[x]形式a”是指可以通过一种或多种表征技术与化合物i的其他结晶形式鉴别和区分的独特结晶形式,所述表征技术包括例如x射线粉末衍射(xrpd)、单晶x射线衍射、ssnmr、差示扫描量热法(dsc)、红外辐射(ir)和/或热重分析(tga)。
[0200]
如本文所用,术语“ssnmr”是指固态核磁共振的分析表征方法。ssnmr光谱可以在环境条件或替代条件下(例如,在275k下)对样品中存在的任何磁性活性同位素进行记录。
小分子活性药物成分的活性同位素的典型实例包括1h、2h、
13
c、
19
f、
31
p、
15
n、
14
n、
35
cl、
11
b、7li、
17
o、
23
na、
79
br和
195
pt。
[0201]
如本文所用,术语“xrpd”是指x-射线粉末衍射的分析表征方法。可以在环境条件下使用衍射仪以透射或反射几何学记录xrpd图案。
[0202]
如本文所用,术语“x射线粉末衍射图”、“x射线粉末衍射图案”和“xrpd图案”可互换地是指实验获得的绘制信号位置(在横坐标上)对信号强度(在纵坐标上)的图案。对于非晶形材料,x射线粉末衍射图可包括一个或多个宽信号;并且对于结晶材料,x射线粉末衍射图可包括一个或多个信号,每一个信号由其以度2θ(
°
2θ)量度的角度值鉴别,绘示在x射线粉末衍射图的横坐标上,其可表示为“在
……°
2θ处的信号”、“在
……
的2θ值处的信号”和/或“在至少
……
个选自
……
的2θ值处的信号”。
[0203]
如本文所用,“信号”或“峰”是指xrpd图案中以计数测量的强度处于局部最大值的点。本领域的普通技术人员将认识到,xrpd图案中的一个或多个信号(或峰)可重叠,并且可例如对肉眼不明显。实际上,本领域的普通技术人员将认识到,一些业内认可的方法能够并且适用于确定信号是否存在于图案中,诸如rietveld精修。
[0204]
如本文所用,“在
……°
2θ处的信号”、“在
……
的2θ值[]处的信号”和/或“在至少
……
个选自
……
的2θ值处的信号”是指如在x-射线粉末衍射实验中所测量和观察到的x-射线反射位置(
°
2θ)。
[0205]
角度值的可重复性在
±
0.2
°
2θ的范围内,即角度值可在所记载的角度值+0.2
°
2θ处、角度值-0.2
°
2θ处或在这两个端点(角度值+0.2
°
2θ和角度值-0.2
°
2θ)之间的任何值处。
[0206]
如本文所用,术语“信号强度”和“峰强度”可互换地指在给定x射线粉末衍射图内的相对信号强度。可影响相对信号或峰强度的因素包括样品厚度和优选定向(例如结晶颗粒不随机分布)。
[0207]
如本文所用,术语“dsc”是指差示扫描量热法的分析方法。
[0208]
如本文所用,术语“tga”是指热重量(或热解重量)分析的分析方法。
[0209]
如本文所用,“结晶水合物”是在晶格中包含化学计量或非化学计量的水的晶体形式。在非化学计量水合物的情况下,结晶水合物中存在的水的量可以至少作为相对湿度(“rh”)的函数而变化。水的存在(或不存在)或不同量的水可能导致x射线衍射图峰位偏移,或者导致峰的出现或消失。水的存在(或不存在)或不同量的水可能导致质子、碳、氟、磷、氮、氯(或其他nmr活性核)固态nmr光谱中的峰移动或甚至新峰的出现。
[0210]
化合物和组合物
[0211]
在一些实施方案中,本公开的至少一种实体是由以下结构式表示的化合物:
[0212][0213]
式i
[0214]
其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐,其中:
[0215]
x1选自s和-cr
2a
并且x2选自s和-cr
2b
,其中:
[0216]
x1和x2之一是s;
[0217]
当x1为s时,则x2为-cr
2b
;并且
[0218]
当x2是s时,则x1是-cr
2a
;
[0219]
r1选自卤素、氰基、c
1-c6烷基、c
1-c6烷氧基、c
3-c6环烷基和苯基,其中:
[0220]
r1的所述c
1-c6烷基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2和c
1-c4烷氧基;
[0221]
r1的所述c
1-c6烷氧基任选地被1至3个独立地选自卤素的基团取代;
[0222]
r1的所述c
3-c6环烷基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷基、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)和-c(=o)n(c
1-c4烷基)2;并且
[0223]
r1的所述苯基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷基、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)和-c(=o)n(c
1-c4烷基)2;
[0224]r2a
选自氢、卤素、氰基、-oh、=o和c
1-c6烷基,其中:
[0225]r2a
的所述c
1-c6烷基任选地被1至3个独立地选自卤素、氰基、-oh和c
1-c4烷氧基的基团取代;
[0226]r2b
选自氢、卤素、氰基、-oh、=o和c
1-c6烷基;
[0227]r3a
选自卤素、氰基、-oh、c
1-c6烷基和=o;其中:
[0228]r3a
的所述c
1-c6烷基任选地被1至3个独立地选自卤素、氰基和-oh的基团取代;
[0229]r3b
选自c
1-c2烷基和=o;其中:
[0230]r3b
的所述c
1-c2烷基任选地被1至3个独立地选自卤素、氰基和-oh的基团取代;
[0231]
当r
3a
选自卤素、氰基、oh、c
1-c6烷基时或当r
3b
选自c
1-c2烷基时,在每次出现时为单键;或者当r
3a
为=o时或当r
3b
为=o时,每次出现时为双键;
[0232]
r4选自c
1-c6烷基、-c(=o)o(c
1-c4烷基)、c
2-c6炔基和其中:
[0233]
r4的所述c
1-c6烷基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)、-c(=o)n(c
1-c4烷基)2、c
3-c6环烷基、5至10元杂环基、苯基和5至10元杂芳基;
[0234]
环a选自c
3-c
12
碳环基、3至12元杂环基、c6和c
10
芳基和5至10元杂芳基,其中环a任选地被1、2、3、4或5个ra基团取代;其中:
[0235]
ra在每次出现时独立地选自卤素、氰基、c
1-c6烷基、c
2-c6烯基、c
1-c6烷氧基、c
1-c6卤代烷基、c
1-c6卤代烯基、c
1-c6卤代烷氧基、-c(=o)nrhri、-nrhri、-nrhc(=o)rk、-nrhc(=o)ork、-nrhc(=o)nr
irj
、-nrhs(=o)
prk
、-ork、-oc(=o)rk、-oc(=o)ork、-oc(=o)nrhri、-[o(ch2)q]ro(c
1-c6烷基)、-s(=o)
prk
、-s(=o)
p
nrhri、-c(=o)ork、c
3-c
12
碳环基、3至12元杂环基、c6和c
10
芳基以及5至10元杂芳基;其中:
[0236]
ra的所述c
1-c6烷基、所述c
1-c6烷氧基和所述c
2-c6烯基各自任选地被1至3个独立地选自以下的基团取代:c6至c
10
芳基(任选地被1至3个rm基团取代:)、5至10元杂环基(任选地被1至3个rm基团取代)、5至10元杂芳基(任选地被1至3个rm基团取代)、氰基、-c(=o)rk、-c(=o)ork、-c(=o)nrhri、-nrhri、-nrhc(=o)rk、-nrhc(=o)ork、-nrhc(=o)nr
irj
、-nrhs(=o)
prk
、-ork、-oc(=o)rk、-oc(=o)ork、-oc(=o)nrhri、-s(=o)
prk
、-s(=o)
p
nrhri和c
3-c6碳环基(任选地被1至3个rm基团取代);
[0237]
ra的所述c
3-c
12
碳环基、所述3至12元杂环基、所述c6和c
10
芳基和所述5至10元杂芳基各自任选地被1至3个独立地选自以下的基团取代:卤素、氰基、c
1-c4烷基、-nrhri和-ork;其中:
[0238]
rh、ri和rj在每次出现时各自独立地选自氢、c
1-c4烷基、c6c
10
芳基和c
3-c6环烷基;其中:
[0239]
rh、ri和rj中任一者的所述c
1-c4烷基任选地被1至3个独立地选自卤素、氰基和-oh的基团取代;
[0240]rk
在每次出现时各自独立地选自氢、c
1-c4烷基、5至10元杂环基和c
3-c6碳环基;其中:
[0241]rk
中任一者的所述c
1-c4烷基任选地被1至3个独立地选自卤素、氰基和-oh的基团取代;
[0242]rm
在每次出现时独立地选自卤素、氰基、氧代、c
1-c6烷基、c
1-c6烷氧基、-s(=o)
prk
和-ork;其中:
[0243]rm
的所述c
1-c6烷基任选地被1至3个独立地选自卤素、氰基和-oh的基团取代;
[0244]
r5选自c
1-c6烷基、-c(=o)o(c
1-c4烷基)、c
3-c
12
碳环基、3至12元杂环基、c6和c
10
芳基以及5至10元杂芳基;其中:
[0245]
r5的所述c
1-c6烷基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)
和-c(=o)n(c
1-c4烷基)2;
[0246]
r5的所述c
3-c
12
碳环基、所述3至12元杂环基、所述c6和c
10
芳基以及所述5至10元杂芳基各自任选地被1至3个独立地选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)(任选地被-oh取代)、-n(c
1-c4烷基)2、c
1-c5烷基(任选地被-oh取代)、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)、-nhc(=o)(c
1-c4烷基)、-c(=o)(c
1-c4烷氧基)和-c(=o)n(c
1-c4烷基)2;
[0247]
k是选自0、1和2的整数;其中
[0248]
当r
3a
选自卤素、氰基、-oh和c
1-c6烷基时,k是1或2;并且
[0249]
当r
3a
为=o时,k为1;
[0250]
m是选自0、1和2的整数,其中:
[0251]
当r
3b
选自c
1-c2烷基时,m是1或2;并且
[0252]
当r
3b
为=o时,m为1;
[0253]
p是选自1和2的整数;并且
[0254]
q和r各自是选自1、2、3和4的整数。
[0255]
在某些实施方案中,本公开的化合物由以下结构式之一表示:
[0256][0257]
其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐,其中:
[0258]r2a
选自氢、卤素、氰基和c
1-c4烷基;其中:
[0259]r2a
的所述c
1-c4烷基任选地被1至3个独立地选自卤素、-oh和c
1-c2烷氧基的基团取代;
[0260]r2b
选自氢、卤素、氰基和c
1-c4烷基;并且
[0261]
k是选自0、1和2的整数;
[0262]
并且本文未具体定义的所有其他变量如前述实施方案所定义。
[0263]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受
的盐中,r4选自c
1-c4烷基和其中:
[0264]
r4的所述c
1-c4烷基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c2烷氧基、c
3-c6环烷基、5至6元杂环基、苯基和5至6元杂芳基;
[0265]
并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0266]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受的盐中,r4选自c
1-c2烷基和其中:
[0267]
r4的所述c
1-c2烷基任选地被1至3个独立地选自卤素、氰基、-oh和5至6元杂环基的基团取代;
[0268]
并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0269]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受的盐中,r4选自-ch3、-ch2oh和(四氢-2h-吡喃-4-基)甲基;并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0270]
在某些实施方案中,本公开的化合物由以下结构式之一表示:
[0271][0272]
其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐,其中:
[0273]
环a在每次出现时选自c
3-c6环烷基、5至10元杂环基、苯基和5至10元杂芳基;其各自任选地被1、2、3、4或5个ra基团取代;
[0274]
并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0275]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受的盐中,环a选自环丙基、5至10元杂环基、苯基和5至9元杂芳基;其各自任选地被1、2、3、4或5个ra基团取代;并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0276]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受的盐中,环a选自环丙基、含有1至3个选自n和o的杂原子的5至10元杂环基、苯基和含有1至3个选自n和o的杂原子的5至9元杂芳基;其各自任选地被1、2、3、4或5个ra基团取代;并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0277]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受的盐中,环a选自环丙基、含有1至3个选自n和o的杂原子的5元杂环基、含有1至3个选自n和o的杂原子的6元杂环基、含有1至3个选自n和o的杂原子的9元杂环基、含有1至3个选自n和o的杂原子的10元杂环基、苯基、含有1至3个选自n和o的杂原子的5元杂芳基、含有1至3个选自n和o的杂原子的6元杂芳基以及含有1至3个选自n和o的杂原子的9元杂芳基;其各自任选地被1、2、3、4或5个ra基团取代;并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0278]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受的盐中,环a选自的盐中,环a选自
[0279]
其各自任选地被1、2、3、4或5个ra基团取代;并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0280]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受的盐中,环a选自的盐中,环a选自
其各自任选地被1、2、3、4或5个ra基团取代;并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0281]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受的盐中,r4选自-ch3和环a;其中环a选自和环a;其中环a选自和环a;其中环a选自其各自任选地被1、2、3、4或5个ra基团取代;并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0282]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受的盐中,r5选自c
1-c4烷基、-c(=o)-o(c
1-c2烷基)、c
3-c6环烷基和5至10元环烷基,其中:
[0283]
r5的所述c
1-c4烷基任选地被1至3个独立地选自卤素、氰基、-oh和c
1-c2烷氧基的基团取代;并且
[0284]
r5的所述c
3-c6环烷基和5至10元杂环基各自任选地被1至3个独立地选自卤素、氰基、-oh、c
1-c2烷基和c
1-c2烷氧基的基团取代;
[0285]
并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0286]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受的盐中,r5选自c
1-c2烷基、c(=o)o(c
1-c2烷基)、环丙基、环丁基和5至6元杂环基;其中:
[0287]
r5的所述c
1-c2烷基任选地被1至3个独立地选自f、cl、br、氰基、-oh和c
1-c2烷氧基的基团取代;并且
[0288]
r5的所述环丙基、所述环丁基和所述5至6元杂环基各自任选地被1至3个独立地选自f、cl、br、氰基、-oh、c
1-c2烷基和c
1-c2烷氧基的基团取代;
[0289]
并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0290]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受的盐中,r5选自-ch3、-ch2ch3、-ch2oh、-c(=o)och3、-ch2och3、-ch(ch3)2、环丙基、二氟环丙基和四氢-2h-吡喃基;并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0291]
在某些实施方案中,本公开的化合物由以下结构式之一表示:
[0292][0293]
其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐;并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0294]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受的盐中,r1选自氢、卤素、氰基、-oh、c
1-c4烷基、c
1-c4烷氧基和c
3-c6环烷基;其中:
[0295]
r1的所述c
1-c4烷基任选地被1至3个独立地选自卤素、氰基、-oh和c
1-c2烷氧基的基团取代;
[0296]
r1的所述c
1-c4烷氧基任选地被1至3个独立选择的卤素基团取代;并且
[0297]
r1的所述c
3-c6环烷基任选地被1至3个独立地选自卤素、氰基、-oh和c
1-c2烷氧基的基团取代;
[0298]
并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0299]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受的盐中,r1选自f、cl、br、c
1-c4烷基和c
3-c6环烷基;其中:
[0300]
r1的所述c
1-c4烷基任选地被1至3个独立地选自卤素和-oh的基团取代;并且
[0301]
r1的所述c
3-c6环烷基任选地被1至3个独立地选自卤素和-oh的基团取代;
[0302]
并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0303]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受的盐中,r1选自f、cl、br、c
1-c4烷基和c
3-c6环烷基;其中:
[0304]
r1的所述c
1-c4烷基任选地被1至3个独立地选自卤素和-oh的基团取代;并且
[0305]
r1的所述c
3-c6环烷基任选地被1至3个独立地选自卤素和-oh的基团取代;
[0306]
并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0307]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受的盐中,r1选自cl、br、-ch3、-cf3、-ch2ch3、-ch(ch3)2、-ch2chf2、-ch2ch(ch3)2、二氟环丁基和环己基;并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0308]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受的盐中,r1为cl;并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0309]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受的盐中,r
3a
选自卤素、-oh和c
1-c4烷基;其中:
[0310]r3a
的所述c
1-c4烷基任选地被1至3个独立地选自卤素和-oh的基团取代;
[0311]
并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0312]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受的盐中,r
3a
选自f、cl、br、-oh和c
1-c2烷基;其中:
[0313]r3a
的所述c
1-c2烷基任选地被1至3个独立地选自f、cl和-oh的基团取代;
[0314]
并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0315]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受的盐中,r
3a
选自f、-oh、-ch3、-chf2和-ch2oh;并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0316]
在某些实施方案中,本公开的化合物由以下结构式之一表示:
[0317][0318]
其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐;并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0319]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受的盐中,ra每次出现时独立地选自卤素、氰基、c
1-c6烷基、c
1-c4烷氧基、c
1-c6卤代烷基、c
1-c6卤代烷氧基、-c(=o)nrhri、-nrhri、-nrhc(=o)rk、-ork、-[o(ch2)q]ro(c
1-c6烷基)、-s(=o)2rk、-s(=o)2nrhri、c
3-c6环烷基、5至10元杂环基、苯基和5至8元杂芳基;其中:
[0320]
ra的所述c
1-c6烷基任选地被1至3个独立地选自以下的基团取代:氰基、-c(=o)
nrhri、-nrhri、-nrhc(=o)rk、-nrhc(=o)ork、-nrhc(=o)nr
irj
、-nrhs(=o)
prk
、-ork、-s(=o)2rk、-s(=o)
p
nrhri和c
3-c6环烷基;
[0321]
ra的所述c
3-c6环烷基、5至10元杂环基、苯基和5至8元杂芳基各自任选地被1至3个独立地选自卤素、c
1-c2烷基和-ork的基团取代;其中:
[0322]
rh、ri和rj在每次出现时各自独立地选自氢、c
1-c2烷基、环丙基和环丁基;其中:
[0323]
rh、ri和rj中任一者的所述c
1-c2烷基任选地被1至3个独立地选自卤素和-oh的基团取代;
[0324]rk
在每次出现时各自独立地选自氢和c
1-c4烷基;其中:
[0325]rk
的所述c
1-c4烷基任选地被1至3个独立地选自卤素和-oh的基团取代;并且
[0326]
q和r各自是选自1、2和3的整数;
[0327]
并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0328]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受的盐中,ra在每次出现时独立地选自卤素、氰基、c
1-c6烷基、c
1-c4烷氧基、c
1-c4卤代烷基、c
1-c4卤代烷氧基、-c(=o)nrhri、-nrhri、-nrhc(=o)rk、-ork、-[o(ch2)q]ro(c
1-c4烷基)、-s(=o)2rk、-s(=o)2nrhri、环丙基、环丁基、5至6元杂环基、苯基和5至6元杂芳基;其中:
[0329]
ra的所述c
1-c6烷基任选地被1至3个独立地选自氰基、-c(=o)nrhri、-nrhri、-ork、环丙基和环丁基的基团取代;
[0330]
ra的环丙基、环丁基、5至6元杂环基、苯基和5至6元杂芳基各自任选地被1至3个独立地选自卤素、-ch3、-oh和-och3的基团取代;其中:
[0331]
rh和ri在每次出现时各自独立地选自氢、-ch3、环丙基和环丁基;其中:
[0332]
rh和ri中任一者的所述-ch3任选地被1至3个独立地选自f、cl和-oh的基团取代;
[0333]rk
在每次出现时各自独立地选自氢和-ch3;其中:
[0334]rk
的所述-ch3任选地被1至3个独立地选自卤素和-oh的基团取代;
[0335]
并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0336]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受的盐中,ra在每次出现时独立地选自f、cl、br、氰基、c
1-c6烷基、c
1-c2烷氧基、c
1-c2卤代烷基、-c(=o)nrhri、-nrhri、-nrhc(=o)rk、-ork、-[o(ch2)q]ro(c
1-c2烷基)、-s(=o)2rk、-s(=o)2nrhri、环丙基、环丁基、5元杂环基、苯基和6元杂芳基;其中:
[0337]
ra的所述c
1-c6烷基任选地被1至3个独立地选自氰基、-c(=o)nrhri、-ork和环丙基的基团取代;
[0338]
ra的所述环丙基、所述环丁基、所述5至6元杂环基、所述苯基和所述5至6元杂芳基各自任选地被1至3个独立地选自卤素、-ch3、-oh和-och3的基团取代;其中:
[0339]
rh和ri在每次出现时各自独立地选自氢、-ch3和环丙基;其中:
[0340]
rh和ri中任一者的所述-ch3任选地被1至3个独立地选自f、cl和-oh的基团取代;
[0341]rk
在每次出现时各自独立地选自氢和-ch3;并且
[0342]
q和r各自是选自1和2的整数;
[0343]
并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0344]
在某些实施方案中,在本公开的化合物、互变异构体、氘化衍生物或药学上可接受的盐中,ra在每次出现时独立地选自f、氰基、-oh、-ch3、-cf3、-ch(ch3)2、-(ch2)2oh、-(ch2)2och3、-ch2ch(oh)c2h5、-ch2c(ch3)(ch2oh)2、-och3、-och2ch3、-[o(ch2)2]2och3、-ch2c(=o)nhch3、-(ch2)2so2ch3、-ch2c(=o)n(ch3)2、-ch2(环丙基)、-c(=o)nh2、-c(=o)nh(环丙基)、-nh2、-nhch3、-n(ch3)2、-nhc(ch3)2ch2oh、-nhc(=o)ch3、-so2ch3、-so2nh2、环丙基、2-甲氧基苯基、n-甲基哌嗪基、四氢-2h-吡喃基、甲基吡唑基、吡啶基和四氢噻吩1,1-二氧化物;并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0345]
在某些实施方案中,本公开的化合物由以下结构式之一表示:
[0346]
[0347][0348]
其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐;并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0349]
在某些实施方案中,本公开的化合物由以下结构式之一表示:
[0350]
[0351][0352]
其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐;并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0353]
在某些实施方案中,本公开的化合物由以下结构式之一表示:
[0354]
[0355][0356]
其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐;并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0357]
在某些实施方案中,本发明的至少一种化合物选自表i中所示的化合物1至220、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。表i中的化合物中的波浪线(即,)绘示两个原子之间的键并指示分子集合如外消旋混合物、顺式/反式异构体或(e)/(z)异构体的混合立体化学的位置。在表i的化合物中,与原子相邻的星号(例如,)表示分子中的手性位置。
[0358]
在某些实施方案中,本发明的至少一种化合物选自表ii中描绘的化合物221至391、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。表ii中的化合物中的波浪线(即,)绘示两个原子之间的键并指示分子集合如外消旋混合物、顺式/反式异构体或(e)/(z)异构体的混合立体化学的位置。在表ii的化合物中,与原子相邻的星号(例如,)表示分子中的手性位置。
[0359]
在某些实施方案中,本发明的至少一种化合物选自表i或ii中描绘的化合物1至
391、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。
[0360]
在某些实施方案中,本发明的至少一种化合物选自表iii中描绘的化合物、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。表iii中的化合物中的波浪线(即,)绘示两个原子之间的键并指示分子集合如外消旋混合物、顺式/反式异构体或(e)/(z)异构体的混合立体化学的位置。在表iii的化合物中,与原子相邻的星号(例如,)表示分子中的手性位置。
[0361]
表i.化合物1至220
[0362]
[0363]
[0364]
[0365]
[0366]
[0367]
[0368]
[0369]
[0370]
[0371]
[0372]
[0373][0374]
表ii.化合物221至391
[0375]
[0376]
[0377]
[0378]
[0379]
[0380]
[0381]
[0382]
[0383]
[0384]
[0385][0386]
表iii.表i和表ii中某些化合物的推断立体化学
[0387]
[0388]
[0389]
[0390]
[0391]
[0392]
[0393]
[0394]
[0395]
[0396]
[0397]
[0398]
[0399][0400]
*使用外消旋起始材料,所描绘的立体化学是相对的,为2种对映异构体的混合物
[0401]13:2混合物
[0402]24.5:1混合物
[0403]32:1混合物
[0404]43:1混合物
[0405]55:1混合物
[0406]63.5:1混合物
[0407]72:1混合物
[0408]
本公开的一些实施方案包括化合物1至391(例如化合物1至220)或式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如,式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)的衍生物、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。在一些实施方案中,所述衍生物是硅衍生物,其中选自以下的化合物中的至少一个碳原子已被硅置换:化合物1至391(例如例如,选自化合物1至220)或式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、
iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如,式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。在一些实施方案中,所述衍生物是硼衍生物,其中选自以下的化合物中的至少一个碳原子已被硼置换:化合物1至391(例如例如,选自化合物1至220)或式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如,式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。在其他实施方案中,所述衍生物是磷衍生物,其中选自以下的化合物中的至少一个碳原子已被磷置换:化合物1至391(例如例如,化合物1至220)或式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如,式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。
[0409]
在一些实施方案中,所述衍生物是硅衍生物,其中选自以下的化合物中的至少一个碳原子已被硅或硅衍生物(例如-si(ch3)
2-或-si(oh)
2-)置换:化合物1至391(例如例如,化合物1至220)或式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如,式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)、其互变异构体、所述化合物或互变异构体的氘化衍生物或上述任何一种的药学上可接受的盐。被硅置换的碳可以是非芳香族碳。在其他实施方案中,氟被硅衍生物(例如,si(ch3)3)置换。在一些实施方案中,本公开的硅衍生物可以包含一个或多个被氘置换的氢原子。在一些实施方案中,选自以下的化合物的硅衍生物可以具有结合到杂环中的硅:化合物1至391(例如例如,化合物1至220)或式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如,式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。
[0410]
在一些实施方案中,所述衍生物是硼衍生物,其中选自以下的化合物中的一个碳原子已被硼或硼衍生物置换:化合物1至391(例如例如,化合物1至220)或式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、
iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如,式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。
[0411]
在一些实施方案中,所述衍生物是磷衍生物,其中选自以下的化合物中的一个碳原子已被磷或磷衍生物置换:化合物1至391(例如例如,化合物1至220)或式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如,式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。
[0412]
本公开的另一方面提供了药物组合物,其包含至少一种根据选自式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的任一个式的化合物(例如,式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)和化合物1至391(例如,化合物1至220)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。在一些实施方案中,所述药物组合物包含至少一种选自以下的化合物:式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0(例如,式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)和化合物1至391(例如,从化合物1至220)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。
[0413]
药物组合物可以还包含至少一种药学上可接受的载体。在一些实施方案中,至少一种药学上可接受的载体选自药学上可接受的媒介物和药学上可接受的佐剂。在一些实施方案中,至少一种药学上可接受的选自药学上可接受的填充剂、崩解剂、表面活性剂、粘合剂和润滑剂。
[0414]
还应理解,本公开的药物组合物可在联合疗法中采用;即,本文描述的药物组合物还可包含至少一种另外的活性治疗剂。或者,药物组合物包含至少一种选自以下的化合物可以作为单独的组合物与包含至少一种其他活性治疗剂的组合物同时、在其之前或之后施用:式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如,式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。在一些实施方案
中,包含至少一种选自化合物1至391(例如,选自化合物1至220)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐的化合物的药物组合物可以作为单独的组合物与包含至少一种其他活性治疗剂的组合物同时、在其之前或之后施用。
[0415]
如上所述,本文公开的药物组合物可以任选地还包含至少一种药学上可接受的载体。至少一种药学上可接受的载体可选自佐剂和媒介物。如本文所使用,至少一种药学上可接受的载体包括适合于所需的特定剂型的任何和所有溶剂、稀释剂、其它液体媒介物、分散助剂、悬浮助剂、表面活性剂、等渗剂、增稠剂、乳化剂、防腐剂、固体粘合剂和润滑剂。remington:the science and practice of pharmacy,第21版,2005,d.b.troy编辑,lippincott williams&wilkins,philadelphia和encyclopedia of pharmaceutical technology,j.swarbrick和j.c.boylan编辑,1988至1999,marcel dekker,new york公开了用于配制药物组合物的各种载体和用于制备其的已知技术。除非任何常规载体与本公开的化合物不相容,诸如通过产生任何不合需要的生物作用或另外以有害方式与药物组合物的任何其他组分相互作用,否则预期其使用在本公开的范围内。合适的药学上可接受的载体的非限制性实例包括但不限于离子交换剂、氧化铝、硬脂酸铝、卵磷脂、血清蛋白(例如,例如、人血清白蛋白)、缓冲物质(例如,例如、磷酸盐、甘氨酸、山梨酸和山梨酸钾)、饱和植物脂肪酸、水、盐和电解质的偏甘油酯混合物(例如,例如,硫酸鱼精蛋白、磷酸氢二钠、磷酸氢钾、氯化钠和锌盐)、胶体二氧化硅、三硅酸镁、聚乙烯吡咯烷酮、聚丙烯酸酯、蜡、聚乙烯-聚氧化丙烯嵌段聚合物、羊毛脂、糖(例如,例如乳糖、葡萄糖和蔗糖)、淀粉(例如,例如玉米淀粉和马铃薯淀粉)、纤维素及其衍生物(例如,例如羧甲基纤维素钠、乙基纤维素和醋酸纤维素)、粉状黄蓍胶、麦芽、明胶、滑石、赋形剂(例如,例如可可脂和栓剂蜡)、油(诸如,例如花生油、棉籽油、红花油、芝麻油、橄榄油、玉米油和大豆油)、二醇(例如,例如,丙二醇和聚乙二醇)、酯(例如例如,油酸乙酯和月桂酸乙酯)、琼脂、缓冲剂(例如例如氢氧化镁和氢氧化铝)、海藻酸、无热原水、等渗盐水、林格氏溶液、乙醇、磷酸盐缓冲溶液、无毒相容性润滑剂(例如例如月桂基硫酸钠和硬脂酸镁)、着色剂、脱模剂、包衣剂、甜味剂、调味剂、芳香剂、防腐剂和抗氧化剂。
[0416]
在本公开的一些实施方案中,本文所述的化合物和药物组合物用于治疗fsgs和/或ndkd。在一些实施方案中,fsgs由apol1介导。在一些实施方案中,ndkd由apol1介导。
[0417]
在一些实施方案中,本公开的方法包括向有需要的患者施用至少一种选自以下的实体:式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如,式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。在一些实施方案中,式i化合物选自化合物1至391(例如,选自化合物1至220)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。在一些实施方案中,所述有需要的患者具有apol1遗传变体,即g1:s342g:i384m和g2:n388del:y389del。
[0418]
本公开的另一个方面提供了抑制apol1活性的方法,其包括使所述apol1与至少一
种选自以下的实体接触:式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物(例如,式i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐。在一些实施方案中,抑制apol1活性的方法包括使所述apol1与至少一种选自化合物1至391(例如,选自化合物1至220)、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐的实体接触。
[0419]
非限制性示例性实施方案1
[0420]
在没有限制的情况下,本公开的一些实施方案包括:
[0421]
1.一种由以下结构式表示的化合物:
[0422][0423]
式i
[0424]
其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐,其中:
[0425]
x1选自s和-cr
2a
并且x2选自s和-cr
2b
,其中:
[0426]
x1和x2之一是s;
[0427]
当x1为s时,则x2为-cr
2b
;并且
[0428]
当x2是s时,则x1是-cr
2a
;
[0429]
r1选自氢、卤素、氰基、-oh、c
1-c6烷基、c
1-c6烷氧基、c
3-c6环烷基和苯基,其中:
[0430]
r1的所述c
1-c6烷基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2和c
1-c4烷氧基;
[0431]
r1的所述c
1-c6烷氧基任选地被1至3个独立地选自卤素的基团取代;
[0432]
r1的所述c
3-c6环烷基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷基、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)和-c(=o)n(c
1-c4烷基)2;并且
[0433]
r1的所述苯基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷基、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)和-c(=o)n(c
1-c4烷基)2;
[0434]r2a
选自氢、卤素、氰基、-oh、=o和c
1-c6烷基,其中:
[0435]r2a
的所述c
1-c6烷基任选地被1至3个独立地选自卤素、氰基、-oh和c
1-c4烷氧基的基团取代;
[0436]r2b
选自氢、卤素、氰基、-oh、=o和c
1-c6烷基;
[0437]r3a
选自卤素、氰基、-oh、c
1-c6烷基和=o;其中:
[0438]r3a
的所述c
1-c6烷基任选地被1至3个独立地选自卤素、氰基和-oh的基团取代;
[0439]r3b
选自c
1-c2烷基和=o;其中:
[0440]r3b
的所述c
1-c2烷基任选地被1至3个独立地选自卤素、氰基和-oh的基团取代;
[0441]
当r
3a
选自卤素、氰基、oh、c
1-c6烷基时或当r
3b
选自c
1-c2烷基时,在每次出现时为单键;或者当r
3a
为=o时或当r
3b
为=o时,每次出现时为双键;
[0442]
r4选自c
1-c6烷基、-c(=o)o(c
1-c4烷基)、c
2-c6炔基和其中:
[0443]
r4的所述c
1-c6烷基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)、-c(=o)n(c
1-c4烷基)2、c
3-c6环烷基、5至10元杂环基、苯基和5至10元杂芳基;
[0444]
环a选自c
3-c
12
碳环基、3至12元杂环基、c6和c
10
芳基和5至10元杂芳基,其中环a任选地被1、2、3、4或5个ra基团取代;其中:
[0445]
ra在每次出现时独立地选自卤素、氰基、c
1-c6烷基、c
2-c6烯基、c
1-c6烷氧基、c
1-c6卤代烷基、c
1-c6卤代烯基、c
1-c6卤代烷氧基、-c(=o)nrhri、-nrhri、-nrhc(=o)rk、-nrhc(=o)ork、-nrhc(=o)nr
irj
、-nrhs(=o)
prk、-ork、-oc(=o)rk、-oc(=o)ork、-oc(=o)nrhri、-[o(ch2)q]ro(c
1-c6烷基)、-s(=o)
prk
、-s(=o)
p
nrhri、-c(=o)ork、c
3-c
12
碳环基、3至12元杂环基、c6和c
10
芳基以及5至10元杂芳基;其中:
[0446]
ra的所述c
1-c6烷基、所述c
1-c6烷氧基和所述c
2-c6烯基各自任选地被1至3个独立地选自以下的基团取代:c6至c
10
芳基(任选地被1至3个rm基团取代:)、5至10元杂环基(任选地被1至3个rm基团取代)、5至10元杂芳基(任选地被1至3个rm基团取代)、氰基、-c(=o)rk、-c(=o)ork、-c(=o)nrhri、-nrhri、-nrhc(=o)rk、-nrhc(=o)ork、-nrhc(=o)nr
irj
、-nrhs(=o)
prk
、-ork、-oc(=o)rk、-oc(=o)ork、-oc(=o)nrhri、-s(=o)
prk
、-s(=o)
p
nrhri和c
3-c6碳环基(任选地被1至3个rm基团取代);
[0447]
ra的所述c
3-c
12
碳环基、所述3至12元杂环基、所述c6和c
10
芳基和所述5至10元杂芳基各自任选地被1至3个独立地选自以下的基团取代:卤素、氰基、c
1-c4烷基、-nrhri和-ork;其中:
[0448]
rh、ri和rj在每次出现时各自独立地选自氢、c
1-c4烷基、c6c
10
芳基和c
3-c6环烷基;其中:
[0449]
rh、ri和rj中任一者的所述c
1-c4烷基任选地被1至3个独立地选自卤素、氰基和-oh的基团取代;
[0450]rk
在每次出现时各自独立地选自氢、c
1-c4烷基、5至10元杂环基和c
3-c6碳环基;其
中:
[0451]rk
中任一者的所述c
1-c4烷基任选地被1至3个独立地选自卤素、氰基和-oh的基团取代;
[0452]rm
在每次出现时独立地选自卤素、氰基、氧代、c
1-c6烷基、c
1-c6烷氧基、-s(=o)
prk
和-ork;其中:
[0453]rm
的所述c
1-c6烷基任选地被1至3个独立地选自卤素、氰基和-oh的基团取代;
[0454]
r5选自c
1-c6烷基、-c(=o)o(c
1-c4烷基)、c
3-c
12
碳环基、3至12元杂环基、c6和c
10
芳基以及5至10元杂芳基;其中:
[0455]
r5的所述c
1-c6烷基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)和-c(=o)n(c
1-c4烷基)2;
[0456]
r5的所述c
3-c
12
碳环基、所述3至12元杂环基、所述c6和c
10
芳基以及所述5至10元杂芳基各自任选地被1至3个独立地选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)(任选地被-oh取代)、-n(c
1-c4烷基)2、c
1-c5烷基(任选地被-oh取代)、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)、-nhc(=o)(c
1-c4烷基)、-c(=o)(c
1-c4烷氧基)和-c(=o)n(c
1-c4烷基)2;
[0457]
k是选自0、1和2的整数;其中
[0458]
当r
3a
选自卤素、氰基、-oh和c
1-c6烷基时,k是1或2;并且
[0459]
当r
3a
为=o时,k为1;
[0460]
m是选自0、1和2的整数,其中:
[0461]
当r
3b
选自c
1-c2烷基时,m是1或2;并且
[0462]
当r
3b
为=o时,m为1;
[0463]
p是选自1和2的整数;并且
[0464]
q和r各自是选自1、2、3和4的整数。
[0465]
2.根据实施方案1所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其所述化合物由以下结构式之一表示:
[0466][0467]
或其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐,其中:
[0468]r2a
选自氢、卤素、氰基和c
1-c4烷基;其中:
[0469]r2a
的所述c
1-c4烷基任选地被1至3个独立地选自卤素、-oh和c
1-c2烷氧基的基团取代;
[0470]r2b
选自氢、卤素、氰基和c
1-c4烷基;并且
[0471]
k是选自0、1和2的整数;
[0472]
并且本文未具体定义的所有其他变量如实施方案1中所定义。
[0473]
3.根据实施方案1或实施方案2所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中r4选自c
1-c4烷基和其中:
[0474]
r4的所述c
1-c4烷基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c2烷氧基、c
3-c6环烷基、5至6元杂环基、苯基和5至6元杂芳基;
[0475]
并且本文未具体定义的所有其他变量如实施方案1或实施方案2中所定义。
[0476]
4.根据实施方案1至3中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中r4选自c
1-c2烷基和其中:
[0477]
r4的c
1-c2烷基任选地被1至3个独立地选自卤素、氰基、-oh和5至6元杂环基的基团取代;
[0478]
并且本文未具体定义的所有其他变量如实施方案1至3中的任一项所定义。
[0479]
5.根据实施方案1至4中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中r4选自-ch3、-ch2oh和(四氢-2h-吡喃-4-基)甲基;并且本文未具体定义的所有其他变量如实施方案1至4中任一项中所定义。
[0480]
6.根据实施方案1至4中任一项所述的化合物,其中所述化合物由以下结构式之一表示:
[0481][0482]
其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐,其中:
[0483]
环a在每次出现时选自c
3-c6环烷基、5至10元杂环基、苯基和5至10元杂芳基;其各自任选地被1、2、3、4或5个ra基团取代;
[0484]
并且本文未具体定义的所有其他变量如实施方案1至5中的任一项所定义。
[0485]
7.根据实施方案1至4和6中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中环a选自环丙基、5至10元杂环基、苯基和5至9元杂芳基;其各自任选地被1、2、3、4或5个ra基团取代;并且本文未具体定义的所有其他变量如实施方案1至6中任一项中所定义。
[0486]
8.根据实施方案1至4、6和7中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中环a选自环丙基、含有1至3个选自n和o的杂原子的5至10元杂环基、苯基和含有1至3个选自n和o的杂原子的5至9元杂芳基;其各自任选地被1、2、3、4或5个ra基团取代;并且本文未具体定义的所有其他变量如实施方案1至7中任一项中所定义。
[0487]
9.根据实施方案1至4和6至8中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中环a选自环丙基、含有1至3个选自n和o的杂原子的5元杂环基、含有1至3个选自n和o的杂原子的6元杂环基、含有1至3个选自n和o的杂原子的9元杂环基、含有1至3个选自n和o的杂原子的10元杂环基、苯基、含有1至3个选自n和o的杂原子的5元杂芳基、含有1至3个选自n和o的杂原子的6元杂芳基和含有1至3个选自n和o的杂原子的9元杂芳基;其各自任选地被1、2、3、4或5个ra基团取代;并且本文未具体定义的所有其他变量如实施方案1至8中任一项中所定义。
[0488]
10.根据实施方案1至4和6至9中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中环a选自
其各自任选地被1、2、3、4或5个ra基团取代;并且本文未具体定义的所有其他变量如实施方案1至9中任一项中所定义。
[0489]
11.根据实施方案1至4和6至10中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中环a选自
其各自任选地被1、2、3、4或5个ra基团取代;并且本文未具体定义的所有其他变量如实施方案1至4和6至10中任一项中所定义。
[0490]
12.根据实施方案1至4和6至11中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中r4选自-ch3和环a;其中环a选自其各自任选地被1、2、3、4或5个ra基团取代;并且本文未具体定义的所有其他变量如实施方案1至4和6至11中任一项中所定义。
[0491]
13.根据实施方案1至12中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中r5选自c
1-c4烷基、c(=o)o(c
1-c2烷基)、c
3-c6环烷基和5至10元环烷基;其中:
[0492]
r5的所述c
1-c4烷基任选地被1至3个独立地选自卤素、氰基、-oh和c
1-c2烷氧基的基团取代;并且
[0493]
r5的所述c
3-c6环烷基和5至10元杂环基各自任选地被1至3个独立地选自卤素、氰基、-oh、c
1-c2烷基和c
1-c2烷氧基的基团取代;
[0494]
并且本文未具体定义的所有其他变量如实施方案1至12中的任一项所定义。
[0495]
14.根据实施方案1至13中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中r5选自c
1-c2烷基、c(=o)o(c
1-c2烷基)、环丙基、环丁基和5至6元杂环基;其中:
[0496]
r5的所述c
1-c2烷基任选地被1至3个独立地选自f、cl、br、氰基、-oh和c
1-c2烷氧基的基团取代;并且
[0497]
r5的所述环丙基、所述环丁基和所述5至6元杂环基各自任选地被1至3个独立地选自f、cl、br、氰基、-oh、c
1-c2烷基和c
1-c2烷氧基的基团取代;
[0498]
并且本文未具体定义的所有其他变量如实施方案1至13中的任一项所定义。
[0499]
15.根据实施方案1至14中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中r5选自-ch3、-ch2ch3、-ch2oh、-c(=o)och3、-ch2och3、-ch(ch3)2、环丙基、二氟环丙基和四氢-2h-吡喃基;并且本文未具体定义的所有其他变量如实施方案1至14中任一项中所定义。
[0500]
16.根据实施方案1至4和6至15中任一项所述的化合物,其中所述化合物由以下结
构式之一表示:
[0501][0502]
其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐;并且本文未具体定义的所有其他变量如实施方案1至4和6至15中任一项中所定义。
[0503]
17.根据实施方案1至16中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中r1选自氢、卤素、氰基、-oh、c
1-c4烷基、c
1-c4烷氧基和c
3-c6环烷基;其中:
[0504]
r1的所述c
1-c4烷基任选地被1至3个独立地选自卤素、氰基、-oh和c
1-c2烷氧基的基团取代;
[0505]
r1的所述c
1-c4烷氧基任选地被1至3个独立选择的卤素基团取代;并且
[0506]
r1的所述c
3-c6环烷基任选地被1至3个独立地选自卤素、氰基、-oh和c
1-c2烷氧基的基团取代;
[0507]
并且本文未具体定义的所有其他变量如实施方案1至16中的任一项所定义。
[0508]
18.根据实施方案1至17中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中r1选自f、cl、br、c
1-c4烷基和c
3-c6环烷基;其中:
[0509]
r1的所述c
1-c4烷基任选地被1至3个独立地选自卤素和-oh的基团取代;并且
[0510]
r1的所述c
3-c6环烷基任选地被1至3个独立地选自卤素和-oh的基团取代;
[0511]
并且本文未具体定义的所有其他变量如实施方案1至17中的任一项所定义。
[0512]
19.根据实施方案1至18中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中r1选自f、cl、br、c
1-c4烷基和c
3-c6环烷基;其中:
[0513]
r1的所述c
1-c4烷基任选地被1至3个独立地选自卤素和-oh的基团取代;
[0514]
并且本文未具体定义的所有其他变量如实施方案1至18中的任一项所定义。
[0515]
20.根据实施方案1至18中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中r1选自cl、br、-ch3、-cf3、-ch2ch3、-ch(ch3)2、-ch2chf2、-ch2ch(ch3)2、二氟环丁基和环己基。
[0516]
21.根据实施方案1至20中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中r1是cl;并且本文未具体定义的所有其他变量如实施方案1至20中任一
项中所定义。
[0517]
22.根据实施方案1至21中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中r
3a
选自卤素、-oh和c
1-c4烷基;其中:
[0518]r3a
的所述c
1-c4烷基任选地被1至3个独立地选自卤素和-oh的基团取代;
[0519]
并且本文未具体定义的所有其他变量如实施方案1至21中的任一项所定义。
[0520]
23.根据实施方案1至22中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中r
3a
选自f、cl、br、-oh和c
1-c2烷基;其中:
[0521]r3a
的所述c
1-c2烷基任选地被1至3个独立地选自f、cl和-oh的基团取代;
[0522]
并且本文未具体定义的所有其他变量如实施方案1至22中的任一项所定义。
[0523]
24.根据实施方案1至23中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中r
3a
选自f、-oh、-ch3、-chf2和ch2oh;并且本文未具体定义的所有其他变量如实施方案1至23中任一项中所定义。
[0524]
25.根据实施方案1至4和6至24中任一项所述的化合物,其中所述化合物由以下结构式之一表示:
[0525][0526]
其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐;并且本文未具体定义的所有其他变量如实施方案1至4和6至24中任一项中所定义。
[0527]
26.根据实施方案1至4和6至25中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中ra在每次出现时独立地选自卤素、氰基、c
1-c6烷基、c
1-c4烷氧基、c
1-c6卤代烷基、c
1-c6卤代烷氧基、-c(=o)nrhri、-nrhri、-nrhc(=o)rk、-ork、-[o(ch2)q]ro(c
1-c6烷基),-s(=o)2rk、-s(=o)2nrhri、c
3-c6环烷基、5至10元杂环基、苯基和5至8元杂芳基;其中:
[0528]
ra的所述c
1-c6烷基任选地被1至3个独立地选自以下的基团取代:氰基、-c(=o)nrhri、-nrhri、-nrhc(=o)rk、-nrhc(=o)ork、-nrhc(=o)nr
irj
、-nrhs(=o)
prk
、-ork、-s(=o)2rk、-s(=o)
p
nrhri和c
3-c6环烷基;
[0529]
ra的所述c
3-c6环烷基、所述5至10元杂环基、所述苯基和所述5至8元杂芳基各自任选地被1至3个独立地选自卤素、c
1-c2烷基和-ork的基团取代;其中:
[0530]
rh、ri和rj在每次出现时各自独立地选自氢、c
1-c2烷基、环丙基和环丁基;其中:
[0531]
rh、ri和rj中任一者的所述c
1-c2烷基任选地被1至3个独立地选自卤素和-oh的基团
取代;
[0532]rk
在每次出现时各自独立地选自氢和c
1-c4烷基;其中:
[0533]rk
的所述c
1-c4烷基任选地被1至3个独立地选自卤素和-oh的基团取代;并且
[0534]
q和r各自是选自1、2和3的整数;
[0535]
并且本文未具体定义的所有其他变量如实施方案1至4和6至25中的任一项所定义。
[0536]
27.根据实施方案1至4和6至26中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中ra在每次出现时独立地选自卤素、氰基、c
1-c6烷基、c
1-c4烷氧基、c
1-c4卤代烷基、c
1-c4卤代烷氧基、-c(=o)nrhri、-nrhri、-nrhc(=o)rk、-ork、-[o(ch2)q]ro(c
1-c4烷基)、-s(=o)2rk、-s(=o)2nrhri、环丙基、环丁基、5至6元杂环基、苯基和5至6元杂芳基;其中:
[0537]
ra的所述c
1-c6烷基任选地被1至3个独立地选自氰基、-c(=o)nrhri、-nrhri、-ork、环丙基和环丁基的基团取代;
[0538]
ra的环丙基、环丁基、5至6元杂环基、苯基和5至6元杂芳基各自任选地被1至3个独立地选自卤素、-ch3、-oh和-och3的基团取代;其中:
[0539]
rh和ri在每次出现时各自独立地选自氢、-ch3、环丙基和环丁基;其中:
[0540]
rh和ri中任一者的所述-ch3任选地被1至3个独立地选自f、cl和-oh的基团取代;
[0541]rk
在每次出现时各自独立地选自氢和-ch3;其中:
[0542]rk
的所述-ch3任选地被1至3个独立地选自卤素和-oh的基团取代;
[0543]
并且本文未具体定义的所有其他变量如实施方案1至4和6至26中的任一项所定义。
[0544]
28.根据实施方案1至4和6至27中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中ra在每次出现时独立地选自f、cl、br、氰基、c
1-c6烷基、c
1-c2烷氧基、c
1-c2卤代烷基、-c(=o)nrhri、-nrhri、-nrhc(=o)rk、-ork、-[o(ch2)q]ro(c
1-c2烷基)、-s(=o)2rk、-s(=o)2nrhri、环丙基、环丁基、5元杂环基、苯基和6元杂芳基;其中:
[0545]
ra的所述c
1-c6烷基任选地被1至3个独立地选自氰基、-c(=o)nrhri、-ork和环丙基的基团取代;
[0546]
ra的环丙基、环丁基、5至6元杂环基、苯基和5至6元杂芳基各自任选地被1至3个独立地选自卤素、-ch3、-oh和-och3的基团取代;其中:
[0547]
rh和ri在每次出现时各自独立地选自氢、-ch3和环丙基;其中:
[0548]
rh和ri中任一者的所述-ch3任选地被1至3个独立地选自f、cl和-oh的基团取代;
[0549]rk
在每次出现时各自独立地选自氢和-ch3;并且
[0550]
q和r各自是选自1和2的整数;
[0551]
并且本文未具体定义的所有其他变量如实施方案1至4和6至27中的任一项所定义。
[0552]
29.根据实施方案1至4和6至28中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中ra在每次出现时独立地选自f、氰基、-oh、-ch3、-cf3、-ch(ch3)2、-(ch2)2oh、-(ch2)2och3、-ch2ch(oh)c2h5、-ch2c(ch3)(ch2oh)2、-och3、-och2ch3、-[o(ch2)2]2och3、-ch2c(=o)nhch3、-(ch2)2so2ch3、-ch2c(=o)n(ch3)2、-ch2(环丙基)、-c(=o)nh2、-c
(=o)nh(环丙基)、-nh2、-nhch3、-n(ch3)2、
[0553]-nhc(ch3)2ch2oh、-nhc(=o)ch3、-so2ch3、-so2nh2、环丙基、2-甲氧基苯基、n-甲基哌嗪基、四氢-2h-吡喃基、甲基吡唑基、吡啶基和四氢噻吩1,1-二氧化物;并且本文未具体定义的所有其他变量如实施方案1至4和6至28中任一项中所定义。
[0554]
30.根据实施方案1所述的化合物,其中所述化合物由以下结构式之一表示:
[0555]
[0556][0557]
其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐;并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0558]
31.一种化合物,其选自表i的化合物、其互变异构体、这些化合物和互变异构体的氘化衍生物以及前述任一者的药学上可接受的盐。
[0559]
32.一种化合物,其选自表ii的化合物、其互变异构体、这些化合物和互变异构体
的氘化衍生物以及前述任一者的药学上可接受的盐。
[0560]
33.一种化合物,其选自表iii的化合物、其互变异构体、这些化合物和互变异构体的氘化衍生物以及前述任一者的药学上可接受的盐。
[0561]
34.一种药物组合物,其包含根据实施方案1至33中任一项所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐以及药学上可接受的载体。
[0562]
35.一种治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病的方法,其包括向有需要的患者施用根据实施方案1至33中任一项所述的至少一种化合物或根据实施方案34所述的药物组合物。
[0563]
36.根据实施方案1至33中任一项所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据实施方案34所述的药物组合物在制备用于治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病的药物中的用途。
[0564]
37.根据实施方案1至33中任一项所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据实施方案34所述的药物组合物,用于治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病。
[0565]
38.一种抑制apol1活性的方法,所述方法包括使所述apol1与根据实施方案1至33中任一项所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或者根据实施方案34所述的药物组合物接触。
[0566]
39.根据实施方案1至33中任一项所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据实施方案34所述的药物组合物在制备用于抑制apol1活性的药物中的用途。
[0567]
40.根据实施方案1至33中任一项所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据实施方案34所述的药物组合物,用于抑制apol1活性。
[0568]
41.一种治疗apol1介导性疾病(例如,apol1介导性肾脏疾病)的方法,其包括向有需要的患者施用根据实施方案1至33中任一项所述的至少一种化合物或根据实施方案34所述的药物组合物。
[0569]
42.根据实施方案41所述的方法,其中所述apol1介导性疾病是癌症。
[0570]
43.根据实施方案41或实施方案42所述的方法,其中所述apol1介导性疾病是胰腺癌。
[0571]
44.根据实施方案1至33中任一项所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据实施方案34所述的药物组合物在制备用于治疗apol1介导性疾病(例如,apol1介导性肾脏疾病)的药物中的用途。
[0572]
45.根据实施方案44所述的用途,其中所述apol1介导性疾病是癌症。
[0573]
46.根据实施方案44或实施方案45所述的用途,其中所述apol1介导性疾病是胰腺癌。
[0574]
47.根据实施方案1至33中任一项所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据实施方案34所述的药物组合物,用于治疗apol1介导性疾病(例如,apol1介导性肾脏疾病)。
[0575]
48.根据实施方案47所述的供使用的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或供使用的药物组合物,其中所述apol1介导性疾病为癌症。
[0576]
49.根据实施方案47或实施方案48所述的供使用的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或供使用的药物组合物,其中所述apol1介导性疾病是胰腺癌。
[0577]
50.一种抑制apol1活性的方法,所述方法包括使所述apol1与根据实施方案1至33中任一项所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或者根据实施方案34所述的药物组合物接触。
[0578]
51.根据实施方案1至33中任一项所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据实施方案34所述的药物组合物在制备用于抑制apol1活性的药物中的用途。
[0579]
52.根据实施方案1至33中任一项所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据实施方案34所述的药物组合物,用于抑制apol1活性。
[0580]
53.一种根据实施方案1至33中任一项所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐的硅衍生物。
[0581]
54.一种药物组合物,其包含根据实施方案53所述的硅衍生物。
[0582]
55.一种治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病的方法,其包括向有需要的患者施用根据实施方案53所述的硅衍生物或根据实施方案54所述的药物组合物。
[0583]
56.根据实施方案53所述的硅衍生物或根据实施方案54所述的药物组合物在制备用于治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病的药物中的用途。
[0584]
57.根据实施方案53所述的硅衍生物或根据实施方案54所述的药物组合物,用于治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病。
[0585]
58.一种治疗apol1介导性疾病(例如,apol1介导性肾脏疾病)的方法,其包括向有需要的患者施用根据实施方案53所述的硅衍生物或根据实施方案54所述的药物组合物。
[0586]
59.根据实施方案58所述的方法,其中所述apol1介导性疾病是癌症。
[0587]
60.根据实施方案58或实施方案59所述的方法,其中所述apol1介导性疾病是胰腺癌。
[0588]
61.根据实施方案53所述的硅衍生物或根据实施方案54所述的药物组合物在制备用于治疗apol1介导性疾病(例如,apol1介导性肾脏疾病)的药物中的用途。
[0589]
62.根据实施方案61所述的用途,其中所述apol1介导性疾病是癌症。
[0590]
63.根据实施方案61或实施方案62所述的用途,其中所述apol1介导性疾病是胰腺癌。
[0591]
64.根据实施方案53所述的硅衍生物或根据实施方案54所述的药物组合物,用于治疗apol1介导性疾病(例如,apol1介导性肾脏疾病)。
[0592]
65.根据实施方案64所述的供使用的硅衍生物或药物组合物,其中所述apol1介导性疾病为癌症。
[0593]
66.根据实施方案64或实施方案65所述的供使用的硅衍生物或药物组合物,其中所述apol1介导性疾病为胰腺癌。
[0594]
67.一种根据实施方案1至33中任一项所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐的硼衍生物。
[0595]
68.一种药物组合物,其包含根据实施方案67所述的硼衍生物。
[0596]
69.一种治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病的方法,其包括向有需要的患者施用根据实施方案67所述的硼衍生物或根据实施方案68所述的药物组合物。
[0597]
70.根据实施方案67所述的硼衍生物或根据实施方案68所述的药物组合物在制备用于治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病的药物中的用途。
[0598]
71.根据实施方案67所述的硼衍生物或根据实施方案68所述的药物组合物,用于治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病。
[0599]
72.一种治疗apol1介导性疾病(例如,apol1介导性肾脏疾病)的方法,其包括向有需要的患者施用根据实施方案67所述的硼衍生物或根据实施方案68所述的药物组合物。
[0600]
73.根据实施方案72所述的方法,其中所述apol1介导性疾病是癌症。
[0601]
74.根据实施方案72或实施方案73所述的方法,其中所述apol1介导性疾病是胰腺癌。
[0602]
75.根据实施方案67所述的硼衍生物或根据实施方案68所述的药物组合物在制备用于治疗apol1介导性疾病(例如,apol1介导性肾脏疾病)的药物中的用途。
[0603]
76.根据实施方案75所述的用途,其中所述apol1介导性疾病是癌症。
[0604]
77.根据实施方案75或实施方案76所述的用途,其中所述apol1介导性疾病是胰腺癌。
[0605]
78.根据实施方案67所述的硼衍生物或根据实施方案68所述的药物组合物,用于治疗apol1介导性疾病(例如,apol1介导性肾脏疾病)。
[0606]
79.根据实施方案78所述的供使用的硼衍生物或药物组合物,其中所述apol1介导性疾病为癌症。
[0607]
80.根据实施方案78或实施方案79所述的供使用的硼衍生物或药物组合物,其中所述apol1介导性疾病为胰腺癌。
[0608]
81.一种根据实施方案1至33中任一项所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐的磷衍生物。
[0609]
82.一种药物组合物,其包含根据实施方案81所述的磷衍生物。
[0610]
83.一种治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病的方法,其包括向有需要的患者施用根据实施方案81所述的磷衍生物或根据实施方案82所述的药物组合物。
[0611]
84.根据实施方案81所述的磷衍生物或根据实施方案82所述的药物组合物在制备用于治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病的药物中的用途。
[0612]
85.根据实施方案81所述的磷衍生物或根据实施方案82所述的药物组合物,用于治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病。
[0613]
86.一种治疗apol1介导性疾病(例如,apol1介导性肾脏疾病)的方法,其包括向有需要的患者施用根据实施方案81所述的磷衍生物或根据实施方案82所述的药物组合物。
[0614]
87.根据实施方案86所述的方法,其中所述apol1介导性疾病是癌症。
[0615]
88.根据实施方案86或实施方案87所述的方法,其中所述apol1介导性疾病是胰腺癌。
[0616]
89.根据实施方案81所述的磷衍生物或根据实施方案82所述的药物组合物在制备用于治疗apol1介导性疾病(例如,apol1介导性肾脏疾病)的药物中的用途。
[0617]
90.根据实施方案89所述的用途,其中所述apol1介导性疾病是癌症。
[0618]
91.根据实施方案89或实施方案90所述的用途,其中所述apol1介导性疾病是胰腺癌。
[0619]
92.根据实施方案81所述的磷衍生物或根据实施方案82所述的药物组合物,用于治疗apol1介导性疾病(例如,apol1介导性肾脏疾病)。
[0620]
93.根据实施方案92所述的供使用的磷衍生物或药物组合物,其中所述apol1介导性疾病为癌症。
[0621]
94.根据实施方案92或实施方案93所述的磷衍生物或药物组合物,其中所述apol1介导性疾病是胰腺癌。
[0622]
95.一种治疗患者的方法;一种用于治疗患者的实体(例如,化合物、互变异构体、氘化衍生物、药学上可接受的盐、硅衍生物、硼衍生物、磷衍生物)或药物组合物;或实体或药物组合物在治疗如本文任一实施方案所述的患者中的用途,其中所述患者具有2个apol1风险等位基因。
[0623]
96.一种治疗患者的方法;一种用于治疗患者的实体(例如,化合物、互变异构体、氘化衍生物、药学上可接受的盐、硅衍生物、硼衍生物、磷衍生物)或药物组合物;或实体或药物组合物在治疗如本文任一实施方案所述的患者中的用途,其中所述患者具有1个apol1风险等位基因。
[0624]
97.根据实施方案1所述的化合物,其中所述化合物由以下结构式之一表示:
[0625][0626]
其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐;并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0627]
98.根据实施方案1所述的化合物,其中所述化合物由以下结构式之一表示:
[0628][0629][0630]
其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐;并且本文未具体定义的所有其他变量如前述实施方案中任一项所定义。
[0631]
99.一种药物组合物,其包含根据实施方案97或实施方案98所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐以及药学上可接受的载体。
[0632]
100.一种治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病的方法,其包括向有需要的患者施用根据实施方案97或实施方案98所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据实施方案99所述的药物组合物。
[0633]
101.根据实施方案97或实施方案98所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据实施方案99所述的药物组合物在制备用于治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病的药物中的用途。
[0634]
102.根据实施方案97或实施方案98所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据实施方案99所述的药物组合物,用于治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病。
[0635]
103.一种抑制apol1活性的方法,其包括使所述apol1与根据实施方案97或实施方案98所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据实施方案99所述的药物组合物接触。
[0636]
104.根据实施方案97或实施方案98所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据实施方案99所述的药物组合物在制备用于抑制apol1活性的药物中的用途。
[0637]
105.根据实施方案97或实施方案98所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据实施方案99所述的药物组合物,用于抑制apol1活性。
[0638]
106.一种治疗apol1介导性疾病(例如,apol1介导性肾脏疾病)的方法,其包括向有需要的患者施用根据实施方案97或实施方案98所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据实施方案99所述的药物组合物。
[0639]
107.根据实施方案106所述的方法,其中所述apol1介导性疾病是癌症。
[0640]
108.根据实施方案106或实施方案107所述的方法,其中所述apol1介导性疾病是胰腺癌。
[0641]
109.根据实施方案97或实施方案98所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据实施方案99所述的药物组合物在制备用于治疗apol1介导性疾病(例如,apol1介导性肾脏疾病)的药物中的用途。
[0642]
110.根据实施方案109所述的用途,其中所述apol1介导性疾病是癌症。
[0643]
111.根据实施方案109或实施方案110所述的用途,其中所述apol1介导性疾病是胰腺癌。
[0644]
112.根据实施方案97或实施方案98所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据实施方案99所述的药物组合物,用于治疗apol1介导性疾病(例如,apol1介导性肾脏疾病)。
[0645]
113.根据实施方案112所述的供使用的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或供使用的药物组合物,其中所述apol1介导性疾病为癌症。
[0646]
114.根据实施方案112或实施方案113所述的供使用的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或供使用的药物组合物,其中所述apol1介导性疾病是胰腺癌。
[0647]
115.一种抑制apol1活性的方法,其包括使所述apol1与根据实施方案97或实施方案98所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据实施方
案99所述的药物组合物接触。
[0648]
116.根据实施方案97或实施方案98所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据实施方案99所述的药物组合物在制备用于抑制apol1活性的药物中的用途。
[0649]
117.根据实施方案97或实施方案98所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据实施方案99所述的药物组合物,用于抑制apol1活性。
[0650]
非限制性示例性实施方案2
[0651]
非限制性地,本公开的一些实施方案/条款包括:
[0652]
1.一种由以下结构式表示的化合物:
[0653][0654]
其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐,其中:
[0655]
x1和x2各自选自s和-cr2,其中:
[0656]
x1和x2之一是s;
[0657]
当x1为s时,则x2为-cr
2b
;并且
[0658]
当x2是s时,则x1是-cr
2a
;
[0659]
r1选自卤素、氰基、c
1-c6烷基、c
1-c6烷氧基、c
3-c6环烷基和苯基;其中:
[0660]
r1的所述c
1-c6烷基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2和c
1-c4烷氧基;
[0661]
r1的所述c
1-c6烷氧基任选地被1至3个独立地选自卤素的基团取代;
[0662]
r1的所述c
3-c6环烷基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷基、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)和-c(=o)n(c
1-c4烷基)2;并且
[0663]
r1的所述苯基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷基、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)和-c(=o)n(c
1-c4烷基)2;
[0664]r2a
选自氢、卤素、氰基、-oh、=o和c
1-c6烷基;其中:
[0665]r2a
的所述c
1-c6烷基任选地被1至3个独立地选自卤素、氰基、-oh和c
1-c4烷氧基的基团取代;
[0666]r2b
选自氢、卤素、氰基、-oh、=o和c
1-c6烷基;
[0667]r3a
选自卤素、氰基、-oh、c
1-c6烷基和=o;其中:
[0668]r3a
的所述c
1-c6烷基任选地被1至3个独立地选自卤素、氰基和-oh的基团取代;
[0669]r3b
选自c
1-c2烷基和=o;其中:
[0670]r3b
的所述c
1-c2烷基任选地被1至3个独立地选自卤素、氰基和-oh的基团取代;
[0671]
当r
3a
选自卤素、氰基、-oh、c
1-c6烷基时或当r
3b
选自c
1-c2烷基时,在每次出现时为单键;或者当r
3a
为=o时或当r
3b
为=o时,每次出现时为双键;
[0672]
r4选自c
1-c6烷基和其中:
[0673]
r4的所述c
1-c6烷基任选地被1至3个独立地选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)、-c(=o)n(c
1-c4烷基)2、c
3-c6环烷基、5至10元杂环基、苯基和5至10元杂芳基;
[0674]
环a选自c
3-c
12
碳环基、3至12元杂环基、c6和c
10
芳基和5至10元杂芳基,其中环a任选地被1、2、3、4或5个ra基团取代;其中:
[0675]
ra在每次出现时独立地选自卤素、氰基、c
1-c6烷基、c
2-c6烯基、c
1-c6烷氧基、c
1-c6卤代烷基、c
1-c6卤代烯基、c
1-c6卤代烷氧基、-c(=o)nrhri、-nrhri、-nrhc(=o)rk、-nrhc(=o)ork、-nrhc(=o)nr
irj
、-nrhs(=o)
prk
、-ork、-oc(=o)rk、-oc(=o)ork、-oc(=o)nrhri、-[o(ch2)q]ro(c
1-c6烷基)、-s(=o)
prk
、-s(=o)
p
nrhri、c
3-c
12
碳环基、3至12元杂环基、c6和c
10
芳基以及5至10元杂芳基;其中:
[0676]
ra的所述c
1-c6烷基和所述c
2-c6烯基任选地被1至3个独立地选自以下的基团取代:氰基、-c(=o)rk、-c(=o)ork、-c(=o)nrhri、-nrhri、-nrhc(=o)rk、-nrhc(=o)ork、-nrhc(=o)nr
irj
、-nrhs(=o)
prk
、-ork、-oc(=o)rk、-oc(=o)ork、-oc(=o)nrhri、-s(=o)
prk
、-s(=o)
p
nrhri和c
3-c6环烷基;
[0677]
ra的所述c
3-c
12
碳环基、所述3至12元杂环基、所述c6和c
10
芳基和所述5至10元杂芳基各自任选地被1至3个选自以下的基团取代:卤素、氰基、c
1-c4烷基、-nrhri和-ork;其中:
[0678]
rh、ri和rj在每次出现时各自独立地选自氢、c
1-c4烷基和c
3-c6环烷基;其中:
[0679]
rh、ri和rj中任一者的所述c
1-c4烷基任选地被1至3个独立地选自卤素、氰基和-oh的基团取代;
[0680]rk
在每次出现时各自独立地选自氢、c
1-c4烷基和c
3-c6环烷基;其中:
[0681]rk
中任一者的所述c
1-c4烷基任选地被1至3个独立地选自卤素、氰基和-oh的基团取代;
[0682]
r5选自c
1-c6烷基、-c(=o)o(c
1-c4烷基)、c
3-c
12
碳环基、3至12元杂环基、c6和c
10
芳基以及5至10元杂芳基;其中:
[0683]
r5的所述c
1-c6烷基任选地被1至3个选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)以及-c(=o)n(c
1-c4烷基)2;
[0684]
r5的所述c
3-c
12
碳环基、所述3至12元杂环基、所述c6和c
10
芳基和所述5至10元杂芳基各自任选地被1至3个选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c4烷基、c
1-c4烷氧基、-c(=o)nh2、-c(=o)nh(c
1-c4烷基)和-c(=o)n(c
1-c4烷基)2;
[0685]
当r
3a
选自卤素、氰基、-oh、c
1-c6烷基时,k是选自0、1和2的整数;或者当r
3a
为=o时,k为选自0和1的整数;
[0686]
当r
3b
选自c
1-c2烷基时,m为选自0、1和2的整数;并且当r
3b
为=o时,m为选自0和1的整数;
[0687]
p是选自1和2的整数;并且
[0688]
q和r各自是选自1、2、3和4的整数。
[0689]
2.根据条款1所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中所述化合物由以下结构式之一表示:
[0690][0691]
或其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐,其中:
[0692]r2a
选自氢、卤素、氰基和c
1-c4烷基;其中:
[0693]r2a
的c
1-c4烷基任选地被1至3个选自卤素、-oh和c
1-c2烷氧基的基团取代;
[0694]r2b
选自氢、卤素、氰基和c
1-c4烷基;并且
[0695]
k是选自0、1和2的整数;
[0696]
并且本文未具体定义的所有其他变量如实施方案1中所定义。
[0697]
3.根据条款1或条款2所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中r4选自c
1-c4烷基和其中:
[0698]
r4的所述c
1-c4烷基任选地被1至3个选自以下的基团取代:卤素、氰基、-oh、-nh2、-nh(c
1-c4烷基)、-n(c
1-c4烷基)2、c
1-c2烷氧基、c
3-c6环烷基、5至6元杂环基、苯基和5至6元杂
芳基;
[0699]
并且本文未具体定义的所有其他变量如条款1或条款2中所定义。
[0700]
4.根据条款1至3中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中r4选自c
1-c2烷基和其中:
[0701]
r4的所述c
1-c2烷基任选地被1至3个选自卤素、氰基、-oh和5至6元杂环基的基团取代;
[0702]
并且本文未具体定义的所有其他变量如条款1至3中任一项所定义。
[0703]
5.根据条款1至4中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中r4选自-ch3、-ch2oh和(四氢-2h-吡喃-4-基)甲基;并且本文未具体定义的所有其他变量如条款1至4中任一项所定义。
[0704]
6.根据条款1至4中任一项所述的化合物,其中所述化合物由以下结构式之一表示:
[0705][0706]
其互变异构体、所述化合物或互变异构体的氘化衍生物、或前述物质的药学上可接受的盐,其中:
[0707]
环a在每次出现时选自c
3-c6环烷基、5至10元杂环基、苯基和5至10元杂芳基;其各自任选地被1、2、3、4或5个ra基团取代;
[0708]
并且本文未具体定义的所有其他变量如条款1至5中任一项所定义。
[0709]
7.根据条款1至4和6中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中环a选自环丙基、5至10元杂环基、苯基和5至9元杂芳基;其各自任选地被1、2、3、4或5个ra基团取代;并且本文未具体定义的所有其他变量如条款1至6中任一项所定义。
[0710]
8.根据条款1至4、6和7中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中环a选自环丙基、含有1至3个选自n和o的杂原子的5至10元杂环基、苯基和
含有1至3个选自n和o的杂原子的5至9元杂芳基;其各自任选地被1、2、3、4或5个ra基团取代;并且本文未具体定义的所有其他变量如条款1至7中任一项所定义。
[0711]
9.根据条款1至4和6至8中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中环a选自环丙基、含有1至3个选自n和o的杂原子的5元杂环基、含有1至3个选自n和o的杂原子的6元杂环基、含有1至3个选自n和o的杂原子的9元杂环基、含有1至3个选自n和o的杂原子的10元杂环基、苯基、含有1至3个选自n和o的杂原子的5元杂芳基、含有1至3个选自n和o的杂原子的6元杂芳基和含有1至3个选自n和o的杂原子的9元杂芳基;其各自任选地被1、2、3、4或5个ra基团取代;并且本文未具体定义的所有其他变量如条款1至8中任一项中所定义。
[0712]
10.根据条款1至4和6至9中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中环a选自
[0713]
其各自任选地被1、2、3、4或5个ra基团取代;并且本文未具体定义的所有其他变量如条款1至9中任一项所定义。
[0714]
11.根据条款1至4和6至10中任一项所述的化合物、互变异构体、氘化衍生物或药
学上可接受的盐,其中环a选自其中环a选自
其各自任选地被1、2、3、4或5个ra取代;并且本文未具体定义的所有其他变量如条款1至4和6至10中任一项所定义。
[0715]
12.根据条款1至4和6至11中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中r4选自-ch3和环a;其中环a选自其各自任选地被1、2、3、4或5个ra基团取代;并且本文未具体定义的所有其他变量如条款1至4和6至11中任一项所定义。
[0716]
13.根据条款1至12中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中r5选自c
1-c4烷基、-c(=o)o(c
1-c2烷基)、c
3-c6环烷基和5至10元环烷基;其中:
[0717]
r5的所述c
1-c4烷基任选地被1至3个选自卤素、氰基、-oh和c
1-c2烷氧基的基团取代;并且
[0718]
r5的所述c
3-c6环烷基和5至10元杂环基各自任选地被1至3个选自卤素、氰基、-oh、c
1-c2烷基和c
1-c2烷氧基的基团取代;
[0719]
并且本文未具体定义的所有其他变量如条款1至12中任一项所定义。
[0720]
14.根据条款1至13中任一项所述的化合物、互变异构体、氘化衍生物或药学上可
接受的盐,其中r5选自c
1-c2烷基、c(=o)o(c
1-c2烷基)、环丙基、环丁基和5至6元杂环基;其中:
[0721]
r5的所述c
1-c2烷基任选地被1至3个选自f、cl、br、氰基、-oh和c
1-c2烷氧基的基团取代;并且
[0722]
r5的所述环丙基、所述环丁基和所述5至6元杂环基各自任选地被1至3个选自f、cl、br、氰基、-oh、c
1-c2烷基和c
1-c2烷氧基的基团取代;
[0723]
并且本文未具体定义的所有其他变量如条款1至13中任一项所定义。
[0724]
15.根据条款1至14中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中r5选自-ch3、-ch2ch3、-ch2oh、-c(=o)och3、-ch2och3、-ch(ch3)2、环丙基、二氟环丙基和四氢-2h-吡喃基;并且本文未具体定义的所有其他变量如条款1至14中任一项所定义。
[0725]
16.根据条款1至4和6至15中任一项所述的化合物,其中所述化合物由以下结构式之一表示:
[0726][0727]
其互变异构体、所述化合物或互变异构体的氘化衍生物或前述化合物的药学上可接受的盐;并且本文未具体定义的所有其他变量如条款1至4和6至15中任一项所定义。
[0728]
17.根据条款1至16中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中r1选自氢、卤素、氰基、-oh、c
1-c4烷基、c
1-c4烷氧基和c
3-c6环烷基;其中:
[0729]
r1的所述c
1-c4烷基任选地被1至3个选自卤素、氰基、-oh和c
1-c2烷氧基的基团取代;
[0730]
r1的所述c
1-c4烷氧基任选地被1至3个卤素基团取代;并且
[0731]
r1的所述c
3-c6环烷基任选地被1至3个选自卤素、氰基、-oh和c
1-c2烷氧基的基团取代;
[0732]
并且本文未具体定义的所有其他变量如条款1至16中任一项所定义。
[0733]
18.根据条款1至17中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中r1选自f、cl、br、c
1-c4烷基和c
3-c6环烷基;其中:
[0734]
r1的所述c
1-c4烷基任选地被1至3个选自卤素和-oh的基团取代;并且
[0735]
r1的所述c
3-c6环烷基任选地被1至3个选自卤素和-oh的基团取代;
c6烷基)、-s(=o)2rk、-s(=o)2nrhri、c
3-c6环烷基、5至10元杂环基、苯基和5至8元杂芳基;其中:
[0753]
ra的所述c
1-c6烷基任选地被1至3个选自以下的基团取代:氰基、-c(=o)nrhri、-nrhri、-nrhc(=o)rk、-nrhc(=o)ork、-nrhc(=o)nr
irj
、-nrhs(=o)
prk
、-ork、-s(=o)2rk、-s(=o)
p
nrhri和c
3-c6环烷基;
[0754]
ra的所述c
3-c6环烷基、5至10元杂环基、苯基和5至8元杂芳基各自任选地被1至3个选自卤素、c
1-c2烷基和-ork的基团取代;其中:
[0755]
rh、ri和rj在每次出现时各自独立地选自氢、c
1-c2烷基、环丙基和环丁基;其中:
[0756]
rh、ri和rj中任一者的所述c
1-c2烷基任选地被1至3个选自卤素和-oh的基团取代;
[0757]rk
在每次出现时各自独立地选自氢和c
1-c4烷基;其中:
[0758]rk
的所述c
1-c4烷基任选地被1至3个选自卤素和-oh的基团取代;并且
[0759]
q和r各自是选自1、2和3的整数;
[0760]
并且本文未具体定义的所有其他变量如条款1至4和6至25中的任一项所定义。
[0761]
27.根据条款1至4和6至26中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中ra在每次出现时独立地选自卤素、氰基、c
1-c6烷基、c
1-c4烷氧基、c
1-c4卤代烷基、c
1-c4卤代烷氧基、-c(=o)nrhri、-nrhri、-nrhc(=o)rk、-ork、-[o(ch2)q]ro(c
1-c4烷基)、-s(=o)2rk、-s(=o)2nrhri、环丙基、环丁基、5至6元杂环基、苯基和5至6元杂芳基;其中:
[0762]
ra的c
1-c6烷基任选地被1至3个选自氰基、-c(=o)nrhri、-nrhri、-ork、环丙基和环丁基的基团取代;
[0763]
ra的所述环丙基、所述环丁基、所述5至6元杂环基、所述苯基和所述5至6元杂芳基各自任选地被1至3个选自卤素、-ch3、-oh和-och3的基团取代;其中:
[0764]
rh和ri在每次出现时各自独立地选自氢、-ch3、环丙基和环丁基;其中:
[0765]
rh和ri中任一者的所述-ch3任选地被1至3个选自f、cl和-oh的基团取代;
[0766]rk
在每次出现时各自独立地选自氢和-ch3;其中:
[0767]rk
的-ch3任选地被1至3个选自卤素和-oh的基团取代;
[0768]
并且本文未具体定义的所有其他变量如条款1至4和6至26中的任一项所定义。
[0769]
28.根据条款1至4和6至27中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中ra在每次出现时独立地选自f、cl、br、氰基、c
1-c6烷基、c
1-c2烷氧基、c
1-c2卤代烷基、-c(=o)nrhri、-nrhri、-nrhc(=o)rk、-ork、-[o(ch2)q]ro(c
1-c2烷基)、-s(=o)2rk、-s(=o)2nrhri、环丙基、环丁基、5元杂环基、苯基和6元杂芳基;其中:
[0770]
ra的所述c
1-c6烷基任选地被1至3个选自氰基、-c(=o)nrhri、-ork和环丙基的基团取代;
[0771]
ra的所述环丙基、所述环丁基、所述5至6元杂环基、所述苯基和所述5至6元杂芳基各自任选地被1至3个选自卤素、-ch3、-oh和-och3的基团取代;其中:
[0772]
rh和ri在每次出现时各自独立地选自氢、-ch3和环丙基;其中:
[0773]
rh和ri中任一者的所述-ch3任选地被1至3个选自f、cl和-oh的基团取代;
[0774]rk
在每次出现时各自独立地选自氢和-ch3;并且
[0775]
q和r各自是选自1和2的整数;
[0776]
并且本文未具体定义的所有其他变量如条款1至4和6至27中的任一项所定义。
[0777]
29.根据条款1至4和6至28中任一项所述的化合物、互变异构体、氘化衍生物或药学上可接受的盐,其中ra在每次出现时独立地选自f、氰基、-oh、-ch3、-cf3、-ch(ch3)2、-(ch2)2oh、-(ch2)2och3、-ch2ch(oh)c2h5、-ch2c(ch3)(ch2oh)2、-och3、-och2ch3、-[o(ch2)2]2och3、-ch2c(=o)nhch3、-(ch2)2so2ch3、-ch2c(=o)n(ch3)2、-ch2(环丙基)、-c(=o)nh2、-c(=o)nh(环丙基)、-nh2、-nhch3、-n(ch3)2、-nhc(ch3)2ch2oh、-nhc(=o)ch3、-so2ch3、-so2nh2、环丙基、2-甲氧基苯基、n-甲基哌嗪基、四氢-2h-吡喃基、甲基吡唑基、吡啶基和四氢噻吩1,1-二氧化物;并且本文未具体定义的所有其他变量如条款1至4和6至28中任一项所定义。
[0778]
30.根据条款1所述的化合物,其中所述化合物由以下结构式之一表示:
[0779]
[0780][0781]
其互变异构体、所述化合物或互变异构体的氘化衍生物或前述化合物的药学上可接受的盐;并且本文未具体定义的所有其他变量如前述条款中任一项所定义。
[0782]
31.一种化合物,其选自表i的化合物、其互变异构体、这些化合物和互变异构体的氘化衍生物以及前述任一者的药学上可接受的盐。
[0783]
32.一种药物组合物,其包含根据条款1至31中任一项所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐以及药学上可接受的载体。
[0784]
33.一种治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病的方法,其包括向有需要的患者施用根据条款1至31中任一项所述的至少一种化合物或根据条款32所述的药物组合物。
[0785]
34.根据条款1至31中任一项所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据条款32所述的药物组合物在制备用于治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病的药物中的用途。
[0786]
35.根据条款1至31中任一项所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据条款32所述的药物组合物,用于治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病。
[0787]
36.一种抑制apol1活性的方法,其包括使所述apol1与根据条款1至31中任一项所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据条款32所述的
药物组合物接触。
[0788]
37.根据条款1至31中任一项所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据条款32所述的药物组合物在制备用于抑制apol1活性的药物中的用途。
[0789]
38.根据条款1至31中任一项所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐或根据条款32所述的药物组合物,用于抑制apol1活性。
[0790]
39.一种根据条款1至31中任一项所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐的硅衍生物。
[0791]
40.一种药物组合物,其包含根据条款39所述的硅衍生物。
[0792]
41.一种治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病的方法,其包括向有需要的患者施用根据条款39所述的硅衍生物或根据条款40所述的药物组合物。
[0793]
42.根据条款39所述的硅衍生物或根据条款40所述的药物组合物在制备用于治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病的药物中的用途。
[0794]
43.根据条款39所述的硅衍生物或根据条款40所述的药物组合物,用于治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病。
[0795]
44.一种根据条款1至31中任一项所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐的硼衍生物。
[0796]
45.一种药物组合物,其包含根据条款44所述的硼衍生物。
[0797]
46.一种治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病的方法,其包括向有需要的患者施用根据条款44所述的硼衍生物或根据条款45所述的药物组合物。
[0798]
47.根据条款44所述的硼衍生物或根据条款45所述的药物组合物在制备用于治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病的药物中的用途。
[0799]
48.根据条款44所述的硼衍生物或根据条款45所述的药物组合物,用于治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病。
[0800]
49.一种根据条款1至31中任一项所述的至少一种化合物、互变异构体、氘化衍生物或药学上可接受的盐的磷衍生物。
[0801]
50.一种药物组合物,其包含根据条款48所述的磷衍生物。
[0802]
51.一种治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病的方法,其包括向有需要的患者施用根据条款48所述的磷衍生物或根据条款49所述的药物组合物。
[0803]
52.根据条款48所述的磷衍生物或根据条款49所述的药物组合物在制备用于治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病的药物中的用途。
[0804]
53.根据条款48所述的磷衍生物或根据条款49所述的药物组合物,用于治疗局灶性节段性肾小球硬化和/或非糖尿病性肾病。
[0805]
实施例
[0806]
为了可以更充分地理解本文描述的公开内容,阐述了以下实施例。应理解,这些实施例仅用于说明性目的,并且不应解释为以任何方式限制本公开。
[0807]
本公开的化合物可以根据标准化学实践或如本文所述来制备。在以下合成方案中以及在用于制备式i、iia、iib、iiia、iiib、iva、ivb、va、vb、i’、iia’、iib’、iiia’、iiib’、iva’、ivb’、va’、vb’、iia”、iib”、iiia”、iiib”、iva”、ivb”、iia
”’
、iib
”’
、iiia
”’
、iiib
”’
、
iva
”’
、ivb
”’
、i0、iia0、iib0、iiia0、iiib0、iva0、ivb0、va0、vb0、i
’0、iia
’0、iib
’0、iiia
’0、iiib
’0、iva
’0、ivb
’0、va
’0和vb
’0的化合物、化合物1至391、其互变异构体、所述化合物或互变异构体的氘化衍生物或前述任一者的药学上可接受的盐的描述中,使用以下缩写:
[0808]
缩写
[0809]
aibn=偶氮双异丁腈
[0810]
arp=即用型测定板(assay ready plate)
[0811]
bbbpy=4,4
′‑
二-叔-丁基-2,2
′‑
联吡啶
[0812]
bf3=三氟化硼
[0813]
bf3.oet2=三氟化硼乙醚化物
[0814]
boc2o=二碳酸二叔丁酯
[0815]
cbzcl=氯甲酸苄酯
[0816]
cdmt=2-氯-4,6-二甲氧基-1,3,5-三嗪
[0817]
dast=二乙氨基三氟化硫
[0818]
dbu=1,8-二氮杂双环[5.4.0]十一碳-7-烯
[0819]
dcm=二氯甲烷
[0820]
dibal-h=氢化二异丁基铝
[0821]
dipea=n,n-二异丙基乙胺或n-乙基-n-异丙基-丙烷-2-胺
[0822]
dmap=二甲基氨基吡啶
[0823]
dma=二甲基乙酰胺
[0824]
dme=二甲氧基乙烷
[0825]
dmem=杜氏改良eagle培养基
[0826]
dmf=二甲基甲酰胺
[0827]
dmpu=n,n
’‑
二甲基丙烯脲
[0828]
dmso=二甲基亚砜
[0829]
dppa=二苯基磷酰基叠氮化物
[0830]
etoac=乙酸乙酯
[0831]
etoh=乙醇
[0832]
et2o=乙醚
[0833]
fbs=胎牛血清
[0834]
flu=荧光值
[0835]
hatu=[二甲基氨基(三唑并[4,5-b]吡啶-3-基氧基)亚甲基]-二甲基-铵(六氟化磷离子)
[0836]
hdmc=n-[(5-氯-3-氧代-1h-苯并三唑-1-基)-4-吗啉基亚甲基]-n-甲基甲胺六氟磷酸盐
[0837]
hepes=4-(2-羟乙基)-1-哌嗪乙磺酸
[0838]
hbss=hank平衡盐溶液
[0839]
ipa=异丙醇
[0840]
ir[df(cf3)ppy]2(dtbbpy)pf6=六氟化磷
[0841]
lda=二异丙基酰胺锂
[0842]
led=发光二极管
[0843]
mecn=乙腈
[0844]
mei=甲基碘
[0845]
meoh=甲醇
[0846]
msoh=甲磺酸
[0847]
mtbe或tbme=甲基叔-丁基醚
[0848]
n-buli=正丁基锂
[0849]
nbs=n-溴琥珀酰亚胺
[0850]
nmm=n-甲基吗啉
[0851]
nmp=n-甲基吡咯烷
[0852]
pbs=磷酸盐缓冲盐水
[0853]
pd(dppf)2cl2=[1,1
′‑
双(二苯基膦基)二茂铁]二氯化钯(ii)
[0854]
pdcl2(pph3)2=双(三苯基膦)二氯化钯(ii)
[0855]
pp=聚丙烯
[0856]
ptsa=对-甲苯磺酸一水合物
[0857]
t3p=2,4,6-三丙基-1,3,5,2,4,6-三氧杂三磷杂环己烷-2,4,6-三氧化物
[0858]
tbaf=四正丁基氟化铵
[0859]
tbscl=叔丁基二甲基甲硅烷基氯
[0860]
tea=三乙胺
[0861]
tet=四环素
[0862]
tfa或tfaa=三氟乙酸
[0863]
tfoh=三氟甲磺酸
[0864]
thf=四氢呋喃
[0865]
2-me-thf=2=甲基四氢呋喃
[0866]
thp=四氢吡喃
[0867]
tmscl=三甲基甲硅烷基氯
[0868]
tmss=三(三甲基甲硅烷基)硅烷
[0869]
实施例1:化合物的合成
[0870]
所有具体和一般的化合物和公开用于制备那些化合物的中间体都被认为是本文公开的公开内容的一部分。
[0871]
合成起始物质
[0872]
制备描述了用于合成化合物1至391的中间体的合成路线。
[0873]
一般方案
[0874]
在一些实施方案中,用于制备式i化合物的方法包括方案1-6中描述的反应。
[0875]
方案1示出了用于制备式i化合物的方法。r1、r3、r4r5、x1、x2、m和k如上文所定义。式1-1的氨基酮可以与式1-2的醛发生反应,得到式1-3的哌啶酮。在一些实施方案中,反应可以在胺催化剂诸如l-脯氨酸的存在下,在碱诸如三乙胺和硫酸镁试剂的存在下发生。式1-3化合物可以使用用于制备哌啶酮的任何合适的方法来制备。使用任何合适的条件进行pictet-spengler反应,可以由式1-3的哌啶酮和式1-4的醇制备式1-3化合物。例如,反应可
以在酸诸如三氟甲基磺酸和溶剂诸如1,4-二噁烷的存在下进行。在一个替代实施方案中,可以使用诸如甲磺酸的酸。反应可以在诸如二氯甲烷的溶剂中,在附加热(例如,40℃)的存在下进行。
[0876]
方案1
[0877][0878]
方案2示出了用于制备式2-3的化合物的方法。pg1是任何合适的氮保护基。例如,在一些实施方案中,pg1是三氟乙酸酯基团。使用任何合适的卞位氧化方法,可以由2-1制备式2-2化合物。例如,在一些实施方案中,反应在气球压力下的氧气、n-羟基邻苯二甲酰胺和二乙酸钴催化剂的存在下进行。在一些实施方案中,反应在乙腈的存在下进行。反应可在附加热(例如,60℃)的存在下进行。使用将酮还原成醇的任何合适的方法,可以由式2-2化合物制备式2-3化合物。例如,可以在还原剂诸如硼烷存在下使用corey
–
bakshi
–
shibata催化剂(cbs催化剂)。在替代实施方案中,可以使用过渡金属催化的转移氢化系统。在手性配体的存在下,过渡金属转移氢化反应可能导致酮的不对称还原。
[0879]
方案2
[0880][0881]
方案3示出了用于制备式3-4的化合物的方法。pg2是任何合适的醇保护基团,例如thp。式3-1的杂环溴化物可以使用任何合适的使卤化物与烷基硼酸盐偶联的方法与式3-2的三氟硼酸盐偶联。例如,在一些实施方案中,反应可以在催化剂体系诸如二环己基-[2-(2,6-二异丙氧基苯基)苯基]磷烷甲磺酸钯(ii)n-甲基-2-苯基-苯胺和碱诸如cs2co3的存在下进行。反应可在附加热(例如,100℃)的存在下进行。在一些实施方案中,反应在溶剂诸如甲苯中进行。任何合适的去除醇保护基团的方法都可以用于制备式3-4化合物。例如,当pg2是thp时,可以使用在溶剂诸如甲醇中的酸诸如对甲苯磺酸。反应可以在室温下进行。
[0882]
方案3
[0883][0884]
方案4示出了由式3-1的芳基卤化物制备式4-5的醇的方法。用于在杂芳基溴化物上进行锂-卤素交换的任何合适的试剂(例如用正丁基锂处理)可以用于原位生成杂芳基有机金属试剂。反应可以在低温(例如,0至-78℃)下在溶剂例如thf或乙醚中进行。在路易斯酸诸如三氟硼乙醚的存在下,将有机金属试剂加成到环氧化物诸如环氧乙烷上,得到式4-2的醇。在一些实施方案中,锂卤素交换反应可以在连续流动条件下进行。
[0885]
在用于制备式4-2化合物的替代性方法中,式4-3的醛可以与试剂诸如式4-4亚基进行维蒂希反应,得到式4-5的烯醇醚。在一些实施方案中,反应在碱诸如叔丁醇钾的存在下在溶剂诸如乙醚中进行。在一些实施方案中,通过用酸诸如hcl处理,式4-5的烯醇醚可以转化为式4-6化合物。在一些实施方案中,可以使用任何合适的用于将醛还原成醇的试剂,例如可以使用在甲醇中的硼氢化钠,由式4-6化合物制备式4-2化合物。
[0886]
方案4
[0887][0888]
方案5示出了用于制备式1-1化合物的方法。pg3是任何合适的氮保护基。式5-1化合物可以用任何合适的氮保护基团进行保护。例如,当pg3是boc基团时,可以使用任何合适的试剂将boc基团加成到胺上。式5-3化合物(weinreb酰胺)可以使用任何合适的酰胺偶联剂由式5-2化合物和n-甲基n-甲氧基胺制备。例如,反应可以在t3p和dipea的存在下在溶剂诸如二氯甲烷中进行。通过添加有机金属试剂诸如甲基碘化镁,可以由式5-3化合物制备式5-5化合物。该反应可以在低温(例如0℃)下在溶剂诸如thf中进行。式1-1化合物可以使用任何合适的方法从式5-5化合物中去除氮保护基团来制备。例如,当pg3是boc时,可以使用hcl于1,4-二噁烷中的溶液。
[0889]
方案5
[0890][0891]
方案6示出了用于由式6-1的n-保护的β-氨基酸制备式1-3化合物的替代性方法。pg4可以是boc或任何合适的氮保护基团。化合物6-2二镁盐可以用试剂诸如cdi在溶剂诸如thf中偶联到式6-1化合物上。将式6-3化合物与式6-4的醛缩合,得到式6-5化合物。在一些实施方案中,该反应可以通过在溶剂诸如二氯甲烷中用酸诸如tfa处理式6-3化合物,随后添加式6-4的醛来进行。式1-3化合物可以由式6-5化合物通过在溶剂诸如二氯甲烷中用酸诸如甲磺酸处理来制备。反应可以在附加热(例如,回流条件)的存在下进行。
[0892]
方案6
[0893]
[0894]
s1的制备
[0895]
2-(3-噻吩基)乙醇(s1)
[0896][0897]
2-(3-噻吩基)乙醇(s1)从商业来源获得。
[0898]
s2的制备
[0899]
2-(5-氯-3-噻吩基)乙醇(s2)
[0900][0901]
步骤1.叔丁基-二甲基-[2-(3-噻吩基)乙氧基]硅烷(c1)的合成
[0902]
向2-(3-噻吩基)乙醇s1(18g,140.4mmol)于dmf(100ml)中的溶液中连续添加咪唑(12g,176.3mmol)和叔丁基-氯-二甲基-硅烷(24g,159.2mmol)。观察到放热。将反应混合物在室温下搅拌3小时。将反应混合物用mtbe(500ml)稀释并用水(200ml)、0.5n hcl(200ml)、水(200ml)和盐水(200ml)洗涤。将有机层干燥,过滤,并在真空中浓缩。将有机层溶解于庚烷并使其穿过硅胶塞;将其用1-5% mtbe/庚烷洗涤。去除溶剂,得到叔丁基-二甲基-[2-(3-噻吩基)乙氧基]硅烷c1(34g,99%)。1h nmr(400mhz,氯仿-d)δ7.28-7.13(m,1h),7.04-6.91(m,2h),3.80(t,j=6.9hz,2h),2.90-2.75(m,2h),0.88(s,9h),-0.00(s,6h)。
[0903]
步骤2.叔丁基-[2-(5-氯-3-噻吩基)乙氧基]-二甲基-硅烷(c2)的合成
[0904]
向冷却至0℃的2,2,6,6-四甲基哌啶(36ml,213.3mmol)于四氢呋喃(200ml)中的溶液中添加己基锂(92ml 2.3m,211.6mmol)。将反应物在-78℃下搅拌30分钟。在20分钟内将叔丁基-二甲基-[2-(3-噻吩基)乙氧基]硅烷c1(34g,138.8mmol)于thf(150ml)中的溶液添加到反应物中。将反应物在-30℃下搅拌45分钟。将反应物冷却至-78℃并分批添加1,1,1,2,2,2-六氯乙烷(54g,228.1mmol)。将反应物升温至室温并搅拌过夜。将反应物用饱和氯化铵(125ml)淬灭,用水(100ml)稀释,用etoac(500ml)萃取,并用etoac(100ml)反萃取。将合并的有机层用0.5n hcl(200ml)、水(300ml)和盐水(200ml)洗涤。将有机层经硫酸钠干燥,过滤,并浓缩,得到粗产物叔丁基-[2-(5-氯-3-噻吩基)乙氧基]-二甲基-硅烷c2。
[0905]
步骤3.2-(5-氯-3-噻吩基)乙醇(s2)的合成
[0906]
向叔丁基-[2-(5-氯-3-噻吩基)乙氧基]-二甲基-硅烷c2(12.5g,42.89mmol)于2-me-thf(120ml)中的溶液中添加tbaf(63ml于thf中的1m,63.00mmol)。将反应物在室温下搅拌过夜。将反应物分配于etoac(400ml)与水(400ml)之间。分离各层,并将有机层用etoac(200ml)萃取,经硫酸钠干燥,过滤,并在真空中浓缩。通过硅胶色谱纯化(梯度:0-50%于庚烷中的etoac),得到产物2-(5-氯-3-噻吩基)乙醇s2(4.5g,58%)。1h nmr(300mhz,氯仿-d)δ6.82(d,j=0.9hz,2h),3.89-3.71(m,2h),2.79(t,j=6.4hz,2h),2.05(s,1h)。lcms m/z 162.91[m+h]
+
。
[0907]
s3的制备
[0908]
2-[5-(三氟甲基)-3-噻吩基]乙醇(s3)
[0909][0910]
步骤1.2-[2-[5-(三氟甲基)-3-噻吩基]乙氧基]四氢吡喃(c5)的合成
[0911]
向4-溴-2-(三氟甲基)噻吩c3(9g,38.96mmol)、二环己基-[2-(2,6-二异丙氧基苯基)苯基]磷烷;甲磺酸酯;n-甲基-2-苯基-苯胺钯(2+)(1.8g,2.117mmol)和三氟(2-四氢吡喃-2-基氧基乙基)硼化钾c4(10g,42.36mmol)的混合物中添加甲苯(75ml)和水(25ml)。使氮气穿过反应物顶部,之后添加cs2co3(40g,122.8mmol)。添加回流冷凝器,并将反应物在100℃下加热48小时。将反应物用etoac(150ml)和水(100ml)稀释。分离两层并将水层用etoac(100ml)萃取。将合并的有机物用盐水洗涤,经硫酸钠干燥,过滤,并在真空中浓缩。通过硅胶色谱纯化(梯度:0-20%于庚烷中的etoac),得到产物2-[2-[5-(三氟甲基)-3-噻吩基]乙氧基]四氢吡喃c5(9g,82%)。1h nmr(300mhz,氯仿-d)δ7.37(t,j=1.3hz,1h),7.22(d,j=1.5hz,1h),4.62(dd,j=4.2,2.8hz,1h),3.96(dt,j=9.6,6.7hz,1h),3.75(ddd,j=11.3,8.0,3.4hz,1h),3.62(dt,j=9.6,6.5hz,1h),3.55-3.41(m,1h),2.93(t,j=6.6hz,2h),1.83(ddd,j=14.2,6.6,3.4hz,1h),1.73(td,j=9.0,4.2hz,1h),1.66-1.50(m,4h)。
[0912]
步骤2.2-[5-(三氟甲基)-3-噻吩基]乙醇(s3)的合成
[0913]
在室温下向2-[2-[5-(三氟甲基)-3-噻吩基]乙氧基]四氢吡喃c5(1.8g,6.100mmol)于meoh(25ml)中的搅拌溶液中添加4-甲基苯磺酸单水合物(1.2g,6.309mmol)。将反应混合物在室温下搅拌1小时。将反应混合物用水(100ml)稀释并用mtbe(2x 100ml)萃取。将合并的有机层用稀的nahco3(10ml nahco3和10ml水)和盐水(10ml)洗涤,经硫酸钠干燥,过滤,并在真空下蒸发,得到粗制化合物。通过硅胶色谱纯化(梯度:0-30%于庚烷中的etoac),得到产物2-[5-(三氟甲基)-3-噻吩基]乙醇s3(820mg,69%)。1h nmr(400mhz,氯仿-d)δ7.35(p,j=1.3hz,1h),7.23(dt,j=1.7,0.9hz,1h),3.85(td,j=7.1,6.5,2.7hz,2h),2.87(td,j=6.4,0.8hz,2h),2.06(d,j=4.3hz,1h)。
[0914]
s3的替代性制备
[0915]
2-[5-(三氟甲基)-3-噻吩基]乙醇(s3)
[0916][0917]
将4-溴-2-(三氟甲基)噻吩c3(50.13g,217.0mmol)于et2o(500ml)中的溶液冷却至-78℃并以适于将温度保持低于-68℃的速率添加nbuli(91ml 2.48m,225.7mmol)。将反应物搅拌20分钟并以将温度保持低于-70℃的速率添加环氧乙烷(14g,317.8mmol)。以将温度保持低于-68℃的速率添加bf3.oet2(28ml,226.9mmol)。bf3.oet2添加是高度放热的。将反应物搅拌一小时在-78℃下并且然后倒入到500ml 1n hcl并用500ml et2o萃取。将萃取物用mgso4干燥,过滤,并在真空中蒸发。通过柱色谱纯化(1600g:等度梯度:10% ch3cn-dcm),得到2-[5-(三氟甲基)-3-噻吩基]乙醇s3(22.48g,53%)。1h nmr(300mhz,氯仿-d)δ7.36(t,j=1.3hz,1h),7.24(d,j=1.5hz,1h),3.88(q,j=6.0hz,2h),2.90(t,j=6.3hz,2h),1.55(t,j=5.4hz,1h)ppm。19f nmr(282mhz,氯仿-d)δ-55.36ppm。
[0918]
s4的制备
[0919]
2-(5-乙基-3-噻吩基)乙醇(s4)
[0920][0921]
步骤1.5-溴噻吩-3-甲醛(c7)的合成
[0922]
在0℃下向噻吩-3-甲醛c6(50g,40.717ml,0.4458mol)于dmf(500ml)中的搅拌溶液中添加nbs(119.02g,0.6687mol)。将反应混合物在室温下搅拌16小时。将反应混合物用冰冷水(600ml)淬灭并用etoac(2x 600ml)萃取。合并的有机层经na2so4干燥,过滤并浓缩。通过硅胶色谱纯化(梯度:0-2%于石油醚中的etoac),得到产物5-溴噻吩-3-甲醛c7(39.2g,44%)。1h nmr(400mhz,氯仿-d)δ9.77(s,1h),7.99(d,j=1.2hz,1h),7.505(d,j=1.6hz,1h)。
[0923]
步骤2.2-溴-4-[(e)-2-甲氧基乙烯基]噻吩(c8)的合成
[0924]
在0℃下向(甲氧基甲基)三苯基氯化膦(115.1g,0.3358mol)于乙醚(450.00ml)中的搅拌溶液中逐滴添加叔丁醇钾(1m于thf中)(381ml 1m,0.3810mol).将反应物在0℃下搅拌1小时。添加5-溴噻吩-3-甲醛c7(45g,0.2215mol)于乙醚(90ml)中的溶液,并且然后将反应混合物在室温下搅拌30分钟。在0℃下将反应混合物用nh4cl溶液(900ml)淬灭并用etoac(2x 700ml)萃取。合并的有机层经na2so4干燥,过滤并浓缩。通过硅胶色谱纯化(洗脱液:石油醚),得到产物2-溴-4-[(e)-2-甲氧基乙烯基]噻吩c8(44.1g,82%)。1h nmr(400mhz,氯仿-d)δ7.25(d,j=2hz,1h),7.18(d,j=0.8hz,1h),7.00(d,j=1.8hz,1h),6.91(d,j=12.8hz,1h),6.97(d,j=1.2hz,1h),6.05(d,j=6.8hz,1h),5.72(d,j=12.8hz,1h),5.22(d,j=6.4hz,1h),3.77(d,j=2.8hz,3h),3.64(d,j=5.2hz,3h)。nmr示出了e和z异构体的1:1混合物。
[0925]
步骤3.2-(5-溴-3-噻吩基)乙醛(c9)的合成
[0926]
在0℃下向2-溴-4-[(e)-2-甲氧基乙烯基]噻吩c8(14.1g,0.0602mol)于1,4-二噁烷(141.00ml)中的搅拌溶液中添加hcl(60.200ml 4m于二噁烷中,0.2408mol)。将反应混合物在室温下搅拌30分钟。在0℃下将反应混合物用饱和nahco3淬灭并用etoac萃取。将有机层经na2so4干燥,过滤,并浓缩,得到2-(5-溴-3-噻吩基)乙醛c9(13.1g,89%)。1h nmr(400mhz,氯仿-d)δ9.72(t,j=2.4hz,1h),7.04(s,1h),6.94(d,j=1.2hz,1h),3.66(d,j=1.6hz,2h)。
[0927]
步骤4.2-(5-溴-3-噻吩基)乙醇(c10)的合成
[0928]
在0℃下向2-(5-溴-3-噻吩基)乙醛c9(38.5g,0.1524mol)于meoh(390ml)中的搅拌溶液中添加nabh4(13.3g,0.3515mol)。将反应物搅拌1小时。将反应混合物用冰水(400ml)淬灭并在真空中浓缩,以去除meoh。将粗残余物用水(500ml)稀释并用etoac(3x 300ml)萃取。将分离的有机层经na2so4干燥,过滤,并浓缩。通过使用中性氧化铝的柱色谱纯化(洗脱液:35%于石油醚中的etoac),得到呈淡黄色液体的产物2-(5-溴-3-噻吩基)乙醇c10(30.2g,84%)。1h nmr(300mhz,dmso-d6)δ7.20(t,j=0.9hz,1h),7.10(d,j=1.2hz,1h),4.64(q,j=5.2hz,1h),3.59-3.55(m,2h),2.67(t,j=6.8hz,2h)。
[0929]
步骤5.2-[2-(5-溴-3-噻吩基)乙氧基]四氢吡喃(c11)的合成
[0930]
在室温下向2-(5-溴-3-噻吩基)乙醇c10(8g,0.0328mol)于thf(80.ml)中的搅拌溶液中添加3,4-二氢-2h-吡喃(3.7696g,3.8ml,0.0448mol)和ptsa(259mg,0.0015mol)并且然后将反应混合物在室温下搅拌16小时。将反应混合物用饱和水溶液k2co3(300ml)淬灭并用etoac(2x 600ml)萃取。将有机层经na2so4干燥,过滤,并浓缩。通过硅胶色谱纯化(梯度:0-5%于石油醚中的etoac),得到产物2-[2-(5-溴-3-噻吩基)乙氧基]四氢吡喃c11(10.1g,90%)。1h nmr(400mhz,氯仿-d)δ6.95(d,j=1.6hz,1h),6.92(d,j=0.8,1h),4.59(t,j=2.8hz,1h),3.94-3.74(m,2h),3.60-3.46(m,2h),2.85(q,j=6.4hz,2h),1.80-1.61(m,6h)。lcms m/z 291.03[m+h]
+
。
[0931]
步骤6.2-[2-(5-乙基-3-噻吩基)乙氧基]四氢吡喃(c12)的合成
[0932]
在-76℃下向2-[2-(5-溴四氢噻吩-3-基)乙氧基]四氢吡喃c11(25g,0.0719mol)于thf(250.00ml)中的搅拌溶液中添加n-buli(2.5m于己烷中)(46.1ml 2.5m,0.1153mol)。将反应物搅拌1小时。在-76℃下添加碘乙烷(24.832g,12.8ml,0.1592mol)并且然后将反应
温度缓慢增加到室温,并然后搅拌16小时。将反应混合物用nh4cl溶液(500ml)淬灭,并用etoac(2x 300ml)萃取。合并的有机层经na2so4干燥,过滤并浓缩。通过硅胶色谱纯化(梯度:0-3%于石油醚中的etoac),得到产物2-[2-(5-乙基-3-噻吩基)乙氧基]四氢吡喃c12(13.2g,59%)。lcms m/z 241.21[m+h]
+
.
[0933]
步骤7.2-(5-乙基-3-噻吩基)乙醇(s4)的合成
[0934]
在室温下向2-[2-(5-乙基-3-噻吩基)乙氧基]四氢吡喃c12(4.4g,0.0142mol)于meoh(44ml)中的搅拌溶液中添加ptsa(3.0g,0.0174mol)并将反应物搅拌2小时。将反应混合物用饱和nahco3溶液(150ml)淬灭,用etoac(2x150ml)萃取,经na2so4干燥,过滤,并浓缩。通过使用中性氧化铝的柱色谱纯化(洗脱液:10%于石油醚中的etoac),得到产物2-(5-乙基-3-噻吩基)乙醇s4(1.1g,45%)。1h nmr(400mhz,dmso-d6)δ6.90(d,j=1.2hz,1h),6.71(d,j=1.2hz,1h),4.62-4.58(m,1h),3.59-3.55(m,2h),2.77-2.71(m,2h),2.64(t,j=7.2,2h),1.22-1.85(m,3h)。
[0935]
s5的制备
[0936]
2-(5-乙基-2-噻吩基)乙醇(s5)
[0937][0938]
步骤1.2-(5-乙基-2-噻吩基)乙醇(s5)的合成
[0939]
在45分钟内在0℃下向2-乙基噻吩c13(54g,466.9mmol)于无水thf(1l)中的溶液中添加己烷中的n-buli(255ml 2.2m,561.0mmol)。得到淡黄色/橙色溶液。在添加期间温度范围为0-10℃。将混合物在室温下搅拌30分钟。在冷却至0℃后,在30分钟内添加环氧乙烷(200ml 2.9m,580.0mmol)的溶液。将反应物在0℃下搅拌2小时并且然后升温至室温。将反应混合物用水(700ml)和饱和nh4cl(200ml)淬灭并蒸发thf。将产物用etoac(1x 400ml;2x 150ml)萃取。将合并的有机层经无水硫酸钠干燥,过滤,并在真空中浓缩。使有机层穿过硅胶塞,用dcm(1000ml)、80% etoac/庚烷(2x 200ml)和dcm(2x 250ml)洗涤,得到2-(5-乙基-2-噻吩基)乙醇s5(71.25g,93%)。1h nmr(300mhz,氯仿-d)δ6.69(dt,j=3.4,0.9hz,1h),6.64(dt,j=3.3,1.0hz,1h),3.84(t,j=6.3hz,2h),3.08-2.97(m,2h),2.82(qd,j=7.5,1.0hz,2h),1.31(t,j=7.5hz,4h)。
[0940]
s6的制备
[0941]
2-[5-(三氟甲基)-2-噻吩基]乙醇(s6)
[0942][0943]
步骤1.2-(5-碘-2-噻吩基)乙醇(c15)的合成
[0944]
在0℃下向nis(104.83g,0.4680mol)于dcm(1000ml)中的搅拌溶液中添加2-(2-噻吩基)乙醇c14(50g,0.3900mol)。将反应物升温至室温并搅拌16小时。将反应混合物用dcm(500ml)稀释,用饱和硫代硫酸钠、盐水洗涤,经na2so4干燥,并在真空中浓缩。通过柱色谱纯化(洗脱液:20%于石油醚中的etoac),得到产物2-(5-碘-2-噻吩基)乙醇c15(62g,56%)。1h nmr(400mhz,氯仿-d)δ7.08(d,j=3.6hz,1h),6.57-6.56(m,1h),3.82(q,j=6hz,2h),3.05(q,j=6.4hz,2h)。lcms m/z 254.89[m+h]
+
。
[0945]
步骤2.2-[2-(5-碘-2-噻吩基)乙氧基]四氢吡喃(c16)的合成
[0946]
在室温下向2-(5-碘-2-噻吩基)乙醇c15(15g,0.0525mol)和3,4-二氢-2h-吡喃(6.6284g,0.0788mol)于thf(60ml)中的搅拌溶液中添加ptsa(1.3604g,1.2714ml,0.0079mol)。将反应物在氩气气球压力下搅拌16小时。将反应混合物减压浓缩。通过硅胶色谱纯化(洗脱液:5%于石油醚中的etoac),得到产物2-[2-(5-碘-2噻吩基)乙氧基]四氢吡喃c16(12.8g,68%)。1h nmr(400mhz,dmso-d6)δ7.14(d,j=3.6hz,1h),6.64(d,j=3.6hz,1h),4.59(t,j=3.6hz,1h),3.80-3.76(m,1h),3.74-3.67(m,1h),3.54-3.50(m,1h),3.48-3.41(m,1h),3.03(t,j=6hz,2h),1.75-1.69(m,1h),1.61-1.59(m,1h),1.51-1.42(m,4h)。
[0947]
步骤3.2-[2-[5-(三氟甲基)-2-噻吩基]乙氧基]四氢吡喃(c17)的合成
[0948]
向2-[2-(5-碘-2-噻吩基)乙氧基]四氢吡喃c16(10g,0.0219mol)和2,2-二氟-2-氟磺酰基-乙酸甲酯(12.63g,0.0657mol)于dmf(40ml)中的搅拌溶液中添加溴化铜(i)二甲基硫醚络合物99%(2.241g,0.0109mol)。将反应物在100℃下搅拌16小时。将反应物升温至室温,用etoac(100ml)稀释,过滤,并用etoac(50ml)洗涤。将滤液用冷冻的盐水溶液洗涤,经na2so4干燥,并在减压下浓缩。通过使用中性氧化铝的柱色谱纯化(洗脱液:5%于石油醚中的etoac),得到产物2-[2-[5-(三氟甲基)-2-噻吩基]乙氧基]四氢吡喃c17(2.9g,41%)。1h nmr(400mhz,氯仿-d)δ7.25(s,1h),6.82-6.81(m,1h),4.63(t,j=3.6hz,1h),4.00-3.95(m,1h),3.78-3.75(m,1h),3.64-3.58(m,1h),3.51-3.48(m,1h),3.12(d,j=6.4hz,2h),1.90-1.80(m,1h),1.73-1.64(m,1h),1.65-1.51(m,4h)。gcms:87.26%,m/z:280[m]
+
。
[0949]
步骤4.2-[5-(三氟甲基)-2-噻吩基]乙醇(s6)的合成
[0950]
在室温下向2-[2-[5-(三氟甲基)-2-噻吩基]乙氧基]四氢吡喃c17(5.8g,0.0170mol)于meoh(100ml)中的搅拌溶液中添加ptsa(2.93g,0.0170mol)。将反应物搅拌16小时。将反应混合物减压浓缩。通过使用中性氧化铝的柱色谱纯化(洗脱液:10%于石油醚中的etoac),得到产物2-[5-(三氟甲基)-2-噻吩基]乙醇s6(2.3g,61%)。1h nmr(400mhz,
dmso-d6)δ7.52-7.51(m,1h),6.99-6.98(m,1h),4.92(t,j=4.8hz,1h),3.65-3.61(m,2h),2.98(t,j=6hz,2h)。
19
f nmr(376.22mhz,dmso-d6)δ-53.53(s,3f)。gcms:88.56%m/z:196.0[m]
+
。
[0951]
s7的制备
[0952]
2-[5-(三氟甲基)-2-噻吩基]丙-1-醇(s7)
[0953][0954]
步骤1.2-(2-噻吩基)乙酸乙酯乙醇(c19)的合成
[0955]
在室温下向2-(2-噻吩基)乙酸c18(100g,703.35mmol)于乙醇(2000ml)中的搅拌溶液中添加hcl(水溶液)(50ml 36%(w/v),493.68mmol)。将反应混合物搅拌12小时在70℃下。将混合物浓缩,并将所得粗材料用etoac(1000ml)稀释,用5% na2co3水溶液(3x 200ml)和盐水(200ml)洗涤。将有机层干燥并浓缩,得到所需产物2-(2-噻吩基)乙酸乙酯c19(100g,82%)。1h nmr(氯仿-d,400mhz)δ7.22-7.21(dd,j=1.2hz,j=3.6hz,1h),6.97-6.95(m,2h),4.21-4.16(q,j=7.2hz,2h),3.83(s,2h),1.30-1.26(t,j=7.2hz,3h)。lcms m/z 171.26[m+h]
+
。
[0956]
步骤2.2-(2-噻吩基)丙酸乙酯(c20)的合成
[0957]
在-78℃下向2-(2-噻吩基)乙酸乙酯c19(1.36g,7.99mmol)于thf(20ml)中的溶液中添加(二异丙基氨基)锂(8ml 1m,8.000mmol)。15分钟后,添加mei(500μl,8.032mmol)并将反应混合物在-78℃下搅拌2小时。将反应物用饱和nh4cl(50ml)淬灭并用etoac萃取。将有机层干燥并浓缩,得到油状物。通过硅胶色谱纯化(梯度:0至25%于庚烷中的etoac),得到产物2-(2-噻吩基)丙酸乙酯c20(1.04g,71%)。1h nmr(300mhz,氯仿-d)δ7.25-7.17(m,1h),7.02-6.93(m,2h),4.18(d,j=7.2hz,2h),4.02(q,j=7.1hz,1h),1.60(d,j=7.2hz,3h),1.28(t,j=7.1hz,3h)。
[0958]
步骤3.2-(5-碘-2-噻吩基)丙酸乙酯(c21)的合成
[0959]
向2-(2-噻吩基)丙酸乙酯c20(35g,143.99mmol)于乙酸(350ml)中的搅拌溶液中添加n-碘琥珀酰亚胺(38.875g,172.79mmol)。将反应混合物在100℃下搅拌一小时。将混合物浓缩并将所得粗材料用etoac(700ml)稀释,依次用水(300ml)、饱和碳酸氢钠溶液(300ml)、饱和硫代硫酸钠溶液(300ml)和盐水溶液(250ml)洗涤。将有机层经na2so4干燥,过滤,并浓缩,得到粗制产物。通过硅胶色谱纯化(洗脱液:3%于石油醚中的etoac),得到产物2-(5-碘-2-噻吩基)丙酸乙酯c21(30g,42%)。1h nmr(氯仿-d,400mhz)δ7.08(d,j=4hz,1h),6.62(d,j=4hz,1h),4.19-4.13(m,2h),3.98-3.92(m,1h),1.55-1.51(m,3h),1.28-1.24(m,3h)。lcms m/z 309.9[m+h]
+
。
[0960]
步骤4.2-[5-(三氟甲基)-2-噻吩基]丙酸乙酯(c22)的合成
[0961]
在氮气气氛下向2-(5-碘-2-噻吩基)丙酸乙酯c21(5g,9.9629mmol)和2,2-二氟-2-(氟磺酰基)乙酸甲酯(9.57g,49.814mmol)于dmf(50ml)中的搅拌溶液中添加cui(2.2768g,11.955mmol)。将反应混合物在100℃下搅拌12小时。通过过滤混合物并将垫用乙醚(2x 100ml)洗涤。将滤液用冷水(100ml)淬灭。分离两层并将水层用乙醚(2x 50ml)萃取。将合并的有机层用盐水(30ml)洗涤,干燥,并浓缩。通过硅胶色谱纯化(洗脱液:3%于石油醚中的etoac),得到产物2-[5-(三氟甲基)-2-噻吩基]丙酸乙酯c22(2g,58%)。1h nmr(氯仿-d,400mhz)δ7.29-7.26(m,1h),6.92-6.90(m,1h),4.21-4.15(m,2h),3.99-3.96(m,1h),1.57-1.53(m,3h),1.23-1.27(m,3h)。gcms:m/z:252.1[m]
+
[0962]
步骤5.2-[5-(三氟甲基)-2-噻吩基]丙-1-醇(s7)的合成
[0963]
在0℃下向2-[5-(三氟甲基)-2-噻吩基]丙酸乙酯c22(12g,41.701mmol)于thf(250ml)中的搅拌溶液中逐滴添加dibal-h(35.584ml 25%(w/v),62.5mmol)。在0℃下将反应混合物搅拌2小时。在0℃下将混合物缓慢用饱和nh4cl溶液(300ml)淬灭并通过过滤悬浮液并将垫用etoac(2x 200ml)洗涤。将滤液分离成各层。将水层用etoac(2x 200ml)萃取。将合并的有机层用盐水(200ml)洗涤,经na2so4干燥,并浓缩。通过硅胶色谱纯化(洗脱液:3%于石油醚中的etoac),得到粗制产物。使用手性sfc分离将外消旋化合物2-[5-(三氟甲基)-2-噻吩基]丙-1-醇(1.6g,7.3067mmol)与二甲基过度烷基化副产物分离。柱:daicelad-h,30x 250mm;流动相:10%甲醇/己烷混合物(7:3),90%二氧化碳。流速:90g/分钟。2-[5-(三氟甲基)-2-噻吩基]丙-1-醇s7(3.64g)。1h nmr(400mhz,氯仿-d)δ7.52(m,1h),7.00(m,1h),4.97(t,j=5.6hz,1h),3.51(t,j=6.0hz,2h)3.17(m,1h),1.27(d,j=6.8hz,3h)。gcms:m/z:210.0[m]
+
。
[0964]
s8的制备
[0965]
2-甲基-2-[5-(三氟甲基)-2-噻吩基]丙-1-醇(s8)
[0966][0967]
在s7的sfc纯化期间由于上文所述的步骤2中的过度烷基化而获得呈副产物形式的s8。
[0968]
s9、s10和s11的制备
[0969]
2-甲基-2-[5-(氯)-2-噻吩基]丙-1-醇(s9)
[0970]
2-[5-(氯)-2-噻吩基]丙-1-醇(s10[enant-1]、s11[enant-2])
[0971][0972]
步骤1.2-(5-氯-2-噻吩基)丙酸乙酯(c24)的合成
[0973]
向2-(2-噻吩基)丙酸乙酯c20(1g,4.1139mmol)于乙酸(10ml)中的搅拌溶液中添加n-氯琥珀酰亚胺c23(549.34mg,4.1139mmol)。将反应混合物在100℃下搅拌1小时。将混合物浓缩并将所得粗材料用etoac(25ml)稀释,用水(10ml)、饱和碳酸氢钠溶液(10ml)、饱和硫代硫酸钠溶液(10ml)和盐水溶液(10ml)洗涤。将有机层经na2so4干燥,过滤,并浓缩,得到粗制产物。通过硅胶色谱纯化(洗脱液:3%于石油醚中的etoac),得到产物2-(5-氯-2-噻吩基)丙酸乙酯c24(700mg,60%)。1h nmr(氯仿-d,400mhz):δ=6.75-6.73(m,1h),6.71-6.69(m,1h),4.20-4.14(m,2h),3.88-3.73(q,j=6.4hz,1h),1.55-1.53(t,j=2.8hz,3h),1.30-1.221(m,3h)。gcms:m/z:218.0[m]
+
[0974]
步骤2.2-(5-氯-2-噻吩基)-2-甲基-丙-1-醇和2-(5-氯-2-噻吩基)丙-1-醇(s9)和(c25)的合成
[0975]
在0℃下向2-(5-氯-2-噻吩基)丙酸乙酯c24(25g,86.877mmol)于thf(500ml)中的搅拌溶液中逐滴添加dibal-h(74.135ml 25%(w/v),130.32mmol)。在0℃下将反应混合物搅拌2小时。在0℃下将混合物缓慢用饱和nh4cl溶液(300ml)淬灭并通过过滤悬浮液并将垫用etoac(2x 200ml)洗涤。将滤液分离成两层。将水层用etoac(2x 200ml)萃取。将合并的有机层用盐水(200ml)洗涤,经硫酸钠干燥,并浓缩。通过硅胶色谱纯化(洗脱液:3%于石油醚中的etoac),得到s9 2-(5-氯-2-噻吩基)-2-甲基-丙-1-醇(410mg,2%)。1h nmr(氯仿-d,400mhz)δ6.76-6.75(d,j=4hz,1h),6.67-6.65(t,j=4hz,1h),3.54-3.52(d,j=6.8hz,2h),1.47-1.43(t,j=6.8hz,1h),1.34(s,6h)。gcms:m/z:190.0[m]
+
;以及2-(5-氯-2-噻吩基)丙-1-醇c25(12g,72%)。1h nmr(氯仿-d,400mhz)δ6.76-6.75(d,j=3.6hz,1h),6.66-6.65(dd,j=4.4hz,1h),3.71-3.61(m,2h),3.15-3.10(m,1h),1.57-1.52(m,1h),1.34-1.31(t,j=6hz,3h)。gcms:m/z:176.0[m]
+
。注意:在c20的合成期间由于过度烷基化而获得呈副产物形式的二甲基化合物(s9)。
[0976]
步骤3.2-(5-氯-2-噻吩基)丙-1-醇(s10)和(s11)的合成
[0977]
通过手性sfc分离将外消旋化合物2-(5-氯-2-噻吩基)丙-1-醇c25(12g,62.492mmol)分离成组成型对映异构体。柱:daicelad-h,30x250mm;流动相:10%甲醇/己烷混合物(7:3),90%二氧化碳。流速:90g/分钟。2-(5-氯-2-噻吩基)丙-1-醇s10(4g,35%)。1h nmr(氯仿-d,400mhz)δ6.76-6.75(d,j=3.6hz,1h),6.66-6.65(dd,j=3.6hz,1h),3.73-3.61(m,2h),3.17-3.10(m,1h),1.52-1.49(t,j=5.2hz,1h),1.32-1.30
(d,j=6.8hz,3h)。gcms:m/z:176.0[m]
+
;以及2-(5-氯-2-噻吩基)丙-1-醇s11(3.75g,34%)。1h nmr(氯仿-d,400mhz)δ6.76-6.75(d,j=4hz,1h),6.66-6.65(dd,j=3.6hz,1h),3.73-3.61(m,2h),3.15-3.10(q,j=6.8hz,1h),1.51-1.48(t,j=5.6hz,1h),1.33-1.30(d,j=7.2hz,3h)。gcms:m/z:176.0[m]
+
。
[0978]
s12和s13的制备
[0979]
2-(5-乙基-2-噻吩基)丙-1-醇(s12 enant-1)和(s13 enant-2)
[0980][0981]
步骤1.2-(5-乙酰基-2-噻吩基)丙酸乙酯(c26)的合成
[0982]
在0℃下向2-(2-噻吩基)丙酸乙酯c20(80g,336.92mmol)于dcm(1500ml)中的搅拌溶液中逐滴添加乙酰氯(39.671g,35.934ml,505.38mmol),随后添加alcl3(67.388g,505.38mmol)在0℃下。将反应混合物搅拌2小时在0℃下。将混合物缓慢用冰水(1000ml)淬灭,分离两层,并将水层用dcm(2x500ml)萃取。将合并的有机层用盐水(500ml)洗涤,经硫酸钠干燥。通过硅胶色谱纯化(梯度:0-5%于石油醚中的etoac),得到产物2-(5-乙酰基-2-噻吩基)丙酸乙酯c26(60g,73%)。1h nmr(氯仿-d,400mhz)δ7.56-7.54(t,j=4.0hz,1h),6.99-6.98(m,1h),4.20-4.14(m,2h),4.01-3.96(q,j=7.2hz,1h),2.52(s,3h),1.60-1.56(d,j=7.2hz,3h),1.28-1.23(m,3h)。lcms m/z 227.1[m+h]
+
.
[0983]
步骤2.2-(5-乙基-2-噻吩基)丙酸乙酯(c27)的合成
[0984]
在0℃下向2-(5-乙酰基-2-噻吩基)丙酸乙酯c26(60g,245.79mmol)于tfa(400ml)中的搅拌溶液中逐滴添加三乙基-硅烷(42.870g,58.9ml,368.69mmol)。在室温下将反应混合物搅拌4小时。将反应物浓缩并用冰水(500ml)淬灭并用etoac(3x 500ml)萃取。将合并的有机层用盐水(250ml)洗涤,经硫酸钠干燥,并浓缩,得到粗制产物。通过硅胶色谱纯化(梯度:0-3%于石油醚中的etoac),得到产物2-(5-乙基-2-噻吩基)丙酸乙酯c27(50g,82%)。1h nmr(氯仿-d,400mhz)δ6.73-6.72(dd,j=3.6hz,1h),6.62-6.60(m,1h),4.18-4.13(m,2h),3.93-3.88(q,j=7.2hz,1h),2.82-2.78(m,2h),1.55-1.53(d,j=7.2hz,3h)1.30-1.23(m,6h)。lcms m/z 213.2[m+h]
+
。
[0985]
步骤3.2-(5-乙基-2-噻吩基)丙-1-醇(c28)的合成
[0986]
在0℃下向2-(5-乙基-2-噻吩基)丙酸乙酯c27(50g,200.18mmol)于thf(1000ml)中的搅拌溶液中逐滴添加dibal-h(25%于甲苯)(227.75ml 25%(w/v),400.36mmol)。在0℃下将反应混合物搅拌2小时。在0℃下将混合物缓慢用饱和nh4cl溶液(500ml)淬灭并用etoac(2x 500ml)萃取。将合并的有机层用盐水(250ml)洗涤,经硫酸钠干燥,并浓缩。通过硅胶色谱纯化(梯度:0-5%于石油醚中的etoac),得到产物2-(5-乙基-2-噻吩基)丙-1-醇c28(31g,89%)。1h nmr(氯仿-d,400mhz):δ6.69-6.68(d,j=3.6hz,1h),6.64-6.62(m,
1h),3.72-3.60(m,2h),3.18-3.13(q,j=6.8hz,1h),2.83-2.77(m,2h),1.61-1.5(m,1h),1.35-1.28(m,6h)。lcms m/z 171.02[m+h]
+
。
[0987]
步骤4.2-(5-乙基-2-噻吩基)丙-1-醇(s12)和(s13)的合成
[0988]
通过手性sfc分离将外消旋化合物2-(5-乙基-2-噻吩基)丙-1-醇c28(31g,178.06mmol)分离成组成型对映异构体。柱:daicelad-h,30x250mm;流动相:10%甲醇/己烷混合物(7:3),85%二氧化碳。2-(5-乙基-2-噻吩基)丙-1-醇s12(13.45g,43%)。1h nmr(氯仿-d,400mhz):δ=6.69-6.68(d,j=3.2hz,1h),6.63-6.62(d,j=3.2hz,1h),3.73-3.61(m,2h),3.19-3.14(q,j=6.8hz,1h),2.83-2.78(m,2h),1.54-1.47(m,1h),1.35-1.27(m,6h)。lcms m/z 171.1[m+h]
+
;以及2-(5-乙基-2-噻吩基)丙-1-醇s13(11.35g,37%)。1hnmr(氯仿-d,400mhz):δ6.68-6.67(d,j=3.6hz,1h),6.63(d,j=3.6hz,1h),3.73-3.61(m,2h),3.20-3.12(m,1h),2.83-2.77(q,j=7.6hz,2h),1.54-1.45(m,1h),1.33-1.27(m,6h)。lcms m/z 171.1[m+h]
+
.
[0989]
s14的制备
[0990]
2-(5-甲基-3-噻吩基)乙醇(s14)
[0991][0992]
在室温下在密封管中向2-(5-溴-3-噻吩基)乙醇c10(2.5g,0.0098mol)于1,4-二噁烷(16.000ml)中的搅拌溶液中添加k2co3(4.9g,0.036mol)。将反应混合物用氩气脱气10分钟。添加xphos pd g2(457mg,580.83μmol)并再次脱气5分钟。添加三甲基硼氧六环(50%于thf中的溶液)(24.605ml 50%(w/v),0.0980mol)并加热至80℃持续16小时。将反应混合物用水(200ml)稀释并用etoac(3x 150ml)萃取。合并的有机层经na2so4干燥,过滤并减压浓缩。通过柱色谱纯化(洗脱液:20%于石油醚中的etoac),得到呈黄色液体的产物s14 2-(5-甲基-3-噻吩基)乙醇(950mg,66%)。1h nmr(400mhz,dmso-d6)δ6.87(d,j=0.8hz,1h),6.68(s,1h),4.59(t,j=5.2hz,1h),3.58-3.53(m,2h),2.63(t,j=7.2hz,2h),2.38(d,j=0.8hz,3h)。
[0993]
s15的制备
[0994]
2-(5-甲基-2-噻吩基)乙醇(s15)
[0995][0996]
步骤1.2-(5-溴-2-噻吩基)乙醇(c30)的合成
[0997]
在-10℃下将2-(2-噻吩基)乙醇c29(15g,0.1170mol)于dmf(150.00ml)中的溶液逐滴添加到nbs(20.824g,0.1170mol)于dmf中的溶液。将反应物在室温下搅拌16小时。将反应混合物用水(300ml)淬灭并用etoac(3x 200ml)萃取。将合并的有机层用6% koh溶液、冰水(2x 150ml)和盐水(150ml)洗涤。有机层经硫酸钠干燥并浓缩。通过柱色谱纯化(洗脱液:10%于石油醚中的etoac),得到产物2-(5-溴-2-噻吩基)乙醇c30(20.5g,79%)。1h nmr(400mhz,氯仿-d)δ6.89(d,j=3.6hz,1h),6.64-6.28(m,1h),3.82(t,j=6.0hz,2h),2.99(t,j=6.0hz,2h)。
[0998]
步骤2.2-[2-(5-溴-2-噻吩基)乙氧基]四氢吡喃(c31)的合成
[0999]
向2-(5-溴-2-噻吩基)乙醇c30(20g,0.0869mol)和3,4-二氢-2h-吡喃(10.969g,0.1304mol)于thf(80ml)中的搅拌溶液中添加ptsa(603mg,0.5636ml,0.0035mol)并将反应物在室温下搅拌24小时。将反应混合物用etoac稀释,用饱和碳酸氢钠溶液(50ml)、水和盐水洗涤。将有机层分离,经硫酸钠干燥,并浓缩。通过硅胶色谱纯化(梯度:0-5%于石油醚中的etoac),得到2-[2-(5-溴-2-噻吩基)乙氧基]四氢吡喃c31(18.5g,64%)。1hnmr(400mhz,氯仿-d)δ6.86(d,j=3.6hz,1h),6.61-6.60(m,1h),4.62(t,j=3.6hz,1h),3.99-3.50(m,4h),3.05-3.01(m,2h),1.73-1.50(m,6h)。
[1000]
步骤3.2-[2-(5-甲基-2-噻吩基)乙氧基]四氢吡喃(c32)的合成
[1001]
在-78℃下向2-[2-(5-溴-2-噻吩基)乙氧基]四氢吡喃c31(19g,0.0555mol)于thf(380.00ml)中的溶液中逐滴添加n-buli(33.320ml 2.5m,0.0833mol)。在-78℃下将反应物搅拌一小时。在-78℃下逐滴添加碘甲烷(15.755g,6.9101ml,0.1110mol)并使反应混合物在室温下搅拌16小时。将反应混合物用饱和nh4cl溶液淬灭并用水稀释。将水层用etoac(2x 250ml)萃取。通过硅胶色谱纯化(洗脱液:100%石油醚),得到2-[2-(5-甲基-2-噻吩基)乙氧基]四氢吡喃c32(19g,130%)。1h nmr(400mhz,氯仿-d)δ6.61(d,j=3.2hz,1h),6.55-6.54(m,1h),4.63(m,1h),3.96-3.50(m,4h),3.03(t,j=2.8hz,2h),2.42(s,3h),1.72-1.42(m,6h)。
[1002]
步骤4.2-(5-甲基-2-噻吩基)乙醇(s15)的合成
[1003]
在室温下向2-[2-(5-甲基-2-噻吩基)乙氧基]四氢吡喃c32(14g,0.0532mol)于meoh(280.00ml)中的溶液中添加ptsa(10.9g,10.187ml,0.0633mol)。将反应物搅拌24小时。将反应混合物用etoac(500ml)稀释并且然后用水(200ml)洗涤。将有机层用饱和碳酸氢钠水溶液(2x 100ml)洗涤。将水层再次用etoac(2x 100ml)萃取。将合并的有机层经na2so4干燥。通过硅胶色谱纯化(梯度:0-15%于石油醚中的etoac),得到2-(5-甲基-2-噻吩基)乙醇s15(6.56g,82%)。1h nmr(400mhz,dmso-d6)δ6.61(d,j=3.6hz,1h),6.58-6.57(d,j=4.0hz,1h),4.73(t,j=5.2hz,1h),3.58-3.53(m,2h),2.82(t,j=6.8hz,2h),2.36(s,3h)。
[1004]
s16的制备
[1005]
[4-(2-羟基乙基)-2-(三氟甲基)-3-噻吩基]甲基乙酸酯(s16)
[1006][1007]
步骤1.叔丁基-二甲基-[2-[5-(三氟甲基)-3-噻吩基]乙氧基]硅烷(c33)的合成
[1008]
向2-[5-(三氟甲基)-3-噻吩基]乙醇s3(500mg,2.498mmol)于dcm(10ml)中的混合物中添加咪唑(190mg,2.791mmol),随后添加tbscl(420mg,2.787mmol),立即沉淀出白色固体。过滤固体并将有机层用1n hcl(10ml)、盐水(10ml)洗涤,用mgso4干燥,过滤,并浓缩。通过硅胶色谱纯化(梯度:0-30%于庚烷中的etoac),得到产物叔丁基-二甲基-[2-[5-(三氟甲基)-3-噻吩基]乙氧基]硅烷c33,假定为定量的并且未经进一步纯化即使用。
[1009]
步骤2.叔丁基-二甲基-[2-[5-(三氟甲基)-2-三甲基甲硅烷基-3-噻吩基]乙氧基]硅烷(c34)的合成
[1010]
将叔丁基-二甲基-[2-[5-(三氟甲基)-3-噻吩基]乙氧基]硅烷c33于thf(10ml)的混合物冷却至-78℃并添加仲-丁基锂(2.3ml 1.4m,3.220mmol),随后添加tmscl(3ml 1m,3.000mmol)。5分钟后,将黄色混合物用饱和氯化铵水溶液淬灭。将混合物用水(10ml)和mtbe(10ml)稀释。将有机层用盐水洗涤,用mgso4干燥,过滤,并浓缩。通过硅胶色谱纯化(梯度:0-10%于庚烷中的etoac),得到叔丁基-二甲基-[2-[5-(三氟甲基)-2-三甲基甲硅烷基-3-噻吩基]乙氧基]硅烷c34(400mg,42%)。1h nmr(300mhz,氯仿-d)δ7.41(d,j=1.2hz,1h),3.80-3.75(m,2h),2.87(t,j=6.8hz,2h),0.87(s,9h),0.36(s,9h),-0.00(d,j=2.2hz,6h)。
[1011]
步骤3.4-[2-[叔丁基(二甲基)甲硅烷基]氧基乙基]-2-(三氟甲基)-5-三甲基甲硅烷基-噻吩-3-甲醛(c35)的合成
[1012]
向冷却至-78℃的叔丁基-二甲基-[2-[5-(三氟甲基)-2-三甲基甲硅烷基-3-噻吩基]乙氧基]硅烷c34(400mg,1.024mmol)于thf(10ml)中的混合物中添加仲-丁基锂(1.2ml 1.4m,1.680mmol),随后添加dmf(3ml 1m,3.000mmol)。5分钟后,将黄色混合物用饱和氯化铵水溶液淬灭。将混合物用etoac(20ml)和水(20ml)稀释并分离。将有机层用盐水(20ml)洗涤,用mgso4干燥,过滤,并浓缩。通过硅胶色谱纯化(洗脱液:100%庚烷),得到产物4-[2-[叔丁基(二甲基)甲硅烷基]氧基乙基]-2-(三氟甲基)-5-三甲基甲硅烷基-噻吩-3-甲醛c35。将混合物浓缩,用庚烷(5ml)稀释并用水(5ml)洗涤。使有机层穿过相分离器,浓缩,并直接错开到下一步骤中。
[1013]
步骤4.[4-[2-[叔丁基(二甲基)甲硅烷基]氧基乙基]-2-(三氟甲基)-5-三甲基甲
硅烷基-3-噻吩基]甲醇(c36)的合成
[1014]
将4-[2-[叔丁基(二甲基)甲硅烷基]氧基乙基]-2-(三氟甲基)-5-三甲基甲硅烷基-噻吩-3-甲醛c35稀释于meoh(1ml)中并向混合物中添加nabh4(7mg,0.1850mmol)。10分钟后,将混合物浓缩,并再稀释于庚烷(2ml)和水(2ml)中。将有机层分离并将水层再用庚烷萃取。将有机层穿过相分离器并浓缩。通过硅胶色谱纯化(梯度:0-10%于庚烷中的etoac),得到产物c36。1hnmr(300mhz,氯仿-d)δ4.65(d,j=6.3hz,2h),4.00
–
3.72(m,2h),3.34(t,j=6.3hz,1h),2.97(t,j=6.1hz,2h),0.82(s,10h),0.36(s,9h)。
[1015]
步骤5.[4-[2-[叔丁基(二甲基)甲硅烷基]氧基乙基]-2-(三氟甲基)-5-三甲基甲硅烷基-3-噻吩基]甲基乙酸酯(c37)的合成
[1016]
向dcm(4ml)中的[4-[2-[叔丁基(二甲基)甲硅烷基]氧基乙基]-2-(三氟甲基)-5-三甲基甲硅烷基-3-噻吩基]甲醇c36中添加dmap(2mg,0.016mmol)和dipea(50μl,0.2871mmol),随后添加ac2o(30μl,0.3180mmol)。将混合物浓缩,用庚烷(5ml)稀释并用水(5ml)洗涤。将有机层穿过相分离器并浓缩,产生产物,其直接错开到下一步骤。
[1017]
步骤6.[4-(2-羟基乙基)-2-(三氟甲基)-3-噻吩基]甲基乙酸酯(s16)的合成
[1018]
将来自步骤5的[4-[2-[叔丁基(二甲基)甲硅烷基]氧基乙基]-2-(三氟甲基)-5-三甲基甲硅烷基-3-噻吩基]甲基乙酸酯c37用etoac(2ml)稀释并向混合物中添加tbaf的thf溶液(1ml 1m,1.000mmol)并将混合物搅拌。将反应物搅拌48小时。将混合物再用etoac(3ml)稀释,用水洗涤,使其穿过相分离器,并浓缩。通过硅胶色谱纯化(梯度:0-60%于庚烷中的etoac),得到产物[4-(2-羟基乙基)-2-(三氟甲基)-3-噻吩基]甲基乙酸酯s16(35mg,12%)。1hnmr(300mhz,氯仿-d)δ7.26(s,1h),5.14(d,j=1.1hz,2h),3.86(t,j=6.4hz,2h),2.97-2.74(m,2h),2.07(s,3h),1.80(s,1h)。lcms m/z 269.21[m+h]
+
。
[1019]
s17的制备
[1020]
1-甲基三唑-4-甲醛(s17)
[1021][1022]
1-甲基三唑-4-甲醛s17从可商购来源获得
[1023]
s18的制备
[1024]
1-(2-甲基磺酰基乙基)三唑-4-甲醛(s18)
[1025][1026]
步骤1.1-叠氮基-2-甲基磺酰基-乙烷(c40)的合成
[1027]
将2-甲基磺酰基乙醇c38(5g,0.04mol)和二苯基磷酰基叠氮化物c39(8.8614g,0.0322mol)于甲苯(50ml)中的溶液在0℃下搅拌10分钟并在10分钟内在0℃下逐滴添加dbu(5.5g,5.42ml,0.04mol)并将反应物在室温下搅拌16小时。将反应混合物用水(25ml)和etoac(100ml)淬灭并搅拌20分钟。将有机层分离并将水层再次用etoac(2x 100ml)萃取。将
有机层经na2so4干燥并浓缩。通过硅胶色谱纯化(梯度:0-100%于石油醚中的乙酸乙酯),得到1-叠氮基-2-甲基磺酰基-乙烷c40(5.2g,86%)。1h nmr(400mhz,dmso-d6)δ3.77-3.73(t,j=8.8hz,2h),3.44-3.42(t,j=8.8hz,2h),3.03(s,3h)。
[1028]
步骤2.1-(2-甲基磺酰基乙基)三唑-4-甲醛(s18)的合成
[1029]
将3,3-二乙氧基丙-1-炔(555μl,3.897mmol)、1-叠氮基-2-甲基磺酰基-乙烷c40(600mg,4.022mmol)、cuso4(15mg,0.09398mmol)、1-(1-苄基三唑-4-基)-n,n-双[(1-苄基三唑-4-基)甲基]甲胺(100mg,0.1885mmol)和抗坏血酸钠(700mg,3.974mmol)于meoh(12ml)/水(3ml)中的混合物加热至60℃持续2小时。将反应物冷却至室温,浓缩,并稀释于etoac(100ml)和水(50ml)中。分开各层并将水层用etoac(50ml)萃取。将各层合并并干燥,稀释于1n hcl(20ml)中,并搅拌过夜。此时,浓缩溶液,产生1-(2-甲基磺酰基乙基)三唑-4-甲醛(盐酸盐)s18(553mg,59%)。1h nmr(400mhz,甲醇-d4)δ8.07(s,1h),5.58-5.45(m,1h),4.89-4.82(m,2h),3.76-3.67(m,2h),3.24(s,3h)。lcms m/z 204.47[m+h]
+
。
[1030]
s19的制备
[1031]
1-(2-甲基磺酰基乙基)吡唑-4-甲醛(s19)
[1032][1033]
将1h-吡唑-4-甲醛c42(10g,104.1mmol)、11-甲基磺酰基乙烯c41(10ml,114.2mmol)和k2co3(25g,180.9mmol)于thf(200ml)中的溶液在60℃下搅拌。在搅拌过夜后,将混合物冷却至室温并浓缩至干燥。将产物悬浮于乙醚(100ml),研磨产物,并搅拌2小时。过滤产物并干燥过夜,产生11-(2-甲基磺酰基乙基)吡唑-4-甲醛s19(20.28g,83%)。1h nmr(400mhz,dmso-d6)δ9.80(s,1h),8.54(d,j=0.7hz,1h),8.05(d,j=0.7hz,1h),4.64(t,j=6.8hz,2h),3.80-3.67(m,2h),2.96(d,j=0.7hz,3h)。lcms m/z 203.01[m+h]
+
。
[1034]
s20的制备
[1035]
1-[2-[叔丁基(二甲基)甲硅烷基]氧基乙基]吡唑-4-甲醛(s20)
[1036][1037]
步骤1.叔丁基-(2-碘乙氧基)-二甲基-硅烷(c44)的合成
[1038]
在0℃下向2-碘乙醇c43(2g,0.0116mol)和咪唑(1.58g,0.0232mol)于dcm(40ml)中的搅拌溶液中添加叔丁基-氯-二甲基-硅烷(1.9g,0.0126mol)。将反应物升温至室温并搅拌4小时。将反应混合物用dcm(100ml)稀释,用饱和nahco3和盐水洗涤,经na2so4干燥,并在减压下浓缩,得到叔丁基-(2-碘乙氧基)-二甲基-硅烷c44(2.5g,68%)。1h nmr(400mhz,氯仿-d)δ3.83(t,j=6.8hz,2h),3.20(t,j=6.8hz,2h),0.90(s,9h),0.08(s,6h)。
[1039]
步骤2.1-[2-[叔丁基(二甲基)甲硅烷基]氧基乙基]吡唑-4-甲醛(s20)的合成
[1040]
向1h-吡唑-4-甲醛c42(20g,208.1mmol)和k2co3(115g,832.1mmol)于mecn(200ml)
中的溶液中添加叔丁基-(2-碘乙氧基)-二甲基-硅烷c44(65g,227.1mmol)。将反应物加热至80℃。将反应物搅拌5小时。将反应物冷却至50℃并搅拌16小时。使反应混合物达到环境温度,过滤,并将固体用mecn(200ml)洗涤。丢弃固体并将滤液浓缩。将残余物分配于etoac(400ml)与水(400ml)之间。将有机层分离,用水(400ml)和盐水(400ml)洗涤,经mgso4干燥,过滤,并浓缩。通过硅胶色谱纯化(800g柱,0-80%于己烷中的etoac),得到呈淡黄色油状物的产物1-[2-[叔丁基(二甲基)甲硅烷基]氧基乙基]吡唑-4-甲醛s20(46g,87%)。1h nmr(300mhz,氯仿-d)δ9.86(s,1h),7.98(s,2h),4.25(dd,j=5.5,4.5hz,2h),3.96(dd,j=5.5,4.5hz,2h),0.83(s,9h),-0.06(s,6h)。lcms m/z 255.14[m+h]
+
。
[1041]
s21的制备
[1042]
1-[3-[叔丁基(二甲基)甲硅烷基]氧基-2-[[叔丁基(二甲基)甲硅烷基]氧基甲基]-2-甲基-丙基]吡唑-4-甲醛(s21)
[1043][1044]
步骤1.2-(溴甲基)-2-甲基-丙烷-1,3-二醇(c46)的合成
[1045]
在0℃下向(3-甲基氧杂环丁烷-3-基)甲醇c45(10ml,100.3mmol)于thf(70ml)中的混合物中添加溴化氢(14ml 48%(w/w),123.7mmol)。在搅拌24小时后,将混合物浓缩至最小体积,稀释于dcm/meoh中并将过量hbr用饱和碳酸氢钠淬灭。分开各层并将有机层用na2so4干燥,过滤,用甲醇冲洗,并浓缩,产生2-(溴甲基)-2-甲基-丙烷-1,3-二醇c46(13.6682g,74%)。1hnmr(400mhz,甲醇-d4)δ3.47(d,j=1.1hz,6h),0.96(s,3h)。
[1046]
步骤2.[2-(溴甲基)-3-[叔丁基(二甲基)甲硅烷基]氧基-2-甲基-丙氧基]-叔丁基-二甲基-硅烷(c47)的合成
[1047]
向2-(溴甲基)-2-甲基-丙烷-1,3-二醇c46(10g,54.09mmol)于dcm(200ml)中的混合物中添加咪唑(7.7g,113.1mmol),随后添加tbscl(17g,112.8mmol)。5分钟后,混合物沉淀出白色结晶固体。将混合物过滤,用dcm冲洗,并浓缩。将混合物用庚烷(25ml)稀释,以进一步沉淀出咪唑/咪唑hcl,过滤,并固体再用庚烷(10ml)冲洗。将混合物浓缩,沉淀出另外的固体。将混合物用庚烷(50ml)稀释并再浓缩两次,得到[2-(溴甲基)-3-[叔丁基(二甲基)甲硅烷基]氧基-2-甲基-丙氧基]-叔丁基-二甲基-硅烷c47(22.246g,100%)1hnmr(400mhz,氯仿-d)δ3.44(s,4h),3.40(s,2h),0.94(s,3h),0.89(s,18h),0.04(d,j=1.2hz,12h)。
[1048]
步骤3.1-[3-[叔丁基(二甲基)甲硅烷基]氧基-2-[[叔丁基(二甲基)甲硅烷基]氧基甲基]-2-甲基-丙基]吡唑-4-甲醛(s21)的合成
[1049]
向小瓶中添加dmf(20ml)中的1h-吡唑-4-甲醛c42(2g,20.81mmol)、k2co3(4g,28.94mmol)和[2-(溴甲基)-3-[叔丁基(二甲基)甲硅烷基]氧基-2-甲基-丙氧基]-叔丁基-二甲基-硅烷c47(9.5g,23.08mmol)。将混合物加热至130℃。3小时后,将混合物冷却至室温,用水(100ml)和庚烷(100ml)稀释。将各层混合,并将水层用庚烷(2x 100ml)洗涤。将合并的有机层用水(100ml)、盐水(100ml)洗涤并将有机层经na2so4干燥并浓缩。通过硅胶色谱纯化(梯度:0-60% etoac:庚烷),得到产物1-[3-[叔丁基(二甲基)甲硅烷基]氧基-2-[[叔丁基(二甲基)甲硅烷基]氧基甲基]-2-甲基-丙基]吡唑-4-甲醛s21(2.39mg,23%)。1h nmr(400mhz,氯仿-d)δ9.85(s,1h),7.98-7.91(m,2h),4.12(s,2h),3.43-3.29(m,4h),0.91(s,18h),0.84(s,3h),0.05(d,j=0.6hz,12h)。lcms m/z 427.31[m+h]
+
。
[1050]
s22的制备
[1051]
2-[(2-羟基-1,1-二甲基-乙基)氨基]嘧啶-5-甲醛(s22)
[1052][1053]
步骤1.2-[(2-羟基-1,1-二甲基-乙基)氨基]嘧啶-5-羧酸乙酯(c49)的合成
[1054]
向2-氯嘧啶-5-羧酸乙酯c48(25g,0.1340mol)于乙醇(750ml)中的搅拌溶液中添加2-氨基-2-甲基-丙-1-醇(14.333g,15.412ml,0.1608mol),随后在室温下添加dipea(34.637g,46.681ml,0.2680mol)。将反应物在80℃下搅拌8小时。将反应物升温至室温并在减压下浓缩。通过硅胶色谱纯化(洗脱液:70%于石油醚中的etoac),得到2-[(2-羟基-1,1-二甲基-乙基)氨基]嘧啶-5-羧酸乙酯c49(18g,55%)。1h nmr(400mhz,dmso-d6)δ8.70(s,2h),7.39(s,1h),4.86(t,j=6hz,1h),4.25(q,j=6.8hz,2h),3.52(d,j=6hz,2h),1.32(s,6h),1.28(t,j=6.8hz,3h)。lcms m/z 240.27[m+h]
+
。
[1055]
步骤2.2-[[2-[叔丁基(二甲基)甲硅烷基]氧基-1,1-二甲基-乙基]氨基]嘧啶-5-羧酸乙酯(c50)的合成
[1056]
在室温下向2-[(2-羟基-1,1-二甲基-乙基)氨基]嘧啶-5-羧酸乙酯c49(10g,0.0410mol)和叔丁基-氯-二甲基-硅烷(9.2694g,0.0615mol)于dcm(500ml)中的搅拌溶液中添加咪唑(8.3735g,0.1230mol),随后添加dmap(1.0018g,0.0082mol)并搅拌16小时。将反应物在减压下浓缩。将粗材料用水(500ml)和戊烷(500ml)稀释。将有机层分离,用水洗
涤,经na2so4干燥,并在减压下浓缩,得到2-[[2-[叔丁基(二甲基)甲硅烷基]氧基-1,1-二甲基-乙基]氨基]嘧啶-5-羧酸乙酯c50(14.9g,100%)。1h nmr(400mhz,dmso-d6)δ8.70(s,2h),7.46(s,1h),4.25(q,j=7.2hz,2h),3.77(s,2h),1.30(s,6h),1.28(t,j=7.6hz,3h),0.82(s,9h),-0.06(s,6h)。lcms m/z 354.3[m+h]
+
。
[1057]
步骤3.[2-[[2-[叔丁基(二甲基)甲硅烷基]氧基-1,1-二甲基-乙基]氨基]嘧啶-5-基]甲醇(c51)的合成
[1058]
在-78℃下在氮气下向2-[[2-[叔丁基(二甲基)甲硅烷基]氧基-1,1-二甲基-乙基]氨基]嘧啶-5-羧酸乙酯c50(15g,0.0411mol)于thf(600ml)中的搅拌溶液中缓慢添加dibal-h(1m于甲苯中)(205.50ml 1m,0.2055mol)。将反应物在-78℃下搅拌30分钟并且然后升温至室温并搅拌4小时。在0℃下将反应混合物用饱和nh4cl(500ml)淬灭并将化合物用etoac(2x 500ml)萃取。将有机层用1n hcl(100ml)、盐水洗涤,经na2so4干燥,并在减压下浓缩。通过硅胶色谱纯化(洗脱液:50%于石油醚中的etoac),得到[2-[[2-[叔丁基(二甲基)甲硅烷基]氧基-1,1-二甲基-乙基]氨基]嘧啶-5-基]甲醇c51(6g,46%)。1h nmr(400mhz,dmso-d6)δ8.19(s,2h),6.25(s,1h),4.99(t,j=5.6hz,1h),4.27(d,j=5.6hz,2h),3.71(s,2h),1.30(s,6h),0.84(s,9h),-0.03(s,6h)。lcms m/z 312.23[m+h]
+
。
[1059]
步骤4.2-[[2-[叔丁基(二甲基)甲硅烷基]氧基-1,1-二甲基-乙基]氨基]嘧啶-5-甲醛(c52)的合成
[1060]
在室温下向[2-[[2-[叔丁基(二甲基)甲硅烷基]氧基-1,1-二甲基-乙基]氨基]嘧啶-5-基]甲醇c51(120mg,271.55μmol)于dcm(10ml)中的搅拌溶液中添加mno2(851.98mg,0.0098mol)并搅拌6小时。通过过滤反应物并用dcm(10ml)洗涤。将滤液在减压下浓缩,提供2-[[2-[叔丁基(二甲基)甲硅烷基]氧基-1,1-二甲基-乙基]氨基]嘧啶-5-甲醛c52(90mg,99%)。1h nmr(400mhz,dmso-d6)δ9.71(s,1h),8.71(d,j=11.6hz,2h),7.72(s,1h),3.78(s,2h),1.34(s,6h),0.84(s,9h),-0.05(s,6h)。lcms m/z 310.22[m+h]
+
。
[1061]
步骤5.2-[(2-羟基-1,1-二甲基-乙基)氨基]嘧啶-5-甲醛(s22)的合成
[1062]
在室温下向2-[[2-[叔丁基(二甲基)甲硅烷基]氧基-1,1-二甲基-乙基]氨基]嘧啶-5-甲醛c52(2.9g,0.0087mol)于thf(20ml)中的搅拌溶液中添加tbaf(1m于thf)(21.700ml 1m,0.0217mol)并搅拌2小时。将反应物用etoac(100ml)稀释,用盐水溶液洗涤,经na2so4干燥,并在减压下浓缩。将粗制化合物用戊烷洗涤并干燥,得到2-[(2-羟基-1,1-二甲基-乙基)氨基]嘧啶-5-甲醛s22(1.47g,86%)。1h nmr(400mhz,dmso-d6)δ9.72(s,1h),8.71(d,j=13.2hz,2h),7.64(s,1h),4.87(t,j=6hz,1h),3.54(d,j=6hz,2h),1.33(s,6h)。lcms m/z 196.35[m+h]
+
。
[1063]
s23的制备
[1064]
(3s)-3-氨基丁酸(s23)
[1065][1066]
(3s)-3-氨基丁酸(s23)从商业来源获得。
[1067]
s24的制备
[1068]
4-氨基戊-2-酮盐酸盐(s24)
[1069][1070]
4-氨基戊-2-酮盐酸盐(s24)从商业来源获得。
[1071]
s25的制备
[1072]
(4s)-4-氨基戊-2-酮盐酸盐(s25)
[1073][1074]
步骤1.(3s)-3-(叔丁氧基羰基氨基)丁酸(c53)的合成
[1075]
在15分钟内向(3s)-3-氨基丁酸s23(100g,969.7mmol)于二噁烷(600ml)中的溶液中添加naoh水溶液溶液(950ml 1m,950.0mmol),随后添加boc2o(300g,1.375mol)。将反应混合物在室温下搅拌12小时。将反应物分配于mtbe(1l)与水(300ml)之间。分离各层,并将水层再次用mtbe(500ml)萃取。然后将水层用1n hcl酸化直到ph=2并用dcm(3x 600ml)萃取。将合并的有机层用盐水洗涤,经mgso4干燥,过滤,并在真空中浓缩,产生呈白色固体的(3s)-3-(叔丁氧基羰基氨基)丁酸c53(176g,89%)。1h nmr(300mhz,氯仿-d)δ4.92(s,1h),4.04(s,1h),2.56(dd,j=5.5,2.9hz,2h),1.44(s,9h),1.25(d,j=6.8hz,3h)。
[1076]
步骤2.n-[(1s)-3-[甲氧基(甲基)氨基]-1-甲基-3-氧代-丙基]氨基甲酸叔丁酯(c54)的合成
[1077]
向(3s)-3-(叔丁氧基羰基氨基)丁酸c53(160g,787.3mmol)于dcm(1.5l)中的溶液中添加n-甲氧基甲胺(盐酸盐)(81g,830.4mmol),随后在10分钟内添加dipea(560ml,3.215mol)。将反应混合物冷却至0℃并在45分钟内添加t3p(600g 50%(w/w)于etoac,942.9mmol)。在添加后,移除冷却浴并将反应物在室温下搅拌1小时。将反应混合物冷却至10℃并添加1n naoh水溶液(700ml)并将溶液搅拌15分钟。将有机相分离,用饱和氯化铵水溶液(200ml)和盐水(200ml)洗涤,干燥,通过硅胶塞过滤,并在真空中浓缩,得到呈澄清的无色粘性油状物的n-[(1s)-3-[甲氧基(甲基)氨基]-1-甲基-3-氧代-丙基]氨基甲酸叔丁酯c54(180g,93%)。1h nmr(300mhz,氯仿-d)δ5.30(s,1h),4.06(ddd,j=14.3,9.7,6.0hz,1h),3.68(s,3h),3.17(s,3h),2.71(dd,j=15.6,5.2hz,1h),2.54(dd,j=15.7,5.7hz,1h),1.43(s,9h),1.24(d,j=6.8hz,3h)。
[1078]
步骤3.n-[(1s)-1-甲基-3-氧代-丁基]氨基甲酸叔丁酯(c55)的合成
[1079]
在40分钟内在0℃下向n-[(1s)-3-[甲氧基(甲基)氨基]-1-甲基-3-氧代-丙基]氨基甲酸叔丁酯c54(220g,893.2mmol)于thf(4l)中的溶液中添加碘(甲基)镁(900ml 3m,2.700mol)。将所得反应混合物在0℃下搅拌4小时。将反应物用饱和氯化铵溶液(2l)淬灭,
随后用mtbe(1l)和水(2l)淬灭。将混合物搅拌30分钟并将有机层分离。将水相用mtbe(1l)萃取并将合并的有机层用饱和氯化铵溶液(1l)洗涤,经mgso4干燥,过滤,并在真空中浓缩。通过硅胶色谱纯化(梯度:0-70%于庚烷中的etoac),得到呈白色固体的产物n-[(1s)-1-甲基-3-氧代-丁基]氨基甲酸叔丁酯c55(115g,64%)。1h nmr(300mhz,氯仿-d)δ4.83(s,1h),4.12-3.87(m,1h),2.69(dd,j=16.5,5.2hz,1h),2.63-2.47(m,1h),2.15(d,j=2.3hz,3h),1.43(d,j=2.4hz,9h),1.20(dd,j=6.8,2.4hz,3h)。
[1080]
步骤4.(4s)-4-氨基戊-2-酮(盐酸盐)(s25)的合成
[1081]
在3分钟内向n-[(1s)-1-甲基-3-氧代-丁基]氨基甲酸叔丁酯c55(16.3g,80.18mmol)于meoh(30ml)中的溶液中添加氯化氢(50ml 4m于二噁烷中,200.0mmol)。将反应物在室温下搅拌5小时并且然后在减压下浓缩。将残余物与etoh(2x 30ml)共蒸发并在真空下干燥,得到呈粉色粘性油状物的(4s)-4-氨基戊-2-酮(盐酸盐)s25(12g,98%)。1h nmr(300mhz,氯仿-d)δ8.06(s,3h),3.48(d,j=6.8hz,1h),2.88(dd,j=18.0,5.8hz,1h),2.75(dd,j=18.0,7.2hz,1h),2.13(s,3h),1.17(d,j=6.6hz,3h)。
[1082]
s26的制备(方法a)
[1083]
(2s,6s)-2-甲基-6-(1-甲基三唑-4-基)哌啶-4-酮(s26)
[1084][1085]
步骤1.(2s)-2-甲基-6-(1-甲基三唑-4-基)哌啶-4-酮(c56)的合成
[1086]
向(4s)-4-氨基戊-2-酮(盐酸盐)s25(12g,78.48mmol)于etoh(300ml)中的混合物中添加1-甲基三唑-4-甲醛s17(9g,81.01mmol)、l-脯氨酸(2g,17.37mmol)、硫酸镁(12g,99.69mmol)和tea(13ml,93.27mmol)。将反应混合物在室温下搅拌过夜。将混合物过滤并在减压下浓缩。将粗残余物用饱和碳酸氢钠溶液(150ml)淬灭并用dcm(3x 100ml)萃取。将合并的有机层用盐水(50ml)洗涤,经硫酸镁干燥,过滤,并在真空中浓缩。通过硅胶色谱纯化(梯度:0-60%of 20% meoh/dcm于dcm),得到呈5:1顺式与反式的比率的产物(2s)-2-甲基-6-(1-甲基三唑-4-基)哌啶-4-酮c56(6.7g,44%)。另外,来自s25的立体中心的e.r.减小至~85%。
[1087]
c56中的主要(顺式)立体异构体的nmr:1h nmr(300mhz,氯仿-d)δ7.47(s,1h),4.26(dd,j=10.3,4.9hz,1h),4.11(s,3h),3.17(dqd,j=12.2,6.2,3.0hz,1h),2.73-2.56(m,2h),2.47(ddd,j=14.2,3.0,1.6hz,1h),2.21(dd,j=14.2,11.7hz,2h),1.28(d,j=6.2hz,3h)。
[1088]
c56中立体异构体分配的nmr合理化:应注意,c56中的主要组分使用4.26ppm处的峰的nmr偶联常数数据(c5-亚甲基质子)分配为顺式立体异构体。假定c6处的三唑占据最低能量构型中的赤道轴位置。在c4处的轴ch与c5处的ch质子之一(j=10.3hz)之间的偶联指示如由卡普拉斯方程(karplus equation)所定义的180
°
关系。在后续重结晶步骤中去除微量反式产物,得到s26。
[1089]
步骤2.(2s,6s)-2-甲基-6-(1-甲基三唑-4-基)哌啶-4-酮(s26)的合成
[1090]
将呈5:1顺式与反式的比率的(2s)-2-甲基-6-(1-甲基三唑-4-基)哌啶-4-酮c56(6.7g)于mtbe(100ml)中的溶液加热至回流持续30分钟。缓慢添加乙醇,直到所有固体均溶解(20ml)。将溶液回流30分钟并使其缓慢冷却过夜。结晶出固体,将其用mtbe(30ml)稀释,过滤,并在真空下干燥,得到呈白色固体的(2s,6s)-2-甲基-6-(1-甲基三唑-4-基)哌啶-4-酮s26(3.2g,48%),其对映异构体比率为≥85%,一直到所有其他化合物,它们利用s26作为起始材料,除非另外提及(不包括经历sfc纯化的实施例)。1h nmr(300mhz,氯仿-d)δ7.45(s,1h),4.23(dd,j=10.3,4.9hz,1h),4.09(s,3h),3.14(ddp,j=12.2,6.1,3.1hz,1h),2.71-2.52(m,2h),2.44(ddd,j=14.1,3.0,1.5hz,1h),2.27-2.00(m,2h),1.26(d,j=6.2hz,3h)。
[1091]
s26的替代性制备(方法b)
[1092]
((2s,6s)-2-甲基-6-(1-甲基三唑-4-基)哌啶-4-酮(s26)
[1093][1094]
步骤1.双[(3-叔丁氧基-3-氧代-丙酰基)氧基]镁(c109)的合成
[1095]
将3-叔丁氧基-3-氧代-丙酸c108(321.51g,1.907mol)于thf(2l)中的溶液在冰浴中冷却至5℃并添加mg(oet)2(111.33g,953.5mmol)。将反应物在0℃下搅拌30分钟,从冷却浴中取出并在室温下搅拌过夜。通过塞过滤反应物并将塞再用thf洗涤。将澄清的无色滤液在真空中蒸发,得到糊状固体。将固体用1l乙醚研磨并过滤。将滤饼用et2o洗涤并在真空中干燥。将滤液再次在真空中蒸发并然后用小体积的et2o研磨并过滤,得到第二批产物。将所述批次合并并在真空中干燥,得到呈白色固体的双[(3-叔丁氧基-3-氧代-丙酰基)氧基]镁c109(294.49g,90%)。1h nmr(300mhz,甲醇-d4)δ4.92(s,4h),1.48(s,18h)ppm。
[1096]
步骤2.(5s)-5-(叔丁氧基羰基氨基)-3-氧代-己酸叔丁酯(c111)的合成
[1097]
向(3s)-3-(叔丁氧基羰基氨基)丁酸c110(170.15g,837.2mmol)于thf(1.5l)中的溶液中添加cdi(149.8g,923.8mmol)。乳状悬浮液在接下来的几分钟内变澄清。观察到气体逸出。将反应物在室温下搅拌3小时。添加双[(3-叔丁氧基-3-氧代-丙酰基)氧基]镁c109
(172.19g,502.6mmol)。形成另一种乳状悬浮液,其在搅拌30分钟后变澄清。将反应物搅拌48小时。将反应物倒入到1.5l 1n hcl中并用mtbe(1l)萃取。确认ph为大约ph 3。将萃取物用饱和水溶液nahco3洗涤,分离,用mgso4干燥,过滤,并在真空中蒸发,得到(5s)-5-(叔丁氧基羰基氨基)-3-氧代-己酸叔丁酯c111(248.5g,98.5%)。1hnmr(300mhz,氯仿-d)δ4.90(d,j=18.1hz,1h),4.04(dt,j=13.8,6.6hz,1h),3.47-3.22(m,2h),2.76(qd,j=17.0,5.7hz,2h),1.48(s,9h),1.44(s,9h),1.23(d,j=6.8hz,3h)ppm。
[1098]
步骤3.(2s,3r,6s)-6-甲基-2-(1-甲基三唑-4-基)-4-氧代-哌啶-3-羧酸叔丁酯(c112)的合成
[1099]
向(5s)-5-(叔丁氧基羰基氨基)-3-氧代-己酸叔丁酯c111(248.5g,824.5mmol)于dcm(1.5l)中的溶液中添加tfa(240ml,3.115mol)并将反应物搅拌过夜。将反应物在25℃下在真空中蒸发。将保留的固体用500ml戊烷研磨并过滤。将滤饼用戊烷洗涤并从滤饼中挤出大部分溶剂。将饼转移回到反应烧瓶中并溶解于1l dcm中。
[1100]
添加1-甲基三唑-4-甲醛s17(120.7g,1.086mol)。将反应物在室温下搅拌过夜。添加盐水(100ml)并添加6n naoh,直到在振摇漏斗时水层保持碱性。分离有机层并将水层用dcm(1l)萃取。将有机层合并,用mgso4干燥,并经硅胶塞过滤。将塞用10%于etoac中的meoh洗脱。将滤液在真空中蒸发,得到固体,将其用mtbe(500ml)研磨并过滤。将滤饼用mtbe洗涤并在真空中干燥,得到一批产物。将来自研磨的母液浓缩。过滤所沉淀的固体,提供第二批产物。将所述批次合并,得到(2s,3r,6s)-6-甲基-2-(1-甲基三唑-4-基)-4-氧代-哌啶-3-羧酸酯c112(105.45g,43%)。1h nmr(300mhz,氯仿-d)δ7.48(s,1h),4.52(d,j=11.0hz,1h),4.09(s,3h),3.61(dd,j=11.0,1.0hz,1h),3.21(ddd,j=11.7,6.1,2.9hz,1h),2.55(dd,j=13.7,2.9hz,1h),2.37-2.13(m,1h),1.98(s,1h),1.39(s,9h),1.29(d,j=6.3hz,3h)ppm。
[1101]
步骤4.(2s,6s)-2-甲基-6-(1-甲基三唑-4-基)哌啶-4-酮(s26)的合成
[1102]
向(2s,3r,6s)-6-甲基-2-(1-甲基三唑-4-基)-4-氧代-哌啶-3-羧酸叔丁酯c112(70.59g,239.8mmol)于dcm(750ml)中的溶液中添加msoh(62ml,955.4mmol)并将反应物加热至回流6小时。将反应物冷却并倒入到分液漏斗中。添加盐水(大约100ml)。添加6n naoh,直到在振摇后水层保持碱性。将有机层分离并将水溶液用dcm(2x 500ml)萃取。将有机层合并,用mgso4干燥,过滤,并在真空中蒸发,得到(2s,6s)-2-甲基-6-(1-甲基三唑-4-基)哌啶-4-酮s26(43.74g,94%)。1h nmr(300mhz,氯仿-d)δ7.46(s,1h),4.20(dd,j=10.1,5.1hz,1h),4.06(s,3h),3.11(dqd,j=12.3,6.2,3.0hz,1h),2.73-2.48(m,2h),2.40(ddd,j=14.1,3.0,1.5hz,1h),2.25-2.00(m,2h),1.23(d,j=6.2hz,3h)ppm。
[1103]
s27-s29的制备
[1104]
中间体s27-s29(参见表1)在单个步骤中使用适当的醛和对于中间体s26(方法a)所述的方法由中间体s25制备。醛通过上文所述的方法制备或从商业来源获得。关于中间体s26(通过方法a制备),在步骤1中观察到对映异构纯的起始材料的(4s)-4-氨基戊-2-酮(盐酸盐)s25的部分立体化学消除,导致在步骤1中产生未分离的立体异构物混合物。在每种情况下,顺式产物是主要异构体。此混合物通过使用波浪键表示。对方法的任何修改在表1和所附脚注中标识。
[1105]
表1.中间体s27-s29的制备方法、结构和物理化学数据
[1106][1107][1108]
1.将反应物搅拌整个周末(步骤1)
[1109]
2.将粗残余物用水和饱和碳酸氢钠溶液稀释并通过相分离器用dcm(5x)萃取。(步骤1)
[1110]
3.通过硅胶色谱纯化(梯度:0-50%of 20%meoh/dcm于dcm),得到产物。(步骤1)
[1111]
4.经由色谱吹扫微量异构体并且不进行步骤2。
[1112]
5.通过硅胶色谱纯化(梯度:0-100%of 20%meoh/dcm于dcm),得到产物。(步骤1)
[1113]
化合物1
[1114]
(2's,6's,7s)-2-氯-2'-甲基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](1)
[1115][1116]
向(2s,6s)-2-甲基-6-(1-甲基三唑-4-基)哌啶-4-酮s26(1380mg,7.11mmol,s26通过方法a制备)于dcm(30ml)中的溶液中添加2-(5-氯-3-噻吩基)乙醇s2(1100μl,8.894mmol),随后添加msoh(3ml,46.23mmol)。将反应物加热至回流90分钟,此时将其冷却至室温并用2n naoh淬灭,直到ph达到14。将混合物用dcm(20ml)稀释并将有机层分离,用盐
水(30ml)洗涤,经mgso4干燥,并在真空中浓缩。通过硅胶色谱纯化(梯度:0-25%of 20%meoh/dcm于dcm),得到呈淡黄色油状物的比率》8:1的产物(2's,6's,7s)-2-氯-2'-甲基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]1(1162mg,48%)。据推断所观察到的微量异构体是化合物1的对映异构体,因为通过方法a制备的s26含有微量其他顺式对映异构体。应注意,化合物1的相对立体化学通过noe nmr研究来分配。1h nmr(400mhz,氯仿-d)δ7.42(s,1h),6.58(s,1h),4.41(dd,j=11.8,2.6hz,1h),4.06(s,3h),4.02-3.86(m,2h),3.30(ddt,j=12.7,6.3,3.2hz,1h),2.70-2.49(m,2h),2.35(dt,j=13.6,2.6hz,1h),2.06(dt,j=13.7,2.5hz,1h),1.79(dd,j=13.6,11.8hz,1h),1.42(dd,j=13.7,11.3hz,1h),1.31-1.19(m,1h),1.12(d,j=6.4hz,3h)。lcmsm/z 339.0[m+h]
+
。
[1117]
化合物1(hcl盐)的替代性制备
[1118]
(2's,6's,7s)-2-氯-2'-甲基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]盐酸盐(1)
[1119]
向于dcm(5ml)中的(2s,6s)-2-甲基-6-(1-甲基三唑-4-基)哌啶-4-酮s26(205mg,1.055mmol)添加2-(5-氯-3-噻吩基)乙醇s2(150μl,1.213mmol),随后添加msoh(300μl,4.623mmol)。将混合物加热至回流持续10分钟,此时将其冷却至室温并用2n naoh淬灭,直到ph达到14。将混合物用dcm(5ml)稀释并将有机层分离并在真空中浓缩。通过硅胶色谱纯化(梯度:0-25%of 20% meoh/dcm于dcm),得到产物,立即将其溶解于少量dcm中并用hcl(100μl 4m于二噁烷中,0.4000mmol)处理。将混合物在真空中浓缩并将残余物与dcm(5ml)共沸并干燥,产生呈淡黄色固体的(2's,6's,7s)-2-氯-2'-甲基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](盐酸盐)1(171.6mg,43%)。1h nmr(300mhz,dmso-d6)δ9.46(s,1h),9.24(d,j=8.3hz,1h),8.29(s,1h),6.95(s,1h),4.67(t,j=11.1hz,1h),4.09(s,3h),3.95(t,j=5.4hz,2h),3.72(s,1h),2.61(t,j=5.3hz,2h),2.46-2.32(m,2h),2.25(d,j=15.1hz,1h),2.01-1.86(m,1h),1.29(d,j=6.5hz,3h)。lcms m/z 339.0[m+h]
+
[1120]
化合物2
[1121]
(2's,6's,7s)-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](2)
[1122][1123]
向(2s,6s)-2-甲基-6-(1-甲基三唑-4-基)哌啶-4-酮s26(250mg,1.287mmol)和2-[5-(三氟甲基)-3-噻吩基]乙醇s3(350mg,1.748mmol)于dcm(5ml)中的溶液中添加msoh(500μl,7.705mmol)并将反应物加热至40℃。在16小时后,添加另外的msoh(200μl,3.082mmol)并将反应物继续加热过夜。将混合物用水(4ml)和dcm(5ml)稀释并用naoh水溶液(2ml 6m,12.00mmol)淬灭。将混合物分离,用dcm(2x 5ml)萃取,使其穿过相分离器,并将有机物在真空中浓缩。通过硅胶色谱纯化(梯度:0-10%于dcm中的meoh),得到呈白色固体的(2's,6's,7s)-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[4,5-二氢噻吩并[2,
3-c]吡喃-7,4'-哌啶]2(445mg,93%)。应注意,化合物2的相对立体化学通过noe nmr研究来分配。1h nmr(300mhz,氯仿-d)δ7.46(s,1h),7.14(s,1h),4.47(d,j=11.6hz,1h),4.08(d,j=3.3hz,3h),4.00(s,2h),3.36(s,1h),2.72(d,j=5.6hz,2h),2.41(d,j=14.2hz,1h),2.12(d,j=13.7hz,1h),1.86(t,j=12.7hz,1h),1.49(d,j=12.8hz,1h),1.15(d,j=6.3hz,3h)。lcms m/z 373.07[m+h]
+
[1124]
化合物3-16
[1125]
化合物3-16(参见表2)使用分离的哌啶酮(s26、s29或c56)和对于化合物1和2所述的相关噻吩乙醇由单个oxa-pictet spengler步骤制备。噻吩乙醇和哌啶酮通过上文所述的方法制备或从商业来源获得。在使用s26的实施例中,s26通过方法a制备,因此所用哌啶酮可含有少量其他顺式异构体。对方法的任何修改在表2和所附脚注中标识。
[1126]
表2.化合物3-16的制备方法、结构和物理化学数据。
[1127]
[1128]
[1129]
[1130][1131]
1.将反应物搅拌30分钟。
[1132]
2.完成后,将混合物浓缩并稀释于meoh中。未进行进一步检查。
[1133]
3.通过反相hplc纯化(方法:c18 waters sunfire柱(30x 150mm,5微米).梯度:于具有5mm hcl的h2o中的mecn),得到呈hcl盐形式的产物。
[1134]
4.将有机层通过相分离器收集并在氮气下干燥。
[1135]
5.纯化后,将产物溶于0.6ml水中,冷却,并冻干过夜,得到。
[1136]
6.将反应物搅拌过夜。
[1137]
7.一旦反应完成,就将有机层分离,经na2so4干燥,过滤,并浓缩。
[1138]
8.通过硅胶色谱纯化(梯度:0-20%于dcm中的meoh),得到产物。
[1139]
9.用c56运行反应,富集成来自s26纯化的两种异构体的混合物。分离作为pictet spangler反应的微量产物的呈单个非对映异构体的化合物7。如对于s26的方法a所述,观察到s25立体中心的差向异构化作用,提供了呈对映异构体混合物形式的此化合物。
[1140]
10.50分钟后,将反应物用饱和nahco3溶液淬灭并用dcm(6x)萃取。将合并的有机层经na2so4干燥,过滤,并浓缩。
[1141]
化合物17
[1142]
[(2's,6's,7s)-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-3-基]甲醇(17)
[1143][1144]
步骤1.[(2s,6s)-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-3-基]甲基乙酸酯(c57)的合成
[1145]
向(2s,6s)-2-甲基-6-(1-甲基三唑-4-基)哌啶-4-酮s26(10mg,0.05148mmol)和[4-(2-羟基乙基)-2-(三氟甲基)-3-噻吩基]甲基乙酸酯s16(18mg,0.06710mmol)于dcm(500μl)中的混合物中添加msoh(30μl,0.4623mmol)并将混合物加热至40℃。在搅拌4小时后,将反应物用饱和nahco3溶液淬灭,将各层分离并将混合物浓缩至干燥,得到粗制c57。
[1146]
步骤2.[(2s,6s)-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-3-基]甲醇(17)的合成
[1147]
将粗材料c57用meoh(2ml)稀释并向混合物中添加naoh(20μl 6m,0.1200mmol)。将反应物搅拌5分钟。将混合物浓缩,再稀释于dcm中并用盐水洗涤。使有机层穿过相分离器,并浓缩。硅胶色谱(梯度:0-20%meoh-dcm)得到作为母体的17。
[1148]
将来自脱保护的17(母体)用乙醚(1ml)稀释并添加hcl(13μl 4m于二噁烷中,0.05200mmol),立即沉淀出白色固体。将混合物浓缩,并与乙醚共沸三次,产生[(2s,6s)-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-3-基]甲醇17(盐酸盐)(10.9mg,45%)。1h nmr(300mhz,dmso-d6)δ9.35(d,j=9.8hz,1h),9.04(s,1h),8.26(s,1h),5.32(s,1h),4.73(s,1h),4.48(s,2h),4.09(s,3h),4.01(s,2h),3.62(s,1h),2.73(s,2h),2.34(s,2h),1.91(d,j=13.5hz,1h),1.29(d,j=6.5hz,3h)。lcms m/z 403.13[m+h]
+
.
[1149]
化合物18
[1150]
2-[4-[(2s,6s)-2-氯-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-2'-基]吡唑-1-基]-n,n-二甲基-乙酰胺(18)
[1151][1152]
向(2s,6s)-2-氯-2'-甲基-6'-(1h-吡唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]s31(20mg,0.05865mmol)于dmf(280μl)中的溶液中添加cs2co3(57mg,
0.1749mmol)。在室温下添加2-溴-n,n-二甲基-乙酰胺(7.6μl,0.07050mmol)。将反应物搅拌1小时。将反应物用饱和nahco3溶液淬灭并用etoac(4x)萃取。将合并的有机层经na2so4干燥,过滤,并浓缩。通过反相hplc纯化(方法:c18 waters sunfire柱(30x 150mm,5微米).梯度:于h2o中的mecn),得到2-[4-[(2s,6s)-2-氯-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-2'-基]吡唑-1-基]-n,n-二甲基-乙酰胺18(6.2mg,23%)。1h nmr(300mhz,氯仿-d)δ7.50(s,2h),6.57(s,1h),4.92(s,2h),4.17(dd,j=11.6,2.5hz,1h),3.93(t,j=5.5hz,2h),3.25(d,j=9.1hz,1h),3.06(s,3h),2.97(s,3h),2.60(td,j=5.4,1.8hz,2h),2.25(d,j=13.6hz,1h),2.01(s,1h),1.70(d,j=12.5hz,1h),1.47-1.32(m,1h),1.11(d,j=6.4hz,3h)。lcms m/z 409.19[m+h]
+
.
[1153]
化合物19
[1154]
(2s)-2-氯-2'-甲基-6'-(1-甲基吡唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](19)
[1155][1156]
将(4s)-4-氨基戊-2-酮盐酸盐s25(25mg,0.1817mmol)和tea(30μl,0.2152mmol)于mecn(1.000ml)中的溶液添加到1-甲基吡唑-4-甲醛(22.01mg,0.20mmol)、mgso4(25mg,0.2077mmol)和l-脯氨酸(5mg,0.043mmol)。将所得混合物在室温下搅拌过夜。将反应混合物在40℃下经由genevac蒸发,直至干燥,得到粗制c58。向其中添加2-(5-氯-3-噻吩基)乙醇s2(25μl,0.2080mmol)于二噁烷(750.0μl)中的溶液,随后添加tfoh(80μl,0.90mmol)于二噁烷(750.0μl)中的溶液。将混合物在室温下搅拌30分钟。添加另外的三氟甲磺酸(50μl,0.5650mmol)并继续搅拌10分钟。将反应物置于氮气流下,直到体积减小一半。将剩余溶液用naoh(1.5ml 2m,3.000mmol)淬灭并用dcm(1.500ml)稀释。将所得双相混合物搅拌几分钟并且然后使其穿过相分离器。将有机层用氮气吹泡。通过反相hplc纯化(方法:c18 waters sunfire柱(30x 150mm,5微米).梯度:于具有0.1%三氟乙酸的h2o中的mecn),得到(2s)-2-氯-2'-甲基-6'-(1-甲基吡唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]19呈三氟乙酸盐(6.6mg,11%)。化合物19通过手性sfc分析确定为88%e.r.(方法:ad-h柱(4.6x 100mm).梯度:具有5mm氨的10%meoh和90% co2)。1h nmr(400mhz,dmso-d6)δ8.91(d,j=10.8hz,1h),8.49(d,j=11.3hz,1h),7.86(s,1h),7.59(s,1h),6.94(s,1h),4.49(t,j=11.2hz,1h),3.93(t,j=5.5hz,2h),3.84(s,3h),2.93(td,j=13.9,6.9hz,1h),2.60(t,j=5.5hz,2h),2.35(d,j=17.2hz,1h),2.21(q,j=13.9hz,2h),1.84-1.73(m,1h),1.24(d,j=6.6hz,3h)。lcms m/z 338.17[m+h]
+
[1157]
化合物20
[1158]
2-氯-2'-甲基-6'-(3-吡啶基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](20)
[1159][1160]
将4-氨基戊-2-酮盐酸盐s24(25mg,0.1817mmol)于etoh(1ml)中的溶液添加到吡啶-3-甲醛(19.5mg,17.06μl,0.1817mmol)、mgso4(25mg,0.2077mmol)和l-脯氨酸(5mg,0.04343mmol)。添加tea(30μl,0.2152mmol)并将反应物在室温下搅拌超过3天。将反应混合物在氮气流下蒸发,得到粗制c59。向其中添加2-(5-氯-3-噻吩基)乙醇s2(25μl,0.2075mmol)于二噁烷(750μl)中的溶液,随后添加新鲜制备的tfoh(100μl,1.130mmol)于二噁烷(750μl)中的溶液。将混合物在室温下搅拌30分钟。将反应物置于氮气流下,直到体积减小一半。将剩余溶液用naoh(1.5ml 2m,3.000mmol)淬灭并用dcm(1.500ml)稀释。将所得双相混合物搅拌几分钟并且然后使其穿过相分离器。将有机层用氮气吹泡。通过反相hplc纯化(方法:c18 waters sunfire柱(30x 150mm,5微米).梯度:于具有0.1%三氟乙酸的h2o中的mecn),得到2-氯-2'-甲基-6'-(3-吡啶基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]20呈三氟乙酸盐(34.4mg,56%)。化合物20通过手性sfc分析确定为94%顺式对映异构体和6%反式对映异构体(方法:ad-h柱(4.6x 100mm).梯度:具有5mm氨的10% meoh和90% co2)。1h nmr(300mhz,甲醇-d4)δ8.87(d,j=2.3hz,1h),8.74(dd,j=5.2,1.5hz,1h),8.36-8.27(m,1h),7.77(dd,j=8.1,5.2hz,1h),6.75(s,1h),4.93-4.88(m,1h),4.03(t,j=5.5hz,2h),3.90(dqd,j=13.4,6.7,3.1hz,1h),2.67(t,j=5.6hz,2h),2.51(dt,j=14.5,2.9hz,1h),2.46-2.30(m,2h),1.93(dd,j=14.8,12.2hz,1h),1.41(d,j=6.6hz,3h)。lcms m/z 335.14[m+h]
+
.
[1161]
化合物21
[1162]
(2s)-2-氯-2'-甲基-6'-(2-甲基-4-吡啶基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](21)
[1163][1164]
将(4s)-4-氨基戊-2-酮盐酸盐s25(34.40mg,0.2500mmol)于etoh(1ml)中的溶液添加到2-甲基吡啶-4-甲醛(30.28mg,0.2500mmol)、mgso4(45mg,0.3739mmol)和l-脯氨酸(7mg,0.06080mmol)。添加tea(40μl,0.2870mmol)并将反应物在室温下搅拌过夜。将反应混合物在35-40℃之间经由genevac蒸发,得到粗制c60。向c60添加2-(5-氯-3-噻吩基)乙醇s2(35μl,0.2905mmol)于二噁烷(1ml)中的溶液,随后添加新鲜制备的tfoh(130μl,1.469mmol)于二噁烷(1ml)中的溶液。将混合物在室温下搅拌30分钟。将反应混合物在40℃下经由genevac蒸发。将残余物用naoh(1.7ml 2m,3.400mmol)淬灭并用dcm(1.7ml)稀释。将所得双相混合物搅拌几分钟并且然后使其穿过相分离器。将有机层用氮气吹泡。通过反相hplc纯化(方法:c18 waters sunfire柱(30x 150mm,5微米).梯度:于具有0.1%三氟乙酸的h2o中的mecn),得到呈三氟乙酸盐的(2s)-2-氯-2'-甲基-6'-(2-甲基-4-吡啶基)螺[4,
5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]21(19.3mg,21%)。化合物21通过手性sfc分析确定为77%e.r.(方法:ad-h柱(4.6x 100mm).梯度:具有5mm氨的10%meoh和90% co2)。1h nmr(400mhz,dmso-d6)δ9.28(s,1h),8.87(s,1h),8.55(d,j=5.3hz,1h),7.51(s,1h),7.42(d,j=5.3hz,1h),6.94(s,1h),4.57(t,j=11.5,9.8hz,1h),3.97(t,j=5.4hz,2h),3.6(在水峰下隐藏的1h),3.17-2.84(m,1h),2.61(q,j=5.3hz,2h),2.52(s,3h),2.43-2.13(m,3h),1.90(t,j=13.3hz,1h),1.29(d,j=6.4hz,3h)。lcms m/z 349.14[m+h]+
[1165]
化合物22-172
[1166]
化合物22-172(参见表3)遵循对于化合物19、20或21所述的方法在两步一锅程序中制备为三氟乙酸盐。使用中间体s24或s25、适当的醛和噻吩乙醇s2。醛通过上文所述的方法制备或从商业来源获得。在步骤1反应条件下观察到对映异构纯的起始材料的(4s)-4-氨基戊-2-酮(盐酸盐)s25的部分立体化学消除,导致未分离的2,6-反式哌啶对映异构体的混合物。这由顺式-哌啶酮中间体(如先前在方法a制备s26中所述)和后续2,6反式哌啶最终产物的混合物产生,其中2和6取代基是顺式的,并且2和4取代基是反式的。对方法的任何修改在表3和所附脚注中标识。
[1167]
表3.化合物22-172的制备方法、结构和物理化学数据
[1168]
[1169]
[1170]
[1171]
[1172]
[1173]
[1174]
[1175]
[1176]
[1177]
[1178]
[1179]
[1180]
[1181]
[1182]
[1183]
[1184]
[1185]
[1186]
[1187]
[1188]
[1189]
[1190]
[1191]
[1192]
[1193]
[1194]
[1195][1196]
1.得到呈游离碱形式的产物。
[1197]
2.分离呈具有未知绝对立体化学的非对映异构体的3:2混合物的产物。
[1198]
3.将来自步骤1的混合物在40℃下用氮气吹泡。
[1199]
4.通过硅胶色谱纯化(梯度:0-100%的20%于dcm中的meoh/dcm),得到产物。
[1200]
5.在反应期间使tbs脱保护(步骤2)。
[1201]
6.产物在纯化后是不纯的并通过反相hplc(方法:c18 waters sunfire柱(30x 150mm,5微米).梯度:于具有10mm氢氧化铵的h2o中的mecn)再纯化。
[1202]
7.分离呈具有未知绝对立体化学的非对映异构体的4.5:1混合物的产物。
[1203]
8.将步骤1在室温下搅拌一周。
[1204]
9.将ph用2n naoh小心调节至约ph 7,之后进行dcm萃取。
[1205]
10.分离呈具有未知绝对立体化学的非对映异构体的2:1混合物的产物。
[1206]
11.分离呈具有未知绝对立体化学的非对映异构体的3:1混合物的产物。
[1207]
12.分离呈具有未知绝对立体化学的非对映异构体的5:1混合物的产物。
[1208]
13.分离呈具有未知绝对立体化学的非对映异构体的3.5:1混合物的产物。
[1209]
14.产物在纯化后是不纯的并通过反相hplc(方法:c18 waters sunfire柱(30x 150mm,5微米).梯度:mecn于h2o with 0.2%甲酸)再纯化。得到呈甲酸盐的产物。
[1210]
15.得到呈双-tfa盐的产物。
[1211]
16.分离呈具有未知绝对立体化学的非对映异构体的4:1混合物的产物。
[1212]
17.得到立体异构体的复杂混合物。
[1213]
化合物173
[1214]
(2s)-2-氯-2'-甲基-6'-(1-甲基三唑-4-基)螺[6,7-二氢噻吩并[3,2-c]吡喃-4,4'-哌啶](173)
[1215][1216]
步骤1.1-[(2s)-2-氯-2'-甲基-6'-(1-甲基三唑-4-基)螺[6,7-二氢噻吩并[3,2-c]吡喃-4,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(c61)的合成
[1217]
向2-(5-氯-2-噻吩基)乙醇s2(410mg,2.521mmol)和(2s)-2-甲基-6-(1-甲基三唑-4-基)哌啶-4-酮c56(420mg,2.141mmol)于dcm(8ml)中的溶液中添加甲磺酸(800μl,12.33mmol)。将所得混合物加热至40℃持续40分钟。添加更多甲磺酸(800μl,12.33mmol)并将反应物再继续加热30分钟。将反应物冷却至室温,用水稀释,并用2n naoh溶液碱化。通过相分离器将混合物用dcm(3x 20ml)萃取并将有机层在真空中浓缩,得到粗制(2s)-2-氯-2'-甲基-6'-(1-甲基三唑-4-基)螺[6,7-二氢噻吩并[3,2-c]吡喃-4,4'-哌啶]
[1218]
将粗制(2s)-2-氯-2'-甲基-6'-(1-甲基三唑-4-基)螺[6,7-二氢噻吩并[3,2-c]吡喃-4,4'-哌啶]于具有dipea(600μl,3.445mmol)的dcm(9ml)中的溶液冷却至0℃。在2分钟内缓慢添加tfaa(390μl,2.806mmol),并将反应物在0℃下搅拌。15分钟后,将反应物用饱和碳酸氢钠溶液淬灭并用dcm(3x)萃取。将有机物经硫酸钠干燥并在真空中浓缩。通过硅胶色谱纯化(梯度:0-50%于庚烷中的etoac),得到单个主要产物1-[(2s)-2-氯-2'-甲基-6'-(1-甲基三唑-4-基)螺[6,7-二氢噻吩并[3,2-c]吡喃-4,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c61(450mg,43%)。1h nmr(300mhz,氯仿-d)δ7.58(s,1h),6.91(s,1h),5.57(s,1h),4.40(d,j=7.4hz,1h),4.10(s,3h),3.89(t,j=5.4hz,2h),3.20(dd,j=14.9,6.4hz,1h),2.80-2.61(m,2h),2.45(dd,j=14.8,8.4hz,1h),2.38-2.13(m,1h),2.04(s,1h),1.41-1.12(m,3h)。
[1219]
步骤2.(2s)-2-氯-2'-甲基-6'-(1-甲基三唑-4-基)螺[6,7-二氢噻吩并[3,2-c]吡喃-4,4'-哌啶](173)的合成
[1220]
将1-[(2s)-2-氯-2'-甲基-6'-(1-甲基三唑-4-基)螺[6,7-二氢噻吩并[3,2-c]吡喃-4,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c61(20mg,0.04415mmol)于meoh(1ml)中的溶液用naoh(400μl 2m,0.8000mmol)处理。将溶液加热至50℃持续3小时,此时将其冷却至室温并搅拌过夜。通过相分离器将反应物用dcm(3x)萃取并将有机物在真空中浓缩,得到呈灰白
色膜状物的(2s)-2-氯-2'-甲基-6'-(1-甲基三唑-4-基)螺[6,7-二氢噻吩并[3,2-c]吡喃-4,4'-哌啶]173(14.0mg,91%),其近似e.r.为85%。1h nmr(300mhz,氯仿-d)δ7.41(s,1h),6.61(s,1h),4.40(dd,j=11.8,2.7hz,1h),4.05(s,3h),3.96(td,j=5.7,2.0hz,2h),3.28(dtd,j=12.6,6.3,2.5hz,1h),2.85-2.60(m,2h),2.18(dt,j=13.5,2.6hz,1h),1.89(dt,j=13.7,2.5hz,1h),1.80(dd,j=13.6,11.9hz,1h),1.44(dd,j=13.7,11.4hz,1h),1.11(d,j=6.3hz,3h)。lcms m/z 339.1[m+h]
+
[1221]
s32的制备
[1222]
(2s,4s,6s)-2-甲基-6-(1-甲基三唑-4-基)-1-(2,2,2-三氟乙酰基)-2'-(三氟甲基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮(s32)
[1223][1224]
步骤1.2,2,2-三氟-1-[(2's,6's,7s)-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]乙酮(c62)的合成
[1225]
向溶解于dcm(25ml)中并冷却至-15℃的(2's,6's,7s)-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]2(1260mg,3.352mmol)的溶液中添加dipea(800μl,4.593mmol),随后添加tfaa(550μl,3.957mmol)。5分钟后,将混合物用1n hcl(25ml)淬灭并将各相分离。将有机层用mgso4干燥,过滤,并浓缩。通过硅胶色谱纯化(梯度:0-50%于庚烷中的etoac),得到2,2,2-三氟-1-[(2's,6's,7s)-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]乙酮c62(1444mg,90%)。1h nmr(400mhz,甲醇-d4)δ7.93(s,1h),7.27(d,j=1.3hz,1h),5.63(s,1h),4.46(h,j=7.1hz,1h),4.11(d,j=1.4hz,3h),3.96(td,j=5.6,1.7hz,2h),3.04(s,1h),2.79-2.70(m,3h),2.51(s,1h),2.09(dd,j=14.7,7.3hz,1h),1.23(q,j=9.6,8.4hz,3h)。lcms m/z 469.14[m+h]
+
。
[1226]
步骤2.(2s,4s,6s)-2-甲基-6-(1-甲基三唑-4-基)-1-(2,2,2-三氟乙酰基)-2'-(三氟甲基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮(s32)的合成
[1227]
向2,2,2-三氟-1-[(2's,6's,7s)-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲
基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]乙酮c62(708mg,1.511mmol)于乙腈(10ml)中的混合物中添加n-羟基苯邻二甲酰亚胺(165mg,1.011mmol)和二乙酸钴四水合物(35mg,0.1405mmol)并且然后将混合物用氧气气球真空吹扫三次。将混合物加热至60℃并过滤。一个半小时后,将反应物冷却至室温。将混合物用氮气真空吹扫三次并且然后用mtbe(25ml)和饱和碳酸氢盐水溶液(25ml)稀释。分离各层,并将有机层用nahco3水溶液(2x 50ml)和盐水(50ml)洗涤。将有机层用na2so4干燥,过滤,并浓缩。通过硅胶色谱纯化(梯度:0-50%于庚烷中的etoac),得到(2s,4s,6s)-2-甲基-6-(1-甲基三唑-4-基)-1-(2,2,2-三氟乙酰基)-2'-(三氟甲基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮s32(207mg,26%),1h nmr(300mhz,甲醇-d4)δ7.98(s,1h),7.80(d,j=1.4hz,1h),5.70(s,1h),4.48(s,1h),4.45(s,2h),4.12(s,3h),2.95(dd,j=14.8,9.8hz,1h),2.73(s,1h),2.22(dd,j=14.8,8.4hz,1h),1.29(s,1h),1.19(d,j=14.9hz,3h)。lcms m/z 483.45[m+h]
+
。
[1228]
中间体s33-s36的制备
[1229]
中间体酮s33-s36(参见表4)在两个步骤中使用如对于中间体s32所述的tfaa保护和卞位氧化由相关化合物制备。对方法的任何修改在表4和所附脚注中标识。
[1230]
表4.酮中间体s33-s36的制备方法、结构和物理化学数据
[1231][1232]
1.在0℃下添加tfaa(步骤1)
[1233]
2.将反应物在45℃下搅拌(步骤2)
[1234]
3.将混合物用水(10ml)淬灭并分离各层。将有机层用1n hcl(10ml)、盐水(10ml)洗涤,用硫酸镁干燥,过滤,并浓缩。(步骤1)
[1235]
4.将反应物搅拌18小时(步骤2)
[1236]
5.通过硅胶色谱纯化(0-100% etoac:庚烷),得到产物(步骤1)
[1237]
6.用水淬灭,之后用1n hcl淬灭(步骤1)
[1238]
7.将反应物搅拌45分钟(步骤1)
[1239]
8.将反应物用dcm、水和饱和碳酸氢钠稀释。用dcm萃取(3x)并通过相分离器收集(步骤1)
[1240]
9.将反应物用dcm、水和饱和碳酸氢钠稀释。用dcm萃取(3x)并通过相分离器收集(步骤2)
[1241]
10.通过硅胶色谱纯化(0-100%于庚烷中的etoac),得到产物(步骤2)
[1242]
11.通过硅胶色谱纯化(0-80%于庚烷中的etoac),得到产物(步骤2)
[1243]
12.通过硅胶色谱纯化(0-45%于庚烷中的etoac),得到产物(步骤2)
[1244]
13.将反应物搅拌过夜(步骤2)
[1245]
14.s36具有大约85%e.r.
[1246]
s33的制备
[1247][1248]
步骤1.1-[(2's,6's,7s)-2-氯-2'-甲基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(c154)的合成
[1249]
向冷却至3℃的(2's,6's,7s)-2-氯-2'-甲基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]1(15.0g,43.82mmol)和dipea(10ml,57.41mmol)于dcm(150ml)中的混合物中添加tfaa(6.4ml,46.04mmol)。5分钟后,将混合物用1n hcl(100ml)淬灭,并分离各相。将有机层用盐水(100ml)洗涤,用硫酸镁干燥,过滤,并浓缩。将固体悬浮于tbme(100ml)并加热至回流。30分钟后,将混合物冷却至0℃,并在10分钟后,将材料过滤并用另外的冷tbme冲洗。将产物干燥,产生1-[(2's,6's,7s)-2-氯-2'-甲基-6'-(1-甲基三唑-4-yl)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c154(15.532g,81%)。lcms m/z计算值435.18[m+h]
+
.
[1250]
步骤2.2s,4s,6s)-2'-氯-2-甲基-6-(1-甲基三唑-4-基)-1-(2,2,2-三氟乙酰基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮的合成(s33)
[1251]
向1-[(2's,6's,7s)-2-氯-2'-甲基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(c154)(4.5g,10.24mmol)于乙腈(70ml)中的混合物中添加n-羟基苯邻二甲酰亚胺(1.2g,7.36mmol)和二乙酸钴四水合物(550mg,0.216mmol)并且然后将混合物用氧气气球真空吹扫三次。将混合物加热至45℃并搅拌18小时,之后冷却至室温。将反应物用dcm、水和饱和碳酸氢钠稀释,然后用dcm(3x 150ml)萃取并通过相分离器收集。将有机层用na2so4干燥,过滤,并浓缩。通过硅胶色谱纯化(梯度:0-50%于庚烷中的etoac),得到(2s,4s,6s)-2'-氯-2-甲基-6-(1-甲基三唑-4-基)-1-(2,2,2-三氟乙酰基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮s33(3.50g,68%)。1hnmr(300mhz,氯仿-d)δ7.61(s,1h),7.19(s,1h),5.61(s,1h),4.44(q,j=7.1hz,1h),4.31(s,2h),4.12(s,3h),3.34(dd,j=15.1,6.2hz,1h),2.78(dd,j=15.1,8.3hz,1h),2.70-2.43(m,1h),2.16(s,1h),1.27(d,j=7.3hz,3h)。lcms m/z 449.12[m+h]
+
。
[1252]
化合物174
[1253]
(2's,4s,6's,7s)-2-氯-2'-甲基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(174)
[1254][1255]
步骤1.1-[(2's,4s,6's,7s)-2-氯-4-羟基-2'-甲基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(c63)的合成
[1256]
向(2s,4s,6s)-2'-氯-2-甲基-6-(1-甲基三唑-4-基)-1-(2,2,2-三氟乙酰基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮s33(3.5g,7.025mmol)于dcm(60ml)中添加1,2,3,4,5五甲基环戊烷四氯化铑(2+)(24mg,0.03821mmol)和n-[(1r,2r)-2-氨基-1,2-二苯基-乙基]-4-甲基-苯磺酰胺(27mg,0.074mmol)于dcm(7ml)中的溶液,随后将添加甲酸(1.4ml,37.11mmol)和三乙胺(2.1ml,15.07mmol)的溶液。向烧瓶装备空气球,捕获co2废气副产物。两小时后,将混合物用饱和碳酸氢钠水溶液(150ml)洗涤。将有机相分离,使其穿过相分离
器,并浓缩。硅胶纯化(柱:120g硅胶,梯度:0-45%于庚烷中的etoac),得到呈淡灰白色泡沫的1-[(2's,4s,6's,7s)-2-氯-4-羟基-2'-甲基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c63(3.3g,86%)。1h nmr(300mhz,氯仿-d)δ7.59(s,1h),6.83(s,1h),5.53(s,1h),4.46(dt,j=9.1,3.1hz,2h),4.10(s,3h),4.03-3.80(m,2h),3.10(dd,j=15.1,7.3hz,1h),2.65(ddd,j=15.1,8.1,2.2hz,1h),2.47(s,1h),2.21-2.08(m,1h),2.08(d,j=9.2hz,1h),1.40-1.19(m,3h)。lcms m/z 451.05[m+h]
+
。
[1257]
应注意,醇c63的立体化学使用nmrnoe研究和还原的文献理解使用此催化剂和配体系统来分配。(参考文献:new chiral rhodium and iridium complexes with chiral diamine ligands for asymmetric transfer hydrogenation of aromatic ketones.kunihiko murata,takao ikariya,and ryoji noyori.the journal of organic chemistry 1999 64(7),2186-2187)。
[1258]
步骤2.(2's,4s,6's,7s)-2-氯-2'-甲基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(174)的合成
[1259]
向1-[(2's,4s,6's,7s)-2-氯-4-羟基-2'-甲基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c63(3.33g,100%)于meoh(50ml)中的溶液中添加naoh(40ml 2m,80.00mmol)并将混合物在60℃下搅拌。在40分钟后,将混合物用饱和氯化铵水溶液稀释直到ph 10(约50ml)并用mtbe(5x 100ml)和乙酸乙酯(1x 75ml)萃取。将合并的有机层用饱和nacl水溶液洗涤,经na2so4干燥,并浓缩。将残余物溶于etoh并汽提(3x),得到白色固体。将固体转移到小瓶中并在真空下在55℃下干燥过夜,得到无定形(2's,4s,6's,7s)-2-氯-2'-甲基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇174(2.1817g,87%)。1h nmr(400mhz,甲醇-d4)δ7.82(s,1h),6.88(s,1h),4.46(t,j=3.8hz,1h),4.34-4.28(m,1h),4.08(s,3h),4.04(dd,j=12.2,3.6hz,1h),3.81(dd,j=12.2,4.1hz,1h),3.36-3.25(m,1h),2.39(dt,j=13.8,2.6hz,1h),2.17(dt,j=13.7,2.6hz,1h),1.71(dd,j=13.9,11.9hz,1h),1.45(dd,j=13.7,11.4hz,1h),1.16(d,j=6.4hz,3h)。lcms m/z 355.03[m+h]
+
。
[1260]
化合物175和176
[1261]
(2s)-2-氯-2'-甲基-6'-(1-甲基咪唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(175)[非对映异构体-1]和(176)[非对映异构体-2]
[1262][1263]
步骤1.1-[(2s)-2-氯-4-羟基-2'-甲基-6'-(1-甲基吡唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(c64)[diast-1]和(c65)[diast-2]的合成
[1264]
将于thf(1ml)中的(r)-(+)-2-甲基-cbs-噁唑硼烷溶液(40μl 1m,0.04000mmol)(1m于thf中的溶液)冷却至0℃并用硼烷;四氢呋喃(220μl1m,0.2200mmol)处理。4分钟后,缓慢添加(2s,4s,6s)-2'-氯-2-甲基-6-(1-甲基吡唑-4-基)-1-(2,2,2-三氟乙酰基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮s35(50mg,0.1023mmol)于thf(300μl)中的溶液并将反应物在0℃下搅拌。15分钟后,制成另一个(r)-(+)-2-甲基-cbs-噁唑硼烷溶液(40μl 1m,0.04000mmol)和硼烷;四氢呋喃(220μl 1m,0.2200mmol)的溶液并将其添加到反应物中。30分钟后,将反应物用2n hcl淬灭,移除冰浴,并将混合物剧烈搅拌24小时。通过相分离器将反应物用dcm(3x)萃取。将有机物经由旋转蒸发仪浓缩。通过硅胶色谱纯化(0-60%于庚烷中的etoac),得到外消旋中间体。
[1265]
sfc分离(ad-h柱,使用10% meoh w/5mm氨)得到1-[(2s)2-氯-4-羟基-2'-甲基-6'-(1-甲基吡唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(c64)[diast-1](50mg,101%)1h nmr(300mhz,氯仿-d)δ7.40(d,j=5.6hz,2h),6.88(s,1h),5.52(s,1h),4.52-4.37(m,2h),4.00-3.76(m,5h),2.74-2.48(m,2h),2.27(s,1h),1.81(dd,j=14.9,6.8hz,1h),1.28(s,3h)。lcms m/z 450.07[m+h]
+
;1-[(2s)-2-氯-4-羟基-2'-甲基-6'-(1-甲基吡唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(c65)[diast-2](34mg,62%)1h nmr(300mhz,氯仿-d)δ7.43(s,1h),7.35(s,1h),6.85(s,1h),5.43(s,1h),4.56-4.40(m,2h),4.02-3.79(m,5h),2.71(ddd,j=15.1,8.1,2.3hz,1h),2.37(dd,j=15.1,6.4hz,1h),2.08(d,j=9.2hz,1h),1.95(dd,j=14.6,6.9hz,1h),1.27(d,j=6.6hz,3h)。lcms m/z 450.03[m+h]
+
。
[1266]
步骤2.(2s)-2-氯-2'-甲基-6'-(1-甲基咪唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(175)[diast-1]和(176)[diast-1]的合成
[1267]
将于meoh(2ml)中的1-[(2's,4s,6's,7s)-2-氯-4-羟基-2'-甲基-6'-(1-甲基吡唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(c64)(50mg,0.094mmol)用naoh(1ml 1m,1.000mmol)处理并加热至40℃持续30分钟。添加另外的naoh(1ml 1m,1.000mmol)并将反应物加热至50℃。两个多小时后,将反应物用dcm稀释。将有机相分离,使其穿过相分离器,并浓缩。将材料溶于mtbe(3ml)中并用二噁烷中的氯化氢(28μl 4m,0.1120mmol)逐滴处理。形成白色沉淀。将溶液浓缩并将残余物溶于水中,在-78℃下冷冻,并在整个周末冻干,得到(2s)-2-氯-2'-甲基-6'-(1-甲基吡唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(盐酸盐)(175)[diast-1](37.9mg,98%)。1h nmr(400mhz,甲醇-d4)δ7.72(s,1h),7.58(s,1h),6.92(s,1h),4.52-4.45(m,2h),4.03(dd,j=12.2,3.5hz,1h),3.88(s,3h),3.83(dd,j=12.2,4.0hz,1h),3.69-3.58(m,1h),2.47(dt,j=14.5,2.8hz,1h),2.33(dt,j=14.4,2.8hz,1h),1.98(t,j=13.7hz,1h),1.71(t,j=13.1hz,1h),1.30(d,j=6.6hz,3h)。lcms m/z 353.99[m+h]
+
。
[1268]
将于meoh(1.5ml)中的1-[(2's,4r,6's,7s)-2-氯-4-羟基-2'-甲基-6'-(1-甲基吡唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(c65)(34mg,0.07mmol)用naoh(800μl 1m,0.8mmol)处理并加热至40℃持续30分钟。添加更多naoh(800μl 1m,0.8mmol)并将反应物加热至50℃。两个多小时后,将反应物冷却至室温,用dcm稀释。将有机相分离,使其穿过相分离器,并浓缩。将材料溶于mtbe(1.5ml)并用hcl(22μl 4m,0.08800mmol)逐滴处理。形成白色沉淀。经由旋转蒸发仪浓缩溶液并将残余物溶于水中,在-78℃下冷冻,并冻干过夜,得到呈白色粉末的(2s)-2-氯-2'-甲基-6'-(1-甲基吡唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(盐酸盐)(176)[diast-2](26.7mg,96%)。1h nmr(400mhz,甲醇-d4)δ7.81(s,1h),7.63(s,1h),6.94(s,1h),4.71(dd,j=12.6,2.9hz,1h),4.50(t,j=3.6hz,1h),4.05(dd,j=12.3,3.4hz,1h),3.90(s,3h),3.85(dd,j=12.2,3.9hz,1h),3.71(dtq,j=13.4,6.8,2.9hz,1h),2.53(dt,j=14.4,2.8hz,1h),2.42(dt,j=14.8,2.8hz,1h),2.21(dd,j=14.4,12.6hz,1h),1.72(dd,j=14.8,12.2hz,1h),1.35(d,j=6.6hz,3h)。lcms m/z 354.04[m+h]
+
。
[1269]
化合物177
[1270]
(2's,4r,6's,7s)-2-氯-2'-甲基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇](177)
[1271][1272]
步骤1.(2's,4r,6's,7s)-2-氯-2'-甲基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇](177)的合成
[1273]
向冷却至0℃的四氢呋喃(500μl)中添加(3ar)-1-甲基-3,3-二苯基-3a,4,5,6-四
氢吡咯并[1,2-c][1,3,2]噁唑硼烷((r)-cbs催化剂)(25μl 1m,0.025mmol),随后添加硼烷四氢呋喃(250μl 1m,0.25mmol)。在搅拌5分钟后,逐滴添加(2s,4s,6s)-2'-氯-2-甲基-6-(1-甲基三唑-4-基)-1-(2,2,2-三氟乙酰基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮s33(25mg,0.05570mmol)于thf(1000μl)中的溶液。将混合物在0℃下搅拌1小时。将混合物浓缩,用meoh(1.5ml)稀释并用naoh(100μl 6m,0.6000mmol)淬灭。将混合物升温至50℃并搅拌过夜。通过反相hplc纯化(方法:c18 waters sunfire柱(30x 150mm,5微米).梯度:于具有5mm hcl的h2o中的mecn),得到(2's,4r,6's,7s)-2-氯-2'-甲基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(盐酸盐)177(3.0mg,13%)。1h nmr(300mhz,甲醇-d4)δ8.11(d,j=2.4hz,1h),6.94(s,1h),4.90(d,j=3.1hz,1h),4.51(t,j=3.6hz,1h),4.13(d,j=2.2hz,3h),4.07(dd,j=12.2,3.5hz,1h),3.87(dd,j=12.3,3.9hz,1h),3.74(ddt,j=13.0,9.4,6.4hz,1h),2.78-2.51(m,1h),2.51-2.19(m,2h),1.87(ddd,j=30.8,14.7,12.2hz,1h),1.39(dd,j=6.6,3.1hz,3h)。lcms m/z 355.03[m+h]
+
。
[1274]
化合物178-182
[1275]
化合物178-182(参见表5)在两个或三个步骤中使用如对于化合物174-177所述的还原和脱保护方法由表5中的酮中间体来制备。通过用naoh水解来制成最终化合物。对这些方法的任何修改在表5和所附脚注中标识。
[1276]
表5.化合物178-182的制备方法、结构和物理化学数据
[1277]
[1278][1279]
1.添加甲酸和三乙胺,之后添加酮中间体(步骤1)
[1280]
2.硅胶纯化(0-60%于庚烷中的etoac),得到产物(步骤1)
[1281]
3.不用etoh洗涤产物(步骤2)
[1282]
4.将反应物搅拌15分钟,之后淬灭
[1283]
5.在用naoh和meoh淬灭后在40℃下搅拌四小时(步骤2)
[1284]
6.在两个单独反应中用cbs-(s)催化剂和cbs-(r)催化剂运行步骤1。然而,两个反应以较差d.r.进行,因此将它们合并以制成外消旋物,与在步骤2中一样通过sfc分离所述外消旋物。
[1285]
7.在用dcm萃取后,通过硅胶色谱纯化(梯度:0-20%于dcm中的meoh),得到产物(步骤3)
[1286]
8.将反应物搅拌过夜(步骤1)
[1287]
9.在用naoh和meoh淬灭后在50℃下搅拌两小时(步骤2)
[1288]
10.化合物182含有大约15%通过与存在于s36中的s36对映异构体不同来形成的非对映异构体。
[1289]
化合物181
[1290]
(2s,4s,4's,6s)-2-甲基-6-(1-甲基-1h-1,2,3-三唑-4-基)-2'-(三氟甲基)-4',5'-二氢螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-醇(181),无定形形式
[1291][1292]
步骤1.2,2,2-三氟-1-[(2's,4s,6's,7s)-4-羟基-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]乙酮(c153)的合成向(2s,4s,6s)-2-甲基-6-(1-甲基三唑-4-基)-1-(2,2,2-三氟乙酰基)-2'-(三氟甲基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮s32(2.23g,4.63mmol)于dcm(20ml)中添加1,2,3,4,5五甲基环戊烷四氯化铑(2+)(7mg,0.002mmol)和n-[(1r,2r)-2-氨基-1,2-二苯基-乙基]-4-甲基-苯磺酰胺(8.5mg,0.005mmol)于dcm(2ml)中的溶液,随后将添加甲酸(0.9ml,5.15mmol)和三乙胺(1.3ml,2.01mmol)的溶液。向烧瓶装备空气球,捕获co2废气副产物。三小时后,将混合物用饱和碳酸氢钠水溶液(10ml)洗涤。将有机相分离,使其穿过相分离器,并浓缩。硅胶纯化(柱:40g硅胶,梯度:0-50%于庚烷中的etoac),得到呈白色固体的2,2,2-三氟-1-[(2's,4s,6's,7s)-4-羟基-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]乙酮c153(2.27g,100%)。lcms m/z 485.11[m+h]
+
。
[1293]
步骤2.(2's,4s,6's,7s)-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(181)的合成
[1294]
向2,2,2-三氟-1-[(2's,4s,6's,7s)-4-羟基-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]乙酮c153(2.27g,4.63mmol)于meoh(45ml)中的溶液中添加naoh(8ml 6m,48.00mmol)并将混合物在60℃下搅
拌。75分钟后,将混合物用饱和氯化铵水溶液稀释直到ph10(约40ml)、用水(40ml)稀释,并用mtbe(100ml)萃取。将水层用另外的mtbe(2x 50ml)萃取,并将合并的有机层用饱和nacl水溶液洗涤,经mgso4干燥,并浓缩,得到无定形(2's,4s,6's,7s)-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇181(1.84g,88%)。1h nmr(400mhz,氯仿-d)δ7.48(s,1h),7.39(q,j=1.2hz,1h),4.58(d,j=8.0hz,1h),4.44(dd,j=11.7,2.5hz,1h),4.09(s,4h),4.01(dd,j=12.5,2.7hz,1h),3.43(ddd,j=11.2,6.4,2.5hz,1h),2.48(dt,j=13.8,2.6hz,1h),2.16-2.07(m,2h),1.77(dd,j=13.9,11.8hz,1h),1.63(s,1h),1.28(s,1h),1.18(d,j=6.3hz,3h)。lcms m/z 389.09[m+h]
+
。
[1295]
化合物183
[1296]
(2's,6's,7s)-2-氯-4,4-二氟-2'-甲基-6'-(1-甲基三唑-4-基)螺[5h-噻吩并[2,3-c]吡喃-7,4'-哌啶](183)
[1297][1298]
步骤1.1-[(2's,6's,7s)-2-氯-4,4-二氟-2'-甲基-6'-(1-甲基三唑-4-基)螺[5h-噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(c66)的合成
[1299]
将(2s,4s,6s)-2'-氯-2-甲基-6-(1-甲基三唑-4-基)-1-(2,2,2-三氟乙酰基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮s33(40mg,0.08912mmol)溶解于dcm(100μl)并添加dast(35μl,0.2649mmol)。将反应物加热至40℃并搅拌3小时。添加另外的dast(35μl,0.2649mmol)并搅拌整个周末。将溶液用dcm稀释并倒入到nahco3水溶液中,在0℃下搅拌。将有机相分离,使其穿过相分离器,并浓缩,得到粗制1-[(2's,6's,7s)-2-氯-4,4-二氟-2'-甲基-6'-(1-甲基三唑-4-基)螺[5h-噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c66(41mg,42%)。lcms m/z 468.71[m+h]
+
。
[1300]
步骤2.(2's,6's,7s)-2-氯-4,4-二氟-2'-甲基-6'-(1-甲基三唑-4-基)螺[5h-噻吩并[2,3-c]吡喃-7,4'-哌啶](183)的合成
[1301]
将[(2's,6's,7s)-2-氯-4,4-二氟-2'-甲基-6'-(1-甲基三唑-4-基)螺[5h-噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c66在6m naoh水溶液(10当量)中搅拌五小时。通过旋转蒸发去除溶剂。通过反相hplc纯化(方法:c18waters sunfire柱(30x 150mm,5微米),使用10mm氢氧化铵),得到(2's,6's,7s)-2-氯-4,4-二氟-2'-甲基-6'-(1-甲基三唑-4-基)螺[5h-噻吩并[2,3-c]吡喃-7,4'-哌啶]183(2mg,6%)。lcms m/z 375.11[m+h]
+
。
[1302]
化合物184和185
[1303]
(2's,6's,7s)-2-氯-4-(二氟甲基)-2'-甲基-6'-(1-甲基三唑-4-基)螺[5h-噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(184)[diast-1]和(185)[diast-2]
[1304][1305]
向(2s,4s,6s)-2'-氯-2-甲基-6-(1-甲基三唑-4-基)-1-(2,2,2-三氟乙酰基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮s33(65mg,0.1448mmol)于mecn(2.4ml)中的溶液中添加dmpu(34μl,0.2822mmol),随后添加[溴(二氟)甲基]-三甲基-硅烷(35mg,0.1723mmol)和pph3(40μl,0.1726mmol)。将所得溶液在55℃下加热。四小时后,将反应物冷却至室温。添加koh水溶液(720μl 1m,0.7200mmol)并将反应物搅拌48小时。将反应物用碳酸氢钠和dcm淬灭。将有机相分离,使其穿过相分离器。通过反相hplc纯化(方法:c18 waters sunfire柱(30x 150mm,5微米).梯度:于具有0.1%三氟乙酸的h2o中的mecn)进行纯化。
[1306]
仍存在一些杂质。通过硅胶色谱纯化(梯度:0-20%于dcm中的meoh),得到两种分离的非对映异构体(2's,6's,7s)-2-氯-4-(二氟甲基)-2'-甲基-6'-(1-甲基三唑-4-基)螺[5h-噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(三氟乙酸盐)184[diast-1](4.3mg,6%)。1h nmr(400mhz,甲醇-d4)δ7.94(s,1h),7.01(s,1h),5.97(t,j=55.2hz,1h),4.57(dd,j=12.3,2.9hz,1h),4.10(s,4h),3.81(dt,j=12.6,2.2hz,1h),3.60-3.42(m,1h),2.60(dt,j=14.4,2.7hz,1h),2.18(dt,j=14.1,2.6hz,1h),2.08-1.95(m,1h),1.69(dd,j=14.2,11.8hz,1h),1.26(d,j=6.5hz,3h)。lcms m/z 405.02[m+h]
+
。(2's,6's,7s)-2-氯-4-(二氟甲基)-2'-甲基-6'-(1-甲基三唑-4-基)螺[5h-噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(三氟乙酸盐)185[diast-2](8.5mg,11%)。1h nmr(400mhz,甲醇-d4)δ8.06(s,1h),7.04(d,j=0.9hz,1h),5.98(t,j=55.1hz,1h),4.91-4.86(m,1h),4.20-4.05(m,4h),3.90-3.71(m,2h),2.64-2.33(m,3h),1.85(dd,j=14.8,12.2hz,1h),1.40(d,j=6.6hz,3h)。lcms m/z 405.02[m+h]
+
。
[1307]
s37的制备
[1308]
4-氨基己-2-酮盐酸盐(s37)
[1309][1310]
步骤1.n-[1-(对甲苯基磺酰基)丙基]氨基甲酸叔丁酯(c68)的合成
[1311]
在0℃下向氨基甲酸叔丁酯c67(20g,0.1639mol)和4-甲基苯亚磺酸盐(53g,0.3313mol)于meoh(120ml)和h2o(240ml)中的溶液中逐滴添加丙醛(15.082g,19ml,0.2545mol)和甲酸(76.921g,65ml,1.6211mol)。将反应混合物在室温下搅拌24小时。将反应混合物过滤,用水(100ml)洗涤,并干燥,得到呈灰白色固体的n-[1-(对甲苯基磺酰基)丙基]氨基甲酸叔丁酯c68(46g,85%)。1h nmr(300mhz,dmso-d6)δ7.82(d,j=9.6hz,1h),7.65(d,j=8.4hz,2h),7.48-7.40(m,2h),4.63-4.56(m,1h),2.39(d,j=14.4hz,3h),2.01-1.95(m,1h),1.69-1.61(m,1h),1.17(s,9h),0.91(t,j=7.2hz,3h)。
[1312]
步骤2.2-乙酰基-3-(叔丁氧基羰基氨基)戊酸叔丁酯(c69)的合成
[1313]
在室温下向nah(2.5g,60%(w/w),0.0625mol)于thf(160ml)中的溶液中分批添加n-[1-(对甲苯基磺酰基)丙基]氨基甲酸叔丁酯c68(10g,0.0303mol)并搅拌5分钟并且然后逐滴添加于thf(40ml)中的3-氧代丁酸叔丁酯(5.1001g,5.4ml,0.0319mol)并在同一温度下搅拌2小时。将反应混合物用nh4cl溶液(150ml)淬灭并用dcm(2x 300ml)萃取。将有机层经无水na2so4干燥,在减压下浓缩并干燥,提供呈黄色胶状物的粗制2-乙酰基-3-(叔丁氧基羰基氨基)戊酸叔丁酯c69(11g,98%)。1h nmr(300mhz,dmso-d6)δ6.72-6.56(m,1h),3.97-3.89(m,1h),3.58-3.44(m,1h),2.16(s,3h),1.48-1.15(m,20h),0.85-0.76(m,3h)。lcms m/z 316.27[m+h]
+
。
[1314]
步骤3.4-氨基己-2-酮(s37)的合成
[1315]
将2-乙酰基-3-(叔丁氧基羰基氨基)戊酸叔丁酯c69(11g,0.0296mol)于10%hcl水溶液(120ml 10%(w/v),0.3291mol)中的溶液加热至110℃并搅拌2小时。将反应混合物用乙醚(4x 50ml)萃取。将水性部分在减压下蒸发并干燥,得到呈淡棕色液体的4-氨基己-2-酮s37(盐酸盐)(3.92g,85%)。1h nmr(400mhz,dmso-d6)δ8.00(s,3h),3.38-3.30(m,1h),2.89-2.73(m,2h),2.14(s,3h),1.63-1.49(m,2h),0.91(t,j=10.4hz,3h)。lcms m/z 116.3[m+h]
+
。
[1316]
s38的制备
[1317]
4-氨基-5-甲基-己-2-酮盐酸盐(s38)
[1318][1319]
步骤1.n-[2-甲基-1-(对甲苯基磺酰基)丙基]氨基甲酸叔丁酯(c71)的合成
[1320]
在室温下向2-甲基丙醛(18.652g,23.610ml,0.2535mol)和氨基甲酸叔丁酯c70(20g,0.1690mol)于meoh(200ml)和水(50ml)中的搅拌溶液中添加对-甲苯亚磺酸钠(60.835g,0.3380mol),随后添加甲酸(79.370g,65.057ml,1.6900mol)并且然后将反应混合物在室温下搅拌24小时。将反应混合物过滤,用水(200ml)和乙醚(50ml)洗涤,并在真空下干燥,得到呈灰白色固体的粗制n-[2-甲基-1-(对甲苯基磺酰基)丙基]氨基甲酸叔丁酯c71(45g,73%)。1hnmr(300mhz,dmso-d6)δ7.81(d,j=10.5hz,1h),7.67(d,j=8.1hz,2h),7.39(d,j=8.1hz,2h),4.58(q,j=5.7hz,1h),2.46
–
2.36(m,4h),1.14(s,9h),1.00(s,6h)。
[1321]
步骤2.2-乙酰基-3-(叔丁氧基羰基氨基)-4-甲基-戊酸叔丁酯(c72)的合成
[1322]
在室温下向nah(2.1998g,0.0550mol)于thf(100.00ml)中的搅拌溶液中添加n-[2-甲基-1-(对甲苯基磺酰基)丙基]氨基甲酸叔丁酯c71(10g,0.0275mol)并将反应物搅拌20分钟。添加thf(30.000ml)中的3-氧代丁酸叔丁酯(5.3270g,5.6074ml,0.0330mol)并在室温下搅拌2小时。在10℃下将反应混合物倒入到饱和氯化铵(500ml)并用etoac(2x 200ml)萃取。将合并的有机层经无水na2so4干燥并过滤,在减压下浓缩,得到呈棕色液体的粗制2-乙酰基-3-(叔丁氧基羰基氨基)-4-甲基-戊酸叔丁酯c72(11g,45%)。lcms m/z330.2[m+h]
+
。
[1323]
步骤3.4-氨基-5-甲基-己-2-酮(s38)的合成
[1324]
将2-乙酰基-3-(叔丁氧基羰基氨基)-4-甲基-戊酸叔丁酯c72(15g,0.0319mol)于hcl(290ml 10%(w/v),0.7954mol)中的溶液加热至110℃持续3小时。将反应混合物在减压下浓缩,获得粗制化合物(6.2g)。将反应物用正戊烷(2x100ml)研磨并在真空下干燥,得到呈棕色半固体的4-氨基-5-甲基-己-2-酮s38(盐酸盐)(5.1g,96%)。1h nmr(400mhz,dmso-d6)δ7.96(br s,3h),3.34-3.28(m,1h),2.86-2.75(m,2h),2.15(s,3h),1.94-1.86(m,1h),0.88(s,6h)lcms m/z 130.2[m+h]
+
.
[1325]
s39的制备
[1326]
1-环丁基-3-氧代-丁基)氯化铵(s39)
[1327][1328]
步骤1.n-[环丁基(对甲苯基磺酰基)甲基]氨基甲酸叔丁酯(c74)的合成
[1329]
在0℃下向对-甲苯亚磺酸钠(27.843g,0.1547mol)和氨基甲酸叔丁酯c73(盐酸盐)(12g,0.0773mol)于meoh(120.00ml)和水(240.00ml)中的混合物中添加甲酸(12.20g,10.00ml,0.2598mol)和环丁烷甲醛c77(10g,0.1177mol)。将混合物在氮气下在室温下搅拌48小时。将白色沉淀在真空下过滤,用水(3x 50ml)洗涤,并干燥,得到呈白色固体的n-[环丁基(对甲苯基磺酰基)甲基]氨基甲酸叔丁酯c74(25g,93%)。1h nmr(300mhz,dmso-d6)δ7.82(d,j=9.9hz,1h),7.67(d,j=8.4hz,2h),7.42(d,j=8.1hz,2h),4.72(t,j=9.3hz,1h),2.86-2.82(m,1h),2.37(s,3h),2.01-1.73(m,6h),1.18(s,9h)。
[1330]
步骤2.2-[(叔丁氧基羰基氨基)-环丁基-甲基]-3-氧代-丁酸叔丁酯(c75)的合成
[1331]
在0℃下向n-[环丁基(对甲苯基磺酰基)甲基]氨基甲酸叔丁酯c74(25g,0.0722mol)和3-氧代丁酸叔丁酯(盐酸盐)(15.5g,0.0788mol)于thf(550ml)中的混合物中添加nah(5.35g,60%(w/w),0.133mmol)。将混合物在氮气下在室温下搅拌48小时,并将白色沉淀在真空下过滤,用水(3x 5ml)洗涤,并干燥,得到呈淡黄色油状物的2-[(叔丁氧基羰基氨基)-环丁基-甲基]-3-氧代-丁酸叔丁酯c75(25g,99%)。1h nmr(400mhz,氯仿-d):δ4.30-4.11(m,1h),3.46(d,j=4.0hz,1h),2.49-2.42(m,1h),2.25(s,3h),1.94-1.74(m,6h),1.48-1.44(m,18h)。lcms m/z 342.13[m+1]
+
.
[1332]
步骤3.(1-环丁基-3-氧代-丁基)氯化铵(s39)的合成
[1333]
在室温下向2-[(叔丁氧基羰基氨基)-环丁基-甲基]-3-氧代-丁酸叔丁酯c75(25g,0.0718mol)于水(100ml)中的混合物中添加hcl(100ml 5m,0.5000mol)。将混合物在氮气下在110℃下搅拌16小时。将反应混合物用水(100ml)稀释并用乙醚(3x 100ml)洗涤。将水层在真空下蒸发并干燥,得到呈淡棕色半固体的(1-环丁基-3-氧代-丁基)氯化铵s39(12.81g,100%)。1h nmr(400mhz,dmso-d6)δ7.86(brs,3h),3.40-3.36(m,1h),2.69-2.67(m,2h),2.51-2.43(m,1h),2.14(s,3h),1.97-1.68(m,6h)。lcms m/z 142.2[m+h]
+
。
[1334]
s40的制备
[1335]
4-氨基-5-甲氧基-戊-2-酮盐酸盐(s40)
3.40(m,2h),3.28(s,3h),2.83(t,j=6.4hz,2h),2.13(s,3h)。lcms m/z 132.2[m+h]
+
。
[1345]
s41的制备
[1346]
2-乙基-6-(1-甲基三唑-4-基)哌啶-4-酮(s41)
[1347][1348]
将4-氨基己-2-酮s37(盐酸盐)(298mg,1.965mmol)溶解于etoh(9ml)中并向其中添加tea(280μl,2.009mmol)、1-甲基三唑-4-甲醛s17(225mg,2.025mmol)、l-脯氨酸(47mg,0.4082mmol)和mgso4(255mg,2.119mmol)。将反应在室温下搅拌16小时。将混合物过滤,浓缩,用饱和nahco3水溶液(50ml)淬灭,并用dcm(5x 20ml)萃取。将合并的有机层用mgso4干燥,过滤,并在减压下浓缩。通过硅胶色谱纯化(梯度:0-10%于dcm中的meoh),得到2-乙基-6-(1-甲基三唑-4-基)哌啶-4-酮s41(162mg,38%)。1h nmr(300mhz,氯仿-d)δ7.46(d,j=2.0hz,1h),4.22(ddd,j=10.3,5.0,2.0hz,1h),4.09(d,j=2.1hz,3h),2.93(dtd,j=11.8,6.3,2.9hz,1h),2.77-2.56(m,2h),2.48(ddd,j=14.1,2.9,1.6hz,1h),2.16(dd,j=14.1,11.7hz,1h),1.74-1.50(m,2h),0.98(td,j=7.6,2.0hz,3h)。lcms m/z 209.08[m+h]
+
。在分离期间吹扫少量反式异构体。
[1349]
s42的制备
[1350]
2-异丙基-6-(1-甲基三唑-4-基)哌啶-4-酮(s42)
[1351][1352]
4-氨基-5-甲基-己-2-酮s38(盐酸盐)(500mg,3.018mmol)溶解于etoh(15ml)并向其中添加tea(430μl,3.085mmol)、1-甲基三唑-4-甲醛s17(350mg,3.150mmol)、l-脯氨酸(72mg,0.6254mmol)和mgso4(392mg,3.257mmol)。将反应物在室温下搅拌3天。将混合物过滤,浓缩,并用饱和碳酸氢盐水溶液(50ml)淬灭并用dcm(5x 20ml)萃取。将合并的有机层用mgso4干燥,过滤,并浓缩。粗制1h nmr显示约5.5:1dr。通过硅胶色谱纯化(梯度:0-5%于dcm中的meoh),得到=2-异丙基-6-(1-甲基三唑-4-基)哌啶-4-酮s42(272mg,38%)。1h nmr(300mhz,氯仿-d)δ7.45(s,1h),4.19(dd,j=11.0,4.2hz,1h),4.10(s,3h),2.79(ddd,j=11.8,5.7,2.9hz,1h),2.73-2.54(m,2h),2.46(ddd,j=13.9,2.8,1.7hz,1h),2.21(dd,j=13.9,11.8hz,1h),2.08(s,1h),1.78(dt,j=13.2,6.6hz,1h),0.98(t,j=6.5hz,6h)。在分离期间吹扫少量反式异构体。
[1353]
s43的制备
[1354]
2-环丁基-6-(1-甲基三唑-4-基)哌啶-4-酮(s43)
[1355][1356]
向4-氨基-4-环丁基丁-2-酮s39(盐酸盐)(1260mg,7.092mmol)于etoh(38ml)中的溶液中添加tea(1.0ml,7.175mmol)、1-甲基三唑-4-甲醛s17(835mg,7.516mmol)、l-脯氨酸(168mg,1.459mmol)和mgso4(919mg,7.635mmol)。将反应在室温下搅拌24小时。将混合物过滤,浓缩,并用饱和碳酸氢盐水溶液(50ml)淬灭并用dcm(5x 20ml)萃取。将合并的有机层用mgso4干燥,过滤,并浓缩。通过硅胶色谱纯化(梯度:0-3%于dcm中的meoh),得到2-环丁基-6-(1-甲基三唑-4-基)哌啶-4-酮s43(849mg,48%)。1hnmr(300mhz,氯仿-d)δ7.45(s,1h),4.27-4.16(m,1h),4.09(s,3h),2.92(ddd,j=11.5,8.6,2.8hz,1h),2.67-2.56(m,2h),2.49-2.30(m,2h),2.23-1.70(m,8h)。在分离期间吹扫少量反式异构体。
[1357]
s44的制备
[1358]
2-(甲氧基甲基)-6-(1-甲基三唑-4-基)哌啶-4-酮(s40)
[1359][1360]
向4-氨基-5-甲氧基-戊-2-酮s40(盐酸盐)(750mg,4.474mmol)于etoh(22ml)中的溶液中添加tea(650μl,4.664mmol)、1-甲基三唑-4-甲醛s17(520mg,4.680mmol)、l-脯氨酸(107mg,0.9294mmol)和mgso4(590mg,4.902mmol)。将反应在室温下搅拌16小时。将混合物过滤,浓缩,并用饱和碳酸氢盐水溶液(50ml)淬灭并用dcm(7x 20ml)萃取。将合并的有机层用硫酸钠干燥,过滤,并浓缩。通过硅胶色谱纯化(梯度:0-12%于dcm中的meoh),提供2-(甲氧基甲基)-6-(1-甲基三唑-4-基)哌啶-4-酮s44(373mg,37%)。1h nmr(300mhz,氯仿-d)δ7.47(s,1h),4.24(dd,j=10.1,5.0hz,1h),4.09(s,3h),3.48(dd,j=9.2,3.3hz,1h),3.43-3.34(m,4h),3.25(ddt,j=10.9,7.4,3.8hz,1h),2.71-2.59(m,3h),2.40-2.28(m,2h)。lcms m/z 225.06[m+h]
+
。分离作为5:1dr混合物的产物。
[1361]
化合物186
[1362]
2-氯-2'-乙基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](186)
[1363]
[1364]
向2-乙基-6-(1-甲基三唑-4-基)哌啶-4-酮s41(31mg,0.1414mmol)和2-(5-氯-3-噻吩基)乙醇(26mg,0.1599mmol)s2于dcm(700μl)中的混合物中添加msoh(37μl,0.5702mmol)并将混合物加热至40℃持续4小时并且然后在室温下搅拌72小时。将反应物用饱和nahco3溶液淬灭并用dcm(6x)萃取。将合并的有机层经无水na2so4干燥,过滤并减压浓缩。通过硅胶色谱纯化(梯度:0-10%于dcm中的meoh)。将最终产物溶解于mecn和水中并经历冻干,提供2-氯-2'-乙基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]186(42.2mg,75%)。1h nmr(300mhz,氯仿-d)δ7.43(s,1h),6.58(s,1h),4.40(dd,j=11.8,2.7hz,1h),4.06(s,3h),4.00-3.89(m,2h),3.15-2.99(m,1h),2.74-2.49(m,2h),2.36(dt,j=13.5,2.6hz,1h),2.10(dt,j=13.7,2.5hz,1h),1.96-1.74(m,2h),1.42(qt,j=11.1,4.9hz,3h),0.94(t,j=7.6hz,3h)。lcms m/z 353.05[m+h]
+
。
[1365]
化合物187
[1366]
2'-乙基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](187)
[1367][1368]
向2-乙基-6-(1-甲基三唑-4-基)哌啶-4-酮s41(31mg,0.1414mmol)和2-[5-(三氟甲基)-3-噻吩基]乙醇(31mg,0.1580mmol)s3于dcm(700μl)中的混合物中添加meoh(37μl,0.5702mmol)并将混合物加热至40℃持续3小时。向所述溶液中添加msoh(11μl)并在40℃下搅拌1.5小时并且然后在35℃下搅拌3.5天。向所述溶液中添加msoh(11μl)并在在50℃下搅拌5天,在此时间期间蒸发溶剂。将反应物用饱和nahco3溶液淬灭并用dcm(6x)萃取。将合并的有机层经无水na2so4干燥,过滤,并浓缩。通过反相hplc纯化(方法:c18 waters sunfire柱(30x 150mm,5微米).梯度:于具有0.1%三氟乙酸的h2o中的mecn),提供2'-乙基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](三氟乙酸盐)187(33.5mg,46%)。1h nmr(300mhz,氯仿-d)δ7.89(s,1h),7.16(s,1h),4.94(d,j=10.2hz,1h),4.09(s,3h),3.98(h,j=6.2hz,2h),3.67(s,1h),2.93-2.64(m,3h),2.49-2.34(m,2h),2.19-2.04(m,2h),1.94(s,1h),1.67(dt,j=14.5,7.8hz,1h),1.00(t,j=7.5hz,3h)。lcms m/z 387.16[m+h]
+
。
[1369]
化合物188-193
[1370]
化合物188-193(参见表6)在一个步骤中在与噻吩乙醇中间体s2和s3和哌啶酮(s42-s44)的oxa-pictet spengler反应中制备。对方法的任何修改在表6和所附脚注中标识。
[1371]
表6.化合物188-193的制备方法、结构和生物化学数据
[1372]
[1373][1374]
1.将反应物在40℃下搅拌22小时
[1375]
2.添加另外的msoh(31μl,4当量)并将反应物在40℃下搅拌4小时,然后在室温下搅拌过夜。
[1376]
3.通过反相hplc纯化(方法:c18 waters sunfire柱(30x 150mm,5微米).梯度:于具有0.2%甲酸的h2o中的mecn),得到产物
[1377]
4.将反应物在40℃下搅拌4小时
[1378]
5.通过反相hplc纯化(方法:c18 waters sunfire柱(30x 150mm,5微米).梯度:于具有0.1%三氟乙酸的h2o中的mecn),得到产物
[1379]
6.将另外的msoh(11μl,1.3当量)添加到反应物中并在80℃下搅拌24小时
[1380]
7.将反应物在40℃下搅拌2.5小时
[1381]
8.将反应物在65℃下搅拌4天
[1382]
9.将反应物在60℃下搅拌5天
[1383]
化合物194
[1384]
2-氯-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](194)
[1385][1386]
步骤1.2-氯-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](194)的合成
[1387]
向2,6-二甲基哌啶-4-酮c80(盐酸盐)(40mg,0.2444mmol)和2-(5-氯-3-噻吩基)乙醇s2(39.75mg,30.23μl,0.2444mmol)于dcm(1ml)中的溶液中添加三氟甲磺酸(91.70mg,54.07μl,0.6110mmol)。将反应物在室温下搅拌过夜。将反应物用naoh(2m)淬灭,并且然后用dcm萃取。通过反相hplc纯化(方法:c18 waters sunfire柱(30x 150mm,5微米).梯度:于具有5mm hcl的h2o中的mecn),得到2-氯-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](三氟乙酸盐)194。1h nmr(300mhz,氯仿-d)δ9.87(s,1h),8.98-8.36(m,1h),6.62(s,1h),3.90(t,j=5.4hz,2h),3.61(s,2h),2.64(q,j=5.4hz,2h),2.35-1.79(m,4h),1.35(d,j=6.6hz,6h)。lcms m/z 272.03[m+h]
+
。
[1388]
化合物195和196
[1389]
化合物195和196(参见表7)在单个oxa-pictet spengler步骤中使用如对于化合物194所述的相关哌啶酮和噻吩乙醇來製備。噻吩乙醇通过上文所述的方法制备或从商业来源获得。哌啶酮从商业来源获得。对方法的任何修改在表7和所附脚注中标识。
[1390]
表7.化合物195-196的制备方法、结构和物理化学数据
[1391][1392]
s45的制备
[1393]
1-[(2r,6s)-2',6'-二甲基螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(s45)
[1394][1395]
步骤1.(2r,6s)-2',6'-二甲基螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](c83)的合成
[1396]
在0℃下向(2s,6r)-2,6-二甲基哌啶-4-酮c82(900mg,7.076mmol)于二噁烷(21ml)中的溶液中添加2-(3-噻吩基)乙醇s1(850mg,6.631mmol),随后添加三氟甲磺酸(2g,13.33mmol)。将反应物升温至室温。在搅拌2小时后,反应进行完成。将反应物用饱和碳酸氢钠溶液小心淬灭。将反应混合物分配于dcm与饱和碳酸氢钠水溶液之间。将有机相分离,使其穿过相分离器,并经由旋转蒸发来浓缩,得到粗制(2r,6s)-2',6'-二甲基螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]c83(1.059g,定量)。lcms m/z 238.11[m+h]
+
。
[1397]
步骤2.1-[(2r,6s)-2',6'-二甲基螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(s45)的合成
[1398]
将(2r,6s)-2',6'-二甲基螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]c83溶解于dcm(12.7ml)和boc2o(1.4g,6.415mmol)中并添加dipea(1.7g,13.15mmol)。将反应物搅拌五小时,但仅观察到少量的转化。将反应混合物浓缩至干燥并再溶解于dcm(12.7ml)。在0
℃下向该溶液中添加tea(1.34g,13.24mmol)和tfaa(1.8g,8.570mmol)。将反应物升温至室温并搅拌两小时,直到反应进行完成。将反应物用饱和碳酸氢钠淬灭并用etoac(2x)萃取。将有机物用盐水洗涤,经na2so4干燥,并在真空中浓缩。通过硅胶色谱纯化(柱:24g硅胶,梯度:0-50%于庚烷中的etoac),得到1-[(2r,6s)-2',6'-二甲基螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮s45(1g,67%)。1h nmr(300mhz,氯仿-d)δ7.19(d,j=5.0hz,1h),6.78(d,j=5.0hz,1h),4.47(d,j=7.8hz,2h),3.85(t,j=5.5hz,2h),2.68(t,j=5.5hz,2h),2.50(dd,j=14.4,8.2hz,2h),1.97(dd,j=14.7,6.6hz,2h),1.49(dd,j=20.4,6.6hz,6h)。lcms m/z 334.05[m+h]
+
。
[1399]
在纯化期间也分离了400mg的boc保护的化合物。1h nmr(300mhz,氯仿-d)δ7.18(dd,j=7.6,5.0hz,1h),6.76(d,j=5.0hz,1h),4.36(h,j=7.1hz,2h),3.88(t,j=5.5hz,2h),2.68(t,j=5.5hz,2h),2.49-2.31(m,2h),1.83(dd,j=14.5,7.0hz,2h),1.51(d,j=25.0hz,9h),1.33(d,j=6.8hz,6h)。
[1400]
化合物197
[1401]
(2r,6s)-2-溴-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](197)
[1402][1403]
步骤1.1-[(2r,6s)-2-溴-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(s46)的合成
[1404]
向溶解于mecn(10.7ml)中的1-[(2r,6s)-2',6'-二甲基螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮s45(1g,3.000mmol)中的溶液中添加nbs(610mg,3.427mmol)并将反应物在65℃下搅拌一小时。浓缩溶液并将其溶解于dcm中。将反应物用硫代硫酸钠水溶液和dcm淬灭。将有机相分离,使其穿过相分离器,并在真空中浓缩并过滤出固体nbs。通过硅胶色谱纯化(梯度:0-60%于庚烷中的etoac),得到产物1-[(2r,6s)-2-溴-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮s46(1.12g,76%)。1h nmr(300mhz,氯仿-d)δ6.74(s,1h),4.61-4.32(m,2h),3.83(t,j=5.5hz,2h),2.65-2.40(m,4h),1.88(dd,j=14.9,6.5hz,2h),1.43(d,j=6.9hz,6h)。lcms m/z 412.05[m+h]
+
。
[1405]
步骤2.(2r,6s)-2-溴-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](197)的合成
[1406]
向1-[(2r,6s)-2-溴-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮s46(1.12g,2.716mmol)于dcm(10ml)中的溶液中添加naoh水溶液(1.5ml 2m,3.000mmol)。将反应物加热至40℃。将反应物搅拌30分钟。将有机相分离,使其穿过相分离器,并在真空中浓缩。通过反相hplc纯化(方法:c18 waters sunfire柱
(30x 150mm,5微米).梯度:于具有0.1%三氟乙酸的h2o中的mecn),得到(2r,6s)-2-溴-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](三氟乙酸盐)197(11.9mg,1%)。1h nmr(300mhz,氯仿-d)δ9.43(s,1h),8.79-8.07(m,1h),6.76(s,1h),3.90(t,j=5.4hz,2h),3.64(d,j=8.0hz,2h),2.66(t,j=5.4hz,2h),2.17(d,j=14.4hz,2h),1.93(dd,j=14.5,12.2hz,2h),1.35(d,j=6.6hz,6h)。lcms m/z 316.18[m+h]
+
。
[1407]
化合物198
[1408]
(2r,6s)-2-(3,3-二氟环丁基)-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](198)
[1409][1410]
步骤1.1-[(2r,6s)-2-(3,3-二氟环丁基)-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(c84)的合成
[1411]
在惰性气氛下向ir[df(cf3)ppy]2(dtbbpy)pf6(六氟化磷离子)(3mg,0.002971mmol)、二氯(二甲氧基乙烷)镍(4mg,0.01820mmol)和4-叔丁基-2-(4-叔丁基-2-吡啶基)吡啶(5mg,0.01863mmol)中添加溶解于dme(2ml)中的1-[(2r,6s)-2-溴-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮s46(65mg,0.1577mmol)、双(三甲基甲硅烷基)甲硅烷基-三甲基-硅烷(61μl,0.1963mmol)和2,6-二甲基吡啶(41.64mg,45.01μl,0.3886mmol)的溶液。添加3-溴-1,1-二氟-环丁烷(158μl,1.571mmol)。将所得混合物密封并在sigma synled光反应器中辐照过夜,同时翻滚搅拌。将反应小瓶打开并在氮气流下蒸发。将所得残余物用2ml水和2ml dcm稀释并搅拌几分钟。使双相混合物穿过平行疏水过滤板。将dcm层在真空中浓缩。通过反相hplc纯化(方法:c18 waters sunfire柱(30x 150mm,5微米).梯度:于具有0.1%三氟乙酸的h2o中的mecn),得到tfa保护的中间体c84。lcms m/z 424.18[m+h]
+
。
[1412]
步骤2.(2r,6s)-2-(3,3-二氟环丁基)-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](198)的合成
[1413]
向dcm(2ml)中的1-[(2r,6s)-2-(3,3-二氟环丁基)-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(15.5mg,0.037mmol)c84中添加naoh 6m(10当量)。将反应物加热至40℃并搅拌5小时。将有机相分离,使其穿过相分离器,并在真空中浓缩。通过反相hplc纯化(方法:c18 waters sunfire柱(30x 150mm,5微米).梯度:于具有0.1%三氟乙酸的h2o中的mecn),得到脱保护产物(2r,6s)-2-(3,3-二氟环丁基)-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](三氟乙酸盐)198(5.6mg,8%)。1hnmr(300mhz,氯仿-d)δ6.50(d,j=1.0hz,1h),3.84(t,j=5.4hz,2h),3.73-3.28(m,3h),2.97(tdd,j=14.3,7.3,4.1hz,2h),2.79-2.35(m,5h),2.10(d,j=13.8hz,2h),2.02-1.74(m,2h),1.28(d,j=6.6hz,7h)。
[1414]
化合物199-202
[1415]
化合物199-202(参见表8)由中间体s47使用适当的试剂并使用对于化合物198所述的光氧化还原方法和脱保护方法来制备。烷基溴化物从商业来源获得。对方法的任何修改在表8中标识。
[1416]
表8.化合物199-202的制备方法、结构和物理化学数据
[1417][1418]
化合物203
[1419]
(2r,6s)-2-(2,2-二氟乙基)-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](203)
[1420][1421]
步骤1.(2r,6s)-2',6'-二甲基螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-羧酸叔丁酯(c85)的合成
[1422]
向(2r,6s)-2',6'-二甲基螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]c83于dcm(4ml)中的溶液中添加boc2o(525μl,2.285mmol)和dipea(597μl,3.427mmol)。将反应物搅拌过夜,直到反应进行完成。将反应物用饱和碳酸氢钠淬灭并用etoac(2x)萃取。将有机物用盐水洗涤,经无水na2so4干燥,并在真空中浓缩。通过硅胶色谱纯化(柱:24g硅胶,梯度:0-30%于庚烷中的etoac),得到(2r,6s)-2',6'-二甲基螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-羧酸叔丁酯c85(210mg,54%)。lcms m/z 338.1[m+h]
+
;
[1423]
步骤2.(2r,6s)-2-溴-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-羧酸叔丁酯(c86)的合成
[1424]
向(2r,6s)-2',6'-二甲基螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-羧酸叔丁酯c85(140mg,0.4148mmol)于mecn(1.5ml)中的溶液中添加nbs(84mg,0.4720mmol)。将反应物在65℃下搅拌一小时。将反应物用硫代硫酸钠溶液和dcm淬灭。将有机相分离,使其穿过相分离器,并在真空中浓缩。通过硅胶色谱纯化(梯度:0-30%于庚烷中的etoac),得到产物(2r,6s)-2-溴-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-羧酸叔丁酯c86(120mg,69%)。lcms m/z 416.14[m+h]
+
。
[1425]
步骤3.(2r,6s)-2-(2,2-二氟乙基)-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-羧酸叔丁酯(c87)的合成
[1426]
向小瓶中添加呈固体形式的ir[df(cf3)ppy]2(dtbbpy)pf6(六氟化磷离子)(3mg,0.002971mmol)、二氯(二甲氧基乙烷)镍(4mg,0.01820mmol)和4-叔丁基-2-(4-叔丁基-2-吡啶基)吡啶(5mg,0.01863mmol)。将小瓶用n2吹扫并再填充(3x)。向小瓶中依次添加(2r,6s)-2-溴-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-羧酸叔丁酯c86(70mg,0.1680mmol)、双(三甲基甲硅烷基)甲硅烷基-三甲基-硅烷(51mg,0.2051mmol)、2,6-二甲基吡啶(45mg,0.4200mmol)和dme(2ml)。用n2吹扫(3x)并添加2-溴-1,1-二氟-乙烷(244mg,1.683mmol)。将反应物用merck integrated光反应器辐照7h,royal blue
(450nm)led灯。施用100% led光照功率。搅拌速率为1000rpm。将反应物用饱和碳酸氢钠水溶液和dcm淬灭并通过相分离器收集有机层。将溶剂在真空中浓缩,得到粗制(2r,6s)-2-(2,2-二氟乙基)-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-羧酸叔丁酯c87(8mg,12%)。lcms m/z 402.23[m+h]
+
。
[1427]
步骤4.(2r,6s)-2-(2,2-二氟乙基)-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](203)的合成
[1428]
向(2r,6s)-2-(2,2-二氟乙基)-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-羧酸叔丁酯c87(8mg,0.02mmol)于二噁烷(350μl)中的溶液中添加二噁烷中的hcl(630μl 4m,2.520mmol)。将反应物搅拌3小时。在真空中去除溶剂。通过反相hplc纯化(方法:c18 waters sunfire柱(30x 150mm,5微米).梯度:于具有0.1%三氟乙酸的h2o中的mecn),得到(2r,6s)-2-(2,2-二氟乙基)-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](三氟乙酸盐)203(6.1mg,78%)。1h nmr(300mhz,氯仿-d)δ9.81(s,1h),8.80(s,1h),6.65(s,1h),5.93(tt,j=56.4,4.4hz,1h),3.90(t,j=5.4hz,2h),3.30(td,j=16.7,4.5hz,3h),2.67(t,j=5.5hz,2h),2.30-1.75(m,4h),1.36(d,j=6.5hz,6h)。lcms m/z 302.22[m+h]
+
。
[1429]
化合物204和205
[1430]
(2r,6s)-2-氯-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](204)和(2r,6s)-2-氯-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(205)
[1431][1432]
步骤1.(2r,6s)-2-氯-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](204)的合成
[1433]
向(2s,6r)-2,6-二甲基哌啶-4-酮(盐酸盐)c82(98mg,0.5989mmol)和2-(5-氯-3-噻吩基)乙醇s2(100μl,0.8184mmol)于dcm(2ml)中的溶液中添加msoh(200μl,3.082mmol)并将混合物在40℃下搅拌1小时。将混合物冷却至室温,并将ph用naoh水溶液调节至约ph 14。将水层用另外的dcm(2ml)萃取。使合并的有机层穿过相分离器并在真空中浓缩。通过硅胶色谱纯化(梯度:0-10%于dcm中的meoh),得到产物,将其稀释于乙醚(4ml)中。将hcl(200μl 4m,0.8000mmol)添加到产物的盐中,并过滤固体,用另外的乙醚冲洗并干燥,产生(2r,
6s)-2-氯-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](盐酸盐)204(102mg,55%)。1h nmr(400mhz,dmso-d6)δ9.21(d,j=10.4hz,1h),8.82(d,j=10.2hz,1h),6.93(s,1h),3.89(t,j=5.4hz,2h),3.36(d,j=8.5hz,2h),2.58(t,j=5.4hz,2h),2.17(d,j=14.1hz,2h),1.82(dd,j=14.3,12.2hz,2h),1.27(d,j=6.5hz,6h)。lcms m/z 272.09[m+h]
+
。
[1434]
步骤2.1-[(2r,6s)-2-氯-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(c88)的合成
[1435]
向(2r,6s)-2-氯-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](盐酸盐)204(319mg,1.173mmol)于dcm(6ml)中的溶液中添加tfaa(750mg,3.571mmol)和tea(600mg,5.929mmol)。将反应物在室温下搅拌30分钟。将反应物用碳酸氢钠和dcm淬灭。将有机相分离,使其穿过相分离器,并在真空中浓缩。通过硅胶色谱纯化(梯度:0-50%于庚烷中的etoac),得到产物1-[(2r,6s)-2-氯-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c88(388mg,85%)。lcms m/z 368.08[m+h]
+
。
[1436]
步骤3.(2r,6s)-2'-氯-2,6-二甲基-1-(2,2,2-三氟乙酰基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮(c89)的合成
[1437]
向1-[(2r,6s)-2-氯-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c88于mecn(10ml)中的溶液中添加乙酸钴四水合物(128mg,0.5139mmol)和n-羟基苯邻二甲酰亚胺(168mg,1.030mmol)。将反应物用氧气吹扫并抽真空(3x)并且然后在氧气气球下加热至45℃。将反应物搅拌7小时。将反应物用水和dcm稀释。将有机相分离,使其穿过相分离器,并在真空中浓缩。将材料溶于少量dcm中并固体开始从溶液中析出。倾析出液体。通过硅胶色谱纯化(梯度:0-60%于庚烷中的etoac),得到(2r,6s)-2'-氯-2,6-二甲基-1-(2,2,2-三氟乙酰基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮c89(122mg,31%)。1h nmr(300mhz,氯仿-d)δ7.21(s,1h),4.63-4.39(m,2h),4.29(s,2h),2.69(dd,j=14.4,8.1hz,2h),2.07-1.80(m,2h),1.48(d,j=6.9hz,6h)。lcms m/z 382.03[m+h]
+
。
[1438]
步骤4.1-[(2r,6s)-2-氯-4-羟基-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(c90)的合成
[1439]
向(2r,6s)-2'-氯-2,6-二甲基-1-(2,2,2-三氟乙酰基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮c89(122mg,0.3195mmol)于dcm(2.44ml)和meoh(620μl)中的溶液中添加nabh4(73mg,1.930mmol)。将反应物搅拌45分钟。将反应物用hcl水溶液和dcm淬灭。将有机相分离,使其穿过相分离器,并在真空中浓缩,得到1-[(2r,6s)-2-氯-4-羟基-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c90(122mg,98%)。lcms m/z 384.28[m+h]
+
。
[1440]
步骤5.(2r,6s)-2-氯-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(205)的合成
[1441]
向1-[(2r,6s)-2-氯-4-羟基-2',6'-二甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c90(122mg,0.3195mmol)于meoh(620μl)中的溶液中添加naoh 6m(10当量)。将反应物加热至40℃并搅拌5小时。将反应物用水和dcm淬灭。将有机层通过相分离器分离并在真空中浓缩,得到纯的(2r,6s)-2-氯-2',6'-二甲基-螺[4,5-二
氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇205(55mg,57%)。1h nmr(300mhz,甲醇-d4)。δ6.85(s,1h),4.43(t,j=3.9hz,1h),3.96(dd,j=12.2,3.7hz,1h),3.73(dd,j=12.1,4.2hz,1h),3.22-2.92(m,2h),2.14-1.96(m,2h),1.38-1.16(m,3h),1.08(dd,j=6.5,4.1hz,6h)。lcms m/z 288.06[m+h]
+
。
[1442]
化合物206
[1443]
(2r,6s)-2-氯-2',6'-二环丙基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](206)
[1444][1445]
步骤1.(2r,6s)-2-氯-2',6'-二环丙基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](206)的合成
[1446]
向2,6-二环丙基哌啶-4-酮c91(30mg,0.1674mmol)于dcm(1000μl)中的溶液中添加2-(5-氯-3-噻吩基)乙醇s2(25μl),随后添加msoh(44μl,0.6780mmol)并将混合物在回流下搅拌。5分钟后,将混合物冷却,将ph用naoh水溶液(150μl 6m,0.9000mmol)调节至ph 14并将有机层分离并浓缩。硅胶纯化(梯度:0-20%于dcm中的meoh)得到顺式和反式异构体的混合物。sfc纯化得到(2r,6s)-2-氯-2',6'-二环丙基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]206(5.6mg,10%)。1h nmr(300mhz,甲醇-d4)δ6.66(s,1h),3.84(t,j=5.5hz,2h),2.57(t,j=5.5hz,2h),2.24-2.11(m,2h),2.06(ddd,j=11.5,9.0,2.4hz,2h),1.54(dd,j=13.3,11.5hz,2h),0.87-0.69(m,2h),0.62-0.39(m,4h),0.37-0.21(m,2h),0.21-0.04(m,2h)。lcms m/z 324.02[m+h]+。
[1447]
化合物207
[1448]
(2r,6s)-2',6'-二甲基-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](207)
[1449][1450]
步骤1.(2r,6s)-2',6'-二甲基-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](207)的合成
[1451]
向冷却至0℃的(2s,6r)-2,6-二甲基-4-氧代-哌啶-1-羧酸叔丁酯c92(300mg,1.320mmol)和2-[5-(三氟甲基)-3-噻吩基]乙醇s3(300mg,1.483mmol)于二噁烷(4.5ml)中的溶液中添加三氟甲磺酸(345μl,3.899mmol)。将反应物升温至室温。10分钟后,观察到缩酮形成。将溶液搅拌6小时。将溶液用dcm稀释并用2m na2co3洗涤。将反应物用dcm(3x)萃取,用na2so4干燥,过滤,并在真空中去除dcm,得到粗制(2r,6s)-2',6'-二甲基-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]207(400mg,71%)。lcms m/z 306.06[m+h]
+
。
[1452]
步骤2.(2r,6s)-2',6'-二甲基-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-羧酸叔丁酯(c93)的合成
[1453]
向(2r,6s)-2',6'-二甲基-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]207(400mg,71%)于dcm(8ml)中的溶液中添加boc酸酐(1.5ml,6.529mmol)和dipea(670μl,3.847mmol)。将反应物搅拌四天,直到观察到完全转化。将溶剂在真空中浓缩。通过硅胶色谱纯化(梯度:0-30%于庚烷中的etoac),得到产物(2r,6s)-2',6'-二甲基-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-羧酸叔丁酯c93(440mg,82%)。1h nmr(300mhz,氯仿-d)δ7.11(d,j=1.3hz,1h),4.47-4.25(m,2h),3.89(t,j=5.5hz,2h),2.67(t,j=5.5hz,2h),2.43(ddd,j=15.5,8.5,2.3hz,2h),1.79(dd,j=14.5,6.9hz,2h),1.50(s,9h),1.33(d,j=6.8hz,6h)。
[1454]
步骤3.(2r,6s)-2',6'-二甲基-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](207)的合成
[1455]
向溶解于二噁烷(100μl)的(2r,6s)-2',6'-二甲基-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-羧酸叔丁酯c93(20mg,0.049mmol)的溶液中添加二噁烷中的hcl(100μl 4m,0.4000mmol)。将反应在室温下搅拌1小时。将溶剂在真空中浓缩。通过反相hplc纯化(方法:c18 waters sunfire柱(30x 150mm,5微米).梯度:于具有0.1%三氟乙酸的h2o中的mecn),得到(2r,6s)-2',6'-二甲基-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](三氟乙酸盐)207(6.6mg,32%)。1h nmr(300mhz,氯仿-d)δ9.55(s,
1h),8.67(s,1h),7.16(d,j=1.3hz,1h),3.94(t,j=5.4hz,2h),3.67(s,2h),2.74(t,j=5.4hz,2h),2.38-1.84(m,4h),1.37(d,j=6.6hz,6h)。lcms m/z 306.24[m+h]
+
。
[1456]
化合物208
[1457]
[(2r,6s)-2',6'-二甲基-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-3-基]甲醇(208)
[1458][1459]
步骤1.(2r,6s)-3-甲酰基-2',6'-二甲基-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-羧酸叔丁酯(c94)的合成
[1460]
在-78℃下在n2下向(2r,6s)-2',6'-二甲基-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-羧酸叔丁酯c93(420mg,1.036mmol)于thf(6ml)中的溶液中添加s-buli(1ml 1.4m,1.400mmol)。将反应物搅拌30分钟,随后添加dmf(160μl,2.066mmol)。将反应物搅拌30分钟并升温至室温。将反应物用nh4cl和dcm淬灭。将有机相分离,使其穿过相分离器,并浓缩,得到粗制(2r,6s)-3-甲酰基-2',6'-二甲基-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-羧酸叔丁酯c94。lcms m/z 434.32[m+h]
+
。
[1461]
步骤2.(2r,6s)-3-(羟基甲基)-2',6'-二甲基-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-羧酸叔丁酯(c95)的合成
[1462]
将反应物搅拌30分钟。将反应物用碳酸氢钠水溶液和dcm淬灭。将有机相分离,使其穿过相分离器,并在真空中浓缩,得到粗制(2r,6s)-3-(羟基甲基)-2',6'-二甲基-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-羧酸叔丁酯c95 lcms m/z 436.35[m+h]
+
。
[1463]
步骤3.(2r,6s)-2',6'-二甲基-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-3-基]甲醇(208)的合成
[1464]
向粗制c95于二噁烷(2ml)中的溶液中添加二噁烷中的hcl(2.5ml 4m,10.00mmol)。将反应物搅拌1小时。在真空中去除溶剂。通过反相hplc纯化(方法:c18 waters sunfire柱(30x 150mm,5微米)),得到(2's,6'r)-2',6'-二甲基-2-(三氟甲基)螺
[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-3-基]甲醇208(192mg,55%)。1h nmr(300mhz,氯仿-d)δ4.66(d,j=1.4hz,2h),3.98(t,j=5.5hz,2h),3.17(dtt,j=12.6,6.3,3.1hz,2h),2.75(t,j=5.5hz,2h),2.19-1.97(m,2h),1.35(dd,j=13.5,11.4hz,2h),1.10(d,j=6.4hz,6h)。lcms m/z336.08[m+h]+。
[1465]
化合物209
[1466]
(2's,6'r,7s)-2-氯-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-2'-羧酸甲酯(209)
[1467][1468]
步骤1.(2s)-2-(三苯甲基氨基)丁二酸二甲酯(c97)的合成
[1469]
向(2s)-2-氨基丁二酸二甲酯c96(盐酸盐)(2000mg,10.12mmol)和dipea(3.7ml,21.24mmol)于dcm(40ml)中的混合物中添加[氯(二苯基)甲基]苯(3g,10.76mmol)。混合物于室温搅拌过夜。将混合物用水淬灭,分离各相,并将有机层用mgso4干燥,过滤,并浓缩。通过硅胶色谱纯化(梯度:0-30%于庚烷中的etoac),得到(2s)-2-(三苯甲基氨基)丁二酸二甲酯(3.11g,75%)c97.1h nmr(300mhz,氯仿-d)δ7.59-7.43(m,6h),7.35-7.24(m,6h),7.24-7.13(m,3h),3.70(s,4h),3.28(s,3h),2.95(d,j=10.1hz,1h),2.67(dd,j=14.7,5.4hz,1h),2.53(dd,j=14.7,7.0hz,1h)。lcms m/z 402.23[m+h]
+
。
[1470]
步骤2.(2s)-5-二甲氧基磷酰基-4-氧代-2-(三苯甲基氨基)戊酸甲酯(c98)的合成
[1471]
向冷却至-78℃的[甲氧基(甲基)磷酰基]氧基m乙烷(2ml,18.46mmol)于thf(90ml)中的混合物中逐滴添加仲-丁基锂(13.5ml 1.4m,18.90mmol)的溶液。在此温度下5分钟后,逐滴添加(2s)-2-(三苯甲基氨基)丁二酸二甲酯c97(3100mg,7.683mmol)于thf
(10ml)中的溶液,并将混合物再搅拌10分钟并且然后用acoh(1.3ml,22.86mmol)淬灭。将混合物用水(100ml)和乙醚(100ml)稀释。将有机层去除,并随后用水(100ml)、盐水(100ml)洗涤,用mgso4干燥,过滤,并浓缩。通过硅胶色谱纯化(梯度:0-20%于dcm中的meoh),得到(2s)-5-二甲氧基磷酰基-4-氧代-2-(三苯甲基氨基)戊酸甲酯c98(1580mg,41%)。1h nmr(300mhz,氯仿-d)δ7.56-7.42(m,6h),7.30(t,j=1.6hz,3h),7.28-7.16(m,6h),3.81(s,3h),3.78(s,3h),3.72(s,1h),3.31(s,3h),3.07(d,j=22.6hz,2h),2.91(dd,j=16.7,4.6hz,2h),2.79(dd,j=16.7,6.9hz,1h)。lcms m/z 494.31[m+h]
+
。
[1472]
步骤3.(e,2s)-4-氧代-2-(三苯甲基氨基)庚-5-烯酸甲酯(c99)的合成
[1473]
向(2s)-5-二甲氧基磷酰基-4-氧代-2-(三苯甲基氨基)戊酸甲酯c98(553mg,1.108mmol)于mecn(30ml)中的混合物中添加碳酸钾(170mg,1.230mmol),随后添加乙醛(200μl,3.564mmol)。将混合物升温至50℃并搅拌过夜。将混合物冷却至室温,浓缩,并再稀释于乙酸乙酯(50ml)和水(30ml)和盐水(10ml)中。将有机层分离,用盐水(30ml)洗涤,用mgso4干燥,过滤,并浓缩。通过硅胶色谱纯化(梯度:0-40%于庚烷中的etoac),得到(e,2s)-4-氧代-2-(三苯甲基氨基)庚-5-烯酸甲酯c99(282mg,61%)。lcms m/z 412.27[m+h]
+
。
[1474]
步骤4.(2s,6s)-6-甲基-4-氧代-哌啶-2-羧酸甲酯(c100)和(2s,6r)-6-甲基-4-氧代-哌啶-2-羧酸甲酯(c101)的合成
[1475]
在室温下向(e,2s)-4-氧代-2-(三苯甲基氨基)庚-5-烯酸甲酯c99(187mg,0.4203mmol)于甲醇(30ml)中的混合物中添加盐酸(7500μl 2m,15.00mmol)。将混合物搅拌10分钟。形成中间体(e,2s)-2-氨基-4-氧代-庚-5-烯酸甲酯。向混合物中添加水(15ml)和dipea(4500μl,25.84mmol)并将反应物搅拌15分钟。将混合物用etoac(60ml)、水(60ml)稀释,并分离各层。将水层用etoac(2x 50ml)萃取,并将合并的有机层用mgso4干燥,过滤,并浓缩。通过硅胶色谱纯化(梯度:0-10%于dcm中的meoh),得到作为顺式:反式异构体的2:1混合物的c100和c101(28mg,39%)。
[1476]
c100[diast-1]1h nmr(300mhz,氯仿-d)δ3.75(s,3h),3.64(dd,j=12.2,3.5hz,1h),2.97(dqd,j=12.3,6.2,2.9hz,1h),2.72-2.53(m,2h),2.43-2.34(m,1h),2.08(ddd,j=14.2,11.7,1.0hz,1h),1.24(d,j=6.2hz,3h)。lcms m/z 172.0[m+h]
+
。
[1477]
c101[diast-2]1h nmr(300mhz,氯仿-d)δ4.06(dd,j=6.6,3.7hz,1h),3.73(s,3h)),3.32
–
3.15(m,1h),2.72-2.53(m,2h),2.43-2.34(m,1h),2.08(ddd,j=14.2,11.7,1.0hz,1h),1.17(d,j=6.2hz,3h)。lcms m/z 172.0[m+h]
+
。
[1478]
步骤5.(2's,6's,7s)-2-氯-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-2'-羧酸甲酯(209)的合成
[1479]
向c100和c101(28mg,0.1636mmol)(2:1顺式与反式比率)于dcm(1000μl)中的混合物中添加2-(5-氯-3-噻吩基)乙醇s2(25μl,0.2021mmol),随后添加msoh(50μl,0.7705mmol)并将混合物回流5分钟。将混合物冷却,并将ph用naoh水溶液(150μl 6m,0.90mmol)调节至ph 14并将有机层分离并浓缩。通过硅胶色谱纯化(梯度:0-20%于dcm中的meoh),得到(2's,6's,7s)-2-氯-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-2'-羧酸甲酯209(24mg,68%)。1h nmr(300mhz,氯仿-d)δ6.57(s,1h),3.92(t,j=5.5hz,2h),3.84(dd,j=11.9,2.8hz,1h),3.71(s,3h),3.12(dtd,j=12.7,6.4,2.6hz,1h),2.69-2.51(m,2h),2.40(dt,j=13.7,2.7hz,1h),1.98(dt,j=13.7,2.6hz,1h),1.62
sunfire柱(30x 150mm,5微米).梯度:于具有5mm hcl的h2o中的mecn),得到(2-氯-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-2'-基)甲醇(盐酸盐)210(5.5mg,66%)。1h nmr(300mhz,dmso-d6)δ8.83(s,1h),8.33(s,1h),6.93(s,1h),3.90(d,j=5.5hz,2h),3.66(dd,j=11.8,3.9hz,1h),3.53(dd,j=11.6,5.3hz,1h),3.34(s,2h),2.58(t,j=5.4hz,2h),2.15(t,j=14.4hz,3h),1.78(q,j=12.8hz,2h),1.27(d,j=6.5hz,3h)。lcms m/z 288.14[m+h]
+
。
[1488]
s47的制备
[1489]
(2s,6s)-2-环丙基-6-甲基-哌啶-4-酮(s47)
[1490][1491]
向(4s)-4-氨基戊-2-酮(盐酸盐)s25(150mg,1.090mmol)于et3n(153μl,1.098mmol)和etoh(9ml)中的混合物中添加环丙烷甲醛(90μl,1.204mmol)、l-脯氨酸(25mg,0.2171mmol)和mgso4(130mg,1.080mmol)。将混合物在室温下搅拌2小时。将混合物过滤,浓缩,并稀释于饱和碳酸氢盐水溶液(3ml),用水(7ml)稀释并用dcm(2x 10ml)萃取。将合并的有机层用盐水(5ml)洗涤,用mgso4干燥,过滤,并浓缩。通过硅胶色谱纯化(梯度:0-20%于dcm中的meoh),得到(2s,6s)-2-环丙基-6-甲基-哌啶-4-酮s47(65mg,39%)。1h nmr(300mhz,氯仿-d)δ2.79(dqd,j=12.2,6.1,3.0hz,1h),2.35(ddd,j=14.0,3.1,2.1hz,1h),2.27-2.13(m,2h),2.02(ddd,j=14.0,11.6,1.1hz,1h),1.96-1.85(m,1h),1.80(s,1h),1.12(d,j=6.2hz,3h),0.78(qt,j=8.2,4.9hz,1h),0.59-0.27(m,2h),0.23
‑‑
0.03(m,2h)。lcms m/z 154.05[m+1]
+
.
[1492]
中间体s48-s53的制备
[1493]
中间体s48-s53在单个步骤中使用适当的醛和对于中间体s47所述的方法由中间体s25或s24来制备。醛通过上文所述的方法制备或从商业来源获得。在该方法中,观察到对映异构体纯的起始材料(4s)-4-氨基戊-2-酮(盐酸盐)s25的部分立体化学消除,导致未分离的立体异构体混合物,其中顺式产物是主要异构体。对方法的任何修改在表9和所附脚注中标识。
[1494]
表9.中间体s48-s53的制备方法、结构和物理化学数据
[1495][1496][1497]
1.将反应物搅拌过夜。
[1498]
2.通过硅胶色谱纯化(梯度:0-10%于dcm中的meoh),得到产物。
[1499]
3.在完成后,将混合物浓缩,再稀释于dcm中并过滤。
[1500]
4.在完成后,将混合物倒入到饱和碳酸氢钠水溶液(10ml)中并用水(3ml)和乙酸乙酯(30ml)稀释。
[1501]
5.反应物未纯化,错开到下一个步骤中。
[1502]
化合物211
[1503]
(2s,6s)-2-氯-2'-环丙基-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](211)
[1504][1505]
向(2s,6s)-2-环丙基-6-甲基-哌啶-4-酮s47(65mg,0.4242mmol)于dcm(2ml)中的混合物中添加2-(5-氯-3-噻吩基)乙醇(75μl),随后添加msoh(130μl,2.003mmol)并将混合物回流40分钟。将混合物冷却,将ph用naoh(500μl 6m,3.000mmol)调节至ph 14并将有机层分离并浓缩。通过硅胶色谱纯化(梯度:0-20%于dcm中的meoh),得到游离碱。将游离碱再稀释于乙醚和hcl(100μl 4m于二噁烷中,0.4000mmol)中,在此时沉淀出白色固体。将混合物浓缩,产生(2s,6s)-2-氯-2'-环丙基-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]211(盐酸盐)(15.2mg,11%)。1h nmr(300mhz,甲醇-d4)δ6.74(s,1h),3.92(t,j=5.5hz,2h),3.65-3.51(m,1h),2.75(ddd,j=12.6,9.8,3.0hz,1h),2.63(dd,j=6.1,4.9hz,2h),2.35(ddt,j=23.2,14.7,2.8hz,2h),1.85(dd,j=14.6,12.3hz,1h),1.70(dd,j=14.6,12.2hz,1h),1.36(d,j=6.6hz,3h),0.93(ddt,j=13.1,9.5,4.2hz,1h),0.81-0.64(m,2h),0.64-0.53(m,1h),0.44-0.26(m,1h)。lcms m/z 298.05[m+h]
+
。
[1506]
化合物212-218
[1507]
化合物212-218(参见表10)由单个oxa-pictet spengler步骤使用分离的哌啶酮(参见表9)和如对于化合物211所述的s2来制备。噻吩乙醇和哌啶酮通过上文所述的方法制备或从商业来源获得。如先前在哌啶酮中间体制备中所述的,观察到对映异构体纯的起始材料(4s)-4-氨基戊-2-酮(盐酸盐)s25的部分立体化学消除,导致未分离的立体异构体混合物,其中顺式产物是主要异构体。对方法的任何修改在表10和所附脚注中标识。
[1508]
表10.化合物212-218的制备方法、结构和物理化学数据。
[1509][1510][1511]
1.将反应物ph用饱和水溶液碳酸氢钠调节,将有机层分离,并在真空中去除溶剂。
7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(c106)的合成
[1524]
向(2's,6's,7s)-2-氯-2'-环丙基-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]211(盐酸盐)(30mg,0.08974mmol)于dcm(1ml)中的混合物中添加dipea(50μl,0.2871mmol),随后添加三氟乙酸酸酐(15μl,0.1079mmol)。将反应物搅拌2小时。将混合物用1n hcl淬灭,分离,并浓缩。通过硅胶色谱纯化(梯度:0-30%于庚烷中的etoac),得到1-[(2s,6s)-2-氯-2'-环丙基-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c106(31mg,87%)。1h nmr(300mhz,氯仿-d)δ6.59(s,1h),4.60-3.95(m,1h),3.82(t,j=5.5hz,2h),3.25(s,1h),2.57(t,j=5.5hz,2h),2.50(dd,j=16.1,8.1hz,2h),2.07(d,j=15.1hz,2h),1.62-1.19(m,3h),1.18-0.85(m,1h),0.86-0.38(m,3h),0.32(dq,j=9.2,4.7hz,1h)。lcms m/z 394.04[m+h]
+
。基于nmr,产物是两种旋转异构体的混合物。
[1525]
步骤2.(2s,6s)-2'-氯-2-环丙基-6-甲基-1-(2,2,2-三氟乙酰基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮(s54)的合成
[1526]
向1-[(2s,6s)-2-氯-2'-环丙基-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c106(30mg,0.07617mmol)于乙腈(500μl)中的混合物中添加n-羟基苯邻二甲酰亚胺(8mg,0.04904mmol)和二乙酸钴四水合物(2mg,0.008029mmol)。将混合物用氧气气球真空吹扫三次。将混合物加热至45℃并过滤。6小时后,将反应物冷却至室温。将混合物用氮气真空吹扫三次并且然后用mtbe(3ml)和饱和碳酸氢钠(3ml)稀释。分离各层,并将有机层用水(2x 2ml)和盐水(20ml)洗涤。将有机层用na2so4干燥,过滤,并浓缩。通过硅胶色谱纯化(梯度:0-30%于庚烷中的etoac),得到(2s,6s)-2'-氯-2-环丙基-6-甲基-1-(2,2,2-三氟乙酰基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮(13mg,41%)s54。lcms m/z 408.0[m+h]
+
。
[1527]
化合物220
[1528]
(2's,6's,7s)-2-氯-2'-环丙基-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇)(226)
[1529][1530]
步骤1.1-[(2s,6s)-2-氯-2'-环丙基-4-羟基-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(c107)的合成
[1531]
向(2s,6s)-2'-氯-2-环丙基-6-甲基-1-(2,2,2-三氟乙酰基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮s54于meoh(0.5ml)中的溶液中添加nabh4(1mg,0.02643mmol)。1小时后,将混合物用mtbe(5ml)稀释,用饱和盐水洗涤,并浓缩,得到粗制1-[(2's,6's,7s)-2-氯-2'-环丙基-4-羟基-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,
2,2-三氟-乙酮c107。
[1532]
步骤2.(2s,6s)-2-氯-2'-环丙基-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(220)的合成
[1533]
将粗制1-[(2's,6's,7s)-2-氯-2'-环丙基-4-羟基-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c107用naoh(500μl 6m,3.000mmol)稀释,随后用meoh(0.5ml)稀释并将混合物在50℃下搅拌。40分钟后,将混合物冷却至室温,用dcm(5ml)稀释并将ph用饱和氯化铵水溶液调节至约ph 10。将有机相分离,使其穿过相分离器,并浓缩。通过硅胶色谱纯化(梯度:0-20%于dcm中的meoh),得到(2s,6s)-2-氯-2'-环丙基-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇220(6mg,24%)。1h nmr(300mhz,氯仿-d)δ6.60(s,1h),4.20(q,j=3.1hz,1h),3.74(dd,j=12.3,2.9hz,1h),3.63(dd,j=12.3,2.8hz,1h),3.01-2.71(m,1h),2.12-1.70(m,4h),1.46(t,j=12.2hz,1h),1.37-1.19(m,1h),1.19-1.02(m,1h),0.92(dd,j=6.3,3.4hz,3h),0.61(t,j=10.9hz,1h),0.28(dt,j=9.9,6.2hz,2h),-0.08(dd,j=9.0,4.4hz,2h)。lcms m/z 314.07[m+h]
+
。
[1534]
化合物221
[1535]
(2s,4s,6s)-2'-氯-2-乙炔基-6-甲基-4',5'-二氢螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]
[1536][1537]
步骤1.(s)-7-甲基-1,4-二氧杂-8-氮杂螺[4.5]癸烷-8-羧酸叔丁酯的合成向(2s)-2-甲基-4-氧代-哌啶-1-羧酸叔丁酯c113(40g,183.80mmol)于甲苯(1000ml)中的溶液中添加乙二醇(22.916g,21ml,361.82mmol)和ppts(7.5g,29.248mmol)。将混合物在dean-stark条件下回流72小时。然后将混合物冷却至室温,用饱和碳酸氢钠(300ml)淬灭,并用etoac(2x 500ml)萃取。将合并的有机层经na2so4干燥并浓缩。通过硅胶柱色谱纯化(梯度:0-5%etoac:庚烷),得到呈黄色液体的(7s)-7-甲基-1,4-二氧杂-8-氮杂螺[4.5]癸烷-8-羧酸叔丁酯c114(35g,67%)。1h nmr(300mhz,cdcl3)δ4.49-4.44(m,1h),4.03-3.88(m,5h),3.12-3.02(m,1h),1.90-1.83(m,1h),1.68-1.56(m,3h),1.46(s,9h),1.23(d,j=6.9hz,3h),lcms m/z 258.31[m+1]
+
。
[1538]
步骤2.(7s,9s)-7-乙炔基-9-甲基-1,4-二氧杂-8-氮杂螺[4.5]癸烷-8-羧酸叔丁酯(c115)的合成
[1539]
i.在室温下向(s)-7-甲基-1,4-二氧杂-8-氮杂螺[4.5]癸烷-8-羧酸叔丁酯c114(14g,53.862mmol)于乙醚(210ml)中的溶液中添加tmeda(7.5460g,10ml,63.638mmol)。将反应物冷却至-78℃,并且然后缓慢添加仲-丁基锂(60ml1.4m,84.000mmol)。将反应物在-78℃下搅拌2小时。缓慢添加dmf(7.8635g,8.5ml,105.43mmol),同时将温度保持在-78℃下并搅拌1小时。然后将反应物升温至室温并搅拌1小时。将混合物用饱和氯化铵溶液(100ml)淬灭并用乙酸乙酯(2x 200ml)萃取。浓缩合并的有机层。通过硅胶色谱纯化(梯度:0-50% etoac:庚烷),产生呈淡黄色液体的粗制(9s)-7-甲酰基-9-甲基-1,4-二氧杂-8-氮杂螺
[4.5]癸烷-8-羧酸叔丁酯(20g,65%)。中间体并未通过lcms电离,并且材料是非对映异构体的混合物,如通过1h nmr观察的,因此粗产物用于下一步骤中。
[1540]
ii.在室温下向溶解于meoh(300ml)中的来自前一步骤的粗产物中添加1-重氮-1-二甲氧基磷酰基-丙-2-酮(17g,75.217mmol)和k2co3(15g,106.36mmol)。将反应混合物在室温下搅拌16小时。将反应混合物过滤并用etoac(50ml)洗涤。浓缩滤液。通过硅胶柱色谱纯化(0-5% etoac:庚烷),提供呈无色液体的分离的单个立体异构体(7s,9s)-7-乙炔基-9-甲基-1,4-二氧杂-8-氮杂螺[4.5]癸烷-8-羧酸叔丁酯c115(9.4g,59%)。1h nmr(400mhz,dmso-d6)δ:5.032-5.014(m,1h),4.23-4.19(m,1h),3.97-3.93(m,2h),3.84-3.80(m,2h),3.11-3.10(m,1h),1.95-1.78(m,4h),1.39(s,9h),1.35(d,j=6.8hz,3h),lcms m/z 282.35[m+1]
+
[1541]
步骤3.(2's,6's,7s)-2-氯-2'-乙炔基-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](221)的合成
[1542]
向(7s,9s)-7-乙炔基-9-甲基-1,4-二氧杂-8-氮杂螺[4.5]癸烷-8-羧酸叔丁酯c115(25mg,0.08886mmol)和2-(5-氯-3-噻吩基)乙醇(20μl,0.1986mmol)于dcm(0.5ml)中的混合物中添加msoh(40μl,0.6164mmol)。将混合物加热至40℃持续2小时。使混合物冷却至室温并然后ph用饱和碳酸氢盐水溶液和1n naoh水溶液调节至ph 11。添加另外的dcm(2ml),并将有机层分离并浓缩。通过硅胶色谱纯化(梯度:0-10% meoh:dcm),得到呈黄色油状物的(2's,6's,7s)-2-氯-2'-乙炔基-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]221(14mg,50%)。1h nmr(300mhz,甲醇-d4)δ6.67(s,1h),3.90(t,j=5.5hz,2h),3.84(dt,j=11.8,2.6hz,1h),3.05(dqd,j=13.0,6.4,2.6hz,1h),2.68(d,j=2.3hz,1h),2.59(t,j=5.5hz,2h),2.20(dt,j=13.8,2.7hz,1h),1.97(dt,j=13.8,2.6hz,1h),1.71(dd,j=13.8,11.8hz,1h),1.32(dd,j=13.8,11.5hz,1h),1.07(d,j=6.4hz,3h)。lcms m/z 282.09[m+1]
+
通过noe进行nmr分析证实了2,4-反式2,6-顺式立体化学。
[1543]
s55的制备
[1544]
(2s,4s,6s)-2'-氯-2-乙炔基-6-甲基-1-(2,2,2-三氟乙酰基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'(5'h)-酮
[1545][1546]
步骤1.1-[(2's,6's,7s)-2-氯-2'-乙炔基-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(c116)的合成
[1547]
将(2's,6's,7s)-2-氯-2'-乙炔基-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]221(6.91g,24.5mmol)于dcm(100ml)中的溶液冷却至0℃。添加dipea(8.5ml,48.80mmol),随后添加tfaa(4.3ml,30.93mmol)并将混合物搅拌1分钟。将混合物用1n hcl(50ml)淬灭,并将有机层分离。将有机层用1n hcl水溶液(50ml)洗涤,用mgso4干燥,过滤,并浓缩。通过硅胶色谱纯化(梯度:0-5% etoac:庚烷),提供呈结晶状白色-粉色固体的粗产物。将该材料悬浮于tbme(28ml)并在回流下搅拌,这完全溶解了固体。将溶液冷却至0℃
etoac:庚烷),得到呈白色固体的1-[(2's,4s,6's,7s)-2-氯-2'-乙炔基-4-羟基-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c117(2.27g,85%)。1h nmr(300mhz,氯仿-d)δ6.85(s,1h),5.19(s,1h),4.48(t,j=9.2hz,2h),3.86(qd,j=12.5,3.1hz,2h),2.75-2.46(m,3h),2.37(dd,j=14.6,6.8hz,1h),2.20(d,j=14.7hz,1h),2.01(d,j=9.1hz,1h),1.53(d,j=6.6hz,3h)。lcms m/z 394.13[m+1]
+
[1555]
步骤2.(2's,4s,6's,7s)-2-氯-2'-乙炔基-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(222)的合成
[1556]
向1-[(2's,4s,6's,7s)-2-氯-2'-乙炔基-4-羟基-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c117(1.5g,3.809mmol)于meoh(7.5ml)中的溶液中添加naoh(4ml 6m,24.00mmol)。将混合物在室温下搅拌15分钟,然后过滤并用另外的水冲洗并浓缩,产生呈白色固体的(2's,4s,6's,7s)-2-氯-2'-乙炔基-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇222(1.07g,88%)。1h nmr(300mhz,氯仿-d)δ6.84(s,1h),4.44(d,j=7.8hz,1h),4.04-3.80(m,3h),3.51(s,2h),3.21(dd,j=11.4,5.7hz,1h),2.35(d,j=14.1hz,1h),2.05(d,j=9.3hz,1h),2.00-1.85(m,1h),1.74-1.63(m,1h),1.45(dd,j=13.4,11.3hz,1h),1.13(d,j=6.3hz,3h)。lcms m/z298.07[m+1]
+
.
[1557]
化合物223
[1558]
(2's,6's,7s)-2-氯-2'-乙炔基-4,4-二氟-6'-甲基-螺[5h-噻吩并[2,3-c]吡喃-7,4'-哌啶](223)
[1559][1560]
步骤1.1-[(2's,6's,7s)-2-氯-2'-乙炔基-4,4-二氟-6'-甲基-螺[5h-噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(c118)的合成
[1561]
将(2s,4s,6s)-2'-氯-2-乙炔基-6-甲基-1-(2,2,2-三氟乙酰基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮s55(1690mg,4.314mmol)于dast(5ml,37.84mmol)中的混合物加热至40℃并搅拌过夜。添加另一部分dast(5ml,37.84mmol),并将反应物加热至40℃持续36小时。将混合物冷却至室温并逐滴添加到保持在0℃下的饱和碳酸氢钠(25ml)的搅拌溶液中。将混合物用dcm(25ml)稀释并分离各层。将水层用另外的dcm(25ml)萃取,并将合并的有机层用mgso4干燥,过滤,并浓缩。通过硅胶色谱纯化(梯度:0-40% etoac:庚烷),得到呈橙色油状物的1-[(2's,6's,7s)-2-氯-2'-乙炔基-4,4-二氟-6'-甲基-螺[5h-噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c118(1.34g,73%)。1h nmr(300mhz,氯仿-d)δ6.99(s,1h),5.29(d,j=44.5hz,1h),4.41(q,j=7.7hz,1h),3.99(t,j=10.0hz,2h),2.70(dd,j=15.2,8.3hz,1h),2.60(s,1h),2.50(t,j=8.4hz,2h),2.32(d,j=15.3hz,1h),1.53(d,j=6.5hz,3h)。lcms m/z414.07[m+1]
+
[1562]
步骤2.(2's,6's,7s)-2-氯-2'-乙炔基-4,4-二氟-6'-甲基-螺[5h-噻吩并[2,3-c]吡喃-7,4'-哌啶](223)的合成
[1563]
向1-[(2's,6's,7s)-2-氯-2'-乙炔基-4,4-二氟-6'-甲基-螺[5h-噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c118(1.36g,3.287mmol)于meoh(25ml)中的混合物中添加naoh(2.5ml 6m,15.00mmol)。将反应物加热至50℃持续1小时。将混合物冷却至室温并用tbme(30ml)和水(30ml)稀释。将有机层用盐水洗涤(50ml)并用mgso4干燥,过滤,并浓缩。通过硅胶色谱纯化(梯度:0-10% meoh:dcm),得到呈棕色油状物的(2's,6's,7s)-2-氯-2'-乙炔基-4,4-二氟-6'-甲基-螺[5h-噻吩并[2,3-c]吡喃-7,4'-哌啶]223(970mg,84%)。1hnmr(300mhz,甲醇-d4)δ7.07(s,1h),4.10(t,j=10.4hz,2h),3.85(dd,j=11.7,3.0hz,1h),3.12-2.97(m,1h),2.77-2.68(m,1h),2.30(dt,j=13.8,2.9hz,1h),2.16-2.02(m,1h),1.76(dd,j=13.7,11.8hz,1h),1.44-1.30(m,1h),1.10(d,j=6.4hz,3h)。lcms m/z 317.99[m+1]
+
[1564]
c120的制备
[1565]
n-重氮氨磺酰氟
[1566][1567]
n-重氮氨磺酰氟(c120)的合成
[1568]
向冷却至0℃的nan3(1.95g,30.00mmol)于水(60ml)中的混合物中添加mtbe(60ml),随后添加2,3-二甲基咪唑-3-鎓-1-磺酰基氟(三氟甲磺酸酯)(11.8g,35.95mmol)c119的mecn(3ml)溶液。在剧烈搅拌10分钟后,使各层分离30分钟并去除有机层,使其穿过相分离器并置于锥形瓶中,并老化过夜。此时,用移液管取出烧瓶底部形成的红色溶液并将有机层用dmso(60ml)稀释。观察到双相混合物。添加乙腈(约25ml),直到观察到均匀的混合物。最终总体积为约150ml。在后续叠氮化物形成中使用该反应混合物,而不经纯化或表征。基于文献先例x,据估计产率为约90%的活性试剂。基于此,据估计浓度为约0.18m。
[1569]
化合物224
[1570]
(2's,4s,6's,7s)-2-氯-2'-[1-[[4-(羟基甲基)苯基]甲基]三唑-4-基]-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(224)
[1571]
实施例:
[1572][1573]
步骤1.[4-(叠氮基甲基)苯基]甲醇(c123)的合成
[1574]
向[4-(氨基甲基)苯基]甲醇c121和水溶液khco3(400μl 3 m)的混合物中添加n-重氮氨磺酰氟c120(1.7 ml 0.18 m)的mtbe/dmso/mecn溶液并将反应物在室温下搅拌2小
时。此时,反应基于文献先例
x
被假设为完全的并且未进一步表征并在下一步骤中直接使用[4-(叠氮基甲基)苯基]甲醇c123。
[1575]
步骤2.(2's,4s,6's,7s)-2-氯-2'-[1-[[4-(羟基甲基)苯基]甲基]三唑-4-基]-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(224)的合成
[1576]
在继续迅速搅拌下,将大约三分之一来自步骤1的所形成的双相悬浮液(700μl)添加到(2's,4s,6's,7s)-2-氯-2'-乙炔基-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇222于dmso(0.1 ml)的溶液中,添加350μl ph 5缓冲液水溶液(抗坏血酸钠(350μl 0.125 m):磷酸二钠:柠檬酸1:4:2,基于抗坏血酸钠为0.125 m)和100μl cuso4水溶液(100μl 0.035 m):3-[4-[[双[[1-(3-羟基丙基)三唑-4-基]甲基]氨基]甲基]三唑-1-基]丙-1-醇1:1并将混合物在50℃下搅拌过夜。此时,将反应物冷却至室温并然后在真空中干燥,去除高挥发性溶剂。然后使所得溶液或悬浮液穿过过滤膜,以通过反相hplc纯化。(方法:waters xselect csh c18 obd制备型柱;19 x 100 mm,5微米.梯度:于具有10 mm氢氧化铵的水中的乙腈)。)将含有产物的级分合并并浓缩,产生呈黄色固体的(2's,4s,6's,7s)-2-氯-2'-[1-[[4-(羟基甲基)苯基]甲基]三唑-4-基]-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇224(3.5 mg,21%)。1h nmr(400 mhz,dmso-d6)δ7.97(s,1h),7.30(t,j=5.7 hz,4h),6.96(s,1h),5.50(s,2h),5.39(s,1h),5.20(s,1h),4.47(s,2h),4.38(t,j=4.5 hz,1h),4.10(d,j=11.5 hz,1h),3.92(dd,j=11.9,4.2 hz,1h),3.63(dd,j=11.7,5.1 hz,1h),3.07(s,1h),2.27(s,1h),2.17(d,j=13.3 hz,1h),2.03(d,j=13.3 hz,1h),1.54(t,j=12.5 hz,1h),1.23(t,j=12.3 hz,1h),1.00(d,j=6.3 hz,3h)。lcms m/z461.12[m+1]
+
[1577]
化合物225
[1578]
(4-((4-((2s,4s,6s)-2'-氯-4',4'-二氟-2-甲基-4',5'-二氢螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-6-基)-1h-1,2,3-三唑-1-基)甲基)苯基)甲醇(225)
[1579][1580]
步骤1.[4-(叠氮基甲基)苯基]甲醇(c123)的合成
[1581]
向[4-(氨基甲基)苯基]甲醇c121和水溶液khco3(400μl 3m)的混合物中添加n-重氮氨磺酰氟c120(1.7ml 0.18m)的mtbe/dmso/mecn溶液并将反应物在室温下搅拌2小时。此时,反应基于文献先例
x
被假设为完全的并且未进一步表征并在下一步骤中直接使用[4-(叠氮基甲基)苯基]甲醇c123。
[1582]
步骤2.(4-((4-((2s,4s,6s)-2'-氯-4',4'-二氟-2-甲基-4',5'-二氢螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-6-基)-1h-1,2,3-三唑-1-基)甲基)苯基)甲醇(225)的合成
[1583]
在继续迅速搅拌下,将大约三分之一来自步骤1的所形成的双相悬浮液(700μl)添加到(2's,6's,7s)-2-氯-2'-乙炔基-4,4-二氟-6'-甲基-螺[5h-噻吩并[2,3-c]吡喃-7,
4'-哌啶]223于dmso(0.1ml)的溶液中,添加350μl ph 5缓冲液水溶液(抗坏血酸钠(350μl 0.125m):磷酸二钠:柠檬酸1:4:2,基于抗坏血酸钠为0.125m)和100μl cuso4水溶液(100μl 0.035m):3-[4-[[双[[1-(3-羟基丙基)三唑-4-基]甲基]氨基]甲基]三唑-1-基]丙-1-醇1:1并将混合物在50℃下搅拌过夜。此时,将反应物冷却至室温并然后在真空中干燥,去除高挥发性溶剂。然后使所得溶液或悬浮液穿过过滤膜,以通过反相hplc纯化。(方法:waters xselect csh c18 obd制备型柱;19x 100mm,5微米.梯度:具有10mm氢氧化铵的水中的乙腈)。将含有产物的级分合并并浓缩,产生呈黄色固体的(4-((4-((2s,4s,6s)-2'-氯-4',4'-二氟-2-甲基-4',5'-二氢螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-6-基)-1h-1,2,3-三唑-1-基)甲基)苯基)甲醇225(3.5mg,21%)。1hnmr(400mhz,dmso-d6)δ8.00(s,1h),7.34(s,1h),7.29(d,j=2.3hz,4h),5.52(s,2h),5.20(s,1h),4.47(d,j=4.6hz,2h),4.21(t,j=10.6hz,2h),4.11(d,j=11.5hz,1h),3.02(d,j=18.8hz,1h),2.33(d,j=12.7hz,2h),2.10(d,j=13.4hz,1h),1.66(dd,j=13.5,11.7hz,1h),1.30(dd,j=13.6,11.3hz,1h),1.01(d,j=6.2hz,3h)。lcms m/z 481.12[m+1]
+
[1584]
化合物226-371
[1585]
化合物226-371(参见表11)遵循对于化合物224和225所述的方法制备为母体或三氟乙酸盐。使用222或223和适当的胺,其中所述胺原位转化成叠氮化物,以环加成到所需三唑。
[1586]
表11.化合物226-371的制备方法、结构和物理化学数据
[1587]
[1588]
[1589]
[1590]
[1591]
[1592]
[1593]
[1594]
[1595]
[1596]
[1597]
[1598]
[1599]
[1600]
[1601]
[1602]
[1603]
[1604]
[1605]
[1606]
[1607]
[1608]
[1609]
[1610]
[1611]
[1612]
[1613]
[1614]
[1615]
[1616]
[1617]
[1618]
[1619]
[1620]
[1621]
[1622]
[1623]
[1624]
[1625]
[1626]
[1627]
[1628]
[1629]
[1630]
[1631]
[1632][1633]
x
sharpless,k.b.等人nature,2019,574,86-89
[1634]
s56的制备
[1635]
1-[(2s,6s)-6-甲基-4-氧代-2-哌啶基]环丙烷羧酸甲酯(s56)
[1636][1637]
标准方法c:发生环化以使用蒸发检查制备哌啶酮
[1638]
将(4s)-4-氨基戊-2-酮(盐酸)s25(1.25g,8.2mmol)于etoh(50ml中的搅拌溶液冷却至0℃。然后添加et3n(2.28g,3.2ml,22.0mmol)、mgso4(950mg,7.7mmol)、l-脯氨酸(490mg,4.0mmol)和1-甲酰基环丙烷羧酸甲酯c124(1g,7.6mmol)。30分钟后,使反应混合物缓慢升温至室温并搅拌24小时。将反应混合物在真空下蒸馏,得到呈棕色浓稠油状物的粗
产物。通过硅胶快速柱色谱(用12%于dcm中的meoh洗脱)纯化粗材料,得到呈淡黄色油状物的产物1-[(2s,6s)-6-甲基-4-氧代-2-哌啶基]环丙烷羧酸甲酯s56(600mg,37%产率)。在这些条件下观察到对映异构体纯的起始材料(4s)-4-氨基戊-2-酮(盐酸盐)s25的部分立体化学消除,导致未分离的立体异构体混合物,其中顺式产物是主要异构体。1h nmr(400mhz,dmso-d6)δ3.58(s,3h),2.90-2.86(m,1h),2.81-2.72(m,1h),2.30-2.13(m,3h),2.06-1.97(m,1h),1.10-0.94(m,8h)。lcms m/z 212.1[m+h]
+
.
[1639]
s57的制备
[1640]
n-[1-[(2s,6s)-6-甲基-4-氧代-2-哌啶基]环丙基]氨基甲酸叔丁酯(s57)
[1641][1642]
标准方法d:发生环化以使用水溶液淬灭检查来制备哌啶酮
[1643]
在室温下向(4s)-4-氨基戊-2-酮(盐酸)(1g,6.5mmol)s25于etoh(20ml)中的搅拌溶液中添加l-脯氨酸(150mg,1.3mmol)、mgso4(783mg,6.4mmol)、et3n(718.74mg,1ml,7.0mmol)和n-(1-甲酰基环丙基)氨基甲酸叔丁酯c125(1.2g,6.4mmol)。使反应物质在室温下搅拌16小时。将反应混合物用饱和nahco3(50ml)稀释并用etoac(3x 100ml)萃取。将合并的有机层用0.15n hcl水溶液(3x 50ml)洗涤,并且然后通过使用1n naoh溶液将水层ph调节至约12。将水层用etoac(3x 100ml)萃取,并将合并的有机层经na2so4干燥,过滤,并在真空下蒸发,提供将粗材料。通过柱色谱使用100-200目硅胶纯化粗材料并用4%于dcm中的meoh洗脱,提供n-[1-[(2s,6s)-6-甲基-4-氧代-2-哌啶基]环丙基]氨基甲酸叔丁酯s57(800mg,46%产率)。在这些条件下观察到对映异构体纯的起始材料(4s)-4-氨基戊-2-酮(盐酸盐)s25的部分立体化学消除,导致未分离的立体异构体混合物,其中顺式产物是主要异构体。1h nmr(400mhz,dmso-d6)δ7.11(brs,1h),2.74(brs,1h),2.49(brs,1h),2.18-2.0(m,5h),1.36(s,9h),1.09(d,j=6.0hz,3h),0.75-0.60(m,4h)。lcms m/z 269.2[m+h]
+
.
[1644]
s58的制备
[1645]
n-[1-[(2s,6s)-6-甲基-4-氧代-2-哌啶基]环丙基]氨基甲酸叔丁酯(s58)
[1646][1647]
步骤1.1-(二甲基氨基)-n-甲氧基-n-甲基-环丙烷甲酰胺(c128)
[1648]
在0℃下向1-(二甲基氨基)环丙烷羧酸c126(500mg,0.0038mol)于dcm(10ml)中的搅拌溶液中添加dipea(2.226g,3.00ml,0.0169mol)和n-甲氧基甲胺(盐酸)c127(453mg,0.0046mol)。将反应混合物搅拌10分钟,并在0℃下缓慢添加t3p(3.12g,2.92ml 50%w/w,0.0049mol)。将反应混合物在室温下搅拌16小时。将反应混合物用dcm(25ml)稀释并冷却至0℃。缓慢添加1n naoh溶液(10ml),并将有机层分离并用饱和nh4cl溶液(10ml)洗涤。将有机层经无水硫酸钠干燥,过滤,并在减压下浓缩,获得粗材料。通过柱色谱使用100-200目硅
胶纯化粗材料并用70%于石油醚中的etoac洗脱,得到呈棕色液体的1-(二甲基氨基)-n-甲氧基-n-甲基-环丙烷甲酰胺c128(300mg,44%)。1h nmr(400mhz,dmso-d6)δ3.62(s,3h),3.19(s,3h),2.23(s,6h),0.82-0.80(m,4h)。lcms m/z 173.22[m+h]
+
.
[1649]
步骤2.1-(二甲基氨基)环丙烷甲醛(s58)
[1650]
在0℃下向lah(988mg,0.0255mol)于乙醚(50ml)中的搅拌悬浮液中逐滴添加1-(二甲基氨基)-n-甲氧基-n-甲基-环丙烷甲酰胺c128(2g,0.0102mol)。使反应混合物在0℃下搅拌4小时。将反应物用水(3.2ml)、1n naoh溶液(3.2ml)和水(3.2ml)淬灭。通过垫过滤反应物质并用乙醚(30ml)洗涤。将滤液在真空下蒸发,得到呈无色油状物的粗制1-(二甲基氨基)环丙烷甲醛s58(1.4g,97%)。1h nmr(300mhz,氯仿-d)δ9.01(s,1h),2.63(s,6h),1.28-1.14(m,4h)。
[1651]
中间体s59-s62
[1652]
中间体s59-s62(参见表12)在单个步骤中使用标准方法c或d由中间体s25制备。相应醛通过上文所述的方法制备或从商业来源获得。在步骤1中观察到对映异构纯的起始材料的(4s)-4-氨基戊-2-酮(盐酸盐)s25的部分立体化学消除,导致在步骤1中产生未分离的立体异构物混合物。在每种情况下,顺式产物是主要异构体。对方法的任何修改在表12和所附脚注中标识。
[1653]
表12.中间体s59-s62的结构和物理化学数据
[1654][1655]
注意:
[1656]
1)将检查程序修改为:将反应混合物用饱和nahco3(50ml)稀释并用etoac(4x 300ml)萃取。将合并的有机层经硫酸钠干燥,过滤,并在真空下蒸发,提供将粗材料。
[1657]
化合物372
[1658]
1-[(2's,6's,7s)-2-氯-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-2'-基]环丙烷羧酸甲酯(372)
[1659][1660]
标准方法e:为了制备螺哌啶
[1661]
向1-[(2s,6s)-6-甲基-4-氧代-2-哌啶基]环丙烷羧酸甲酯s56(100mg,0.473mmol)和2-(5-氯-3-噻吩基)乙醇s2(92mg,0.57mmol,1.2当量)于dcm(2.2ml)中的混合物中添加msoh(250μl,3.85mmol,8当量),并将混合物加热至40℃。5小时后,将反应物用饱和nahco3溶液淬灭并用dcm(6x)萃取。将合并的有机层经na2so4干燥,过滤,并浓缩。将粗材料用硅胶柱纯化并用0至100%于庚烷中的etoac洗脱,提供1-[(2's,6's,7s)-2-氯-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-2'-基]环丙烷羧酸甲酯372(123mg,69%)。当制备s56时,由于对映异构体纯度的部分消除,372也被分离为对映异构体的混合物,其中(2's,6's,7s)构型是主要对映异构体。1h nmr(300mhz,氯仿-d)δ6.56(s,1h),3.88(t,j=5.5hz,2h),3.63(s,3h),3.04(dtd,j=12.9,6.5,2.5hz,1h),2.73(dd,j=11.8,2.5hz,1h),2.58(t,j=5.5hz,2h),2.00(ddt,j=17.5,13.4,2.5hz,2h),1.70(dd,j=13.3,11.7hz,2h),1.40-1.16(m,3h),1.11(d,j=6.4hz,3h),1.04-0.92(m,1h),0.77(ddd,j=9.7,5.4,2.6hz,1h)。lcms m/z 356.15[m+h]
+
.
[1662]
化合物373-378
[1663]
化合物373-378(参见表13)在单个步骤中由相应哌啶酮试剂标准方法e制备。当制备哌啶酮试剂时,由于对映异构体纯度的部分消除,化合物被分离为对映异构体的混合物,其中(2's,6's,7s)构型是主要对映异构体。对方法的任何修改在表13和所附脚注中标识。
[1664]
表13.化合物373-378的结构和物理化学数据
[1665]
溶液淬灭并用etoac萃取(4x)。将合并的有机层经na2so4干燥,过滤,并浓缩。通过反相hplc纯化(方法:waters xselect csh c18 obd制备型柱;30x 150mm,5微米.梯度:于具有5mm盐酸的水中的乙腈),得到产物n-[1-[(2's,6's,7s)-2-氯-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-2'-基]环丙基]乙酰胺(盐酸盐)379(4.9mg,27%产率)。当制备373时,由于对映异构体纯度的部分消除,379被分离为对映异构体的混合物,其中(2's,6's,7s)构型是主要对映异构体。1h nmr(300mhz,氯仿-d)δ9.40(s,1h),9.07(s,1h),7.48(s,1h),6.60(s,1h),3.87(s,2h),3.69(s,1h),3.36(s,1h),2.61(d,j=7.0hz,2h),2.29(d,j=14.7hz,1h),2.20-1.97(m,5h),1.86(t,j=13.3hz,1h),1.64(s,3h),1.18(s,1h),1.08(s,1h),0.96(s,1h)。lcms m/z 355.28[m+h]
+
.试验性地分配n-酰基化位点。
[1675]
化合物380
[1676]
[1-[(2's,6's,7s)-2-氯-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-2'-基]环丙基]甲醇(380)
[1677][1678]
步骤1.1-[(2's,6's,7s)-2-氯-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-2'-基]环丙烷羧酸(c129)
[1679]
向1-[(2's,6's,7s)-2-氯-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-2'-基]环丙烷羧酸甲酯372(82mg,0.22mmol)于thf(500μl)中的混合物中添加lioh(26mg,1.1mmol)的水(500μl)溶液。将反应混合物加热至50℃持续2小时,并添加另外的thf(1ml),以帮助溶解起始材料。在加热几乎3天后,将反应物冷却至室温。将反应混合物然后浓缩,以去除溶剂并将其再溶解于dmso中。通过反相hplc纯化(方法:waters xselect csh c18 obd制备型柱;30x 150mm,5微米.梯度:于具有5mm盐酸的水中的乙腈),分离为1-[(2's,6's,7s)-2-氯-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-2'-基]环丙烷羧酸c129(58mg,77%产率)。当制备372时,由于对映异构体纯度的部分消除,c129被分离为对映异构体的混合物,其中(2's,6's,7s)构型是主要对映异构体。1h nmr(300mhz,dmso-d6)δ12.98(s,1h),6.93(s,1h),3.89(t,j=5.4hz,2h),3.48(s,2h),2.58(s,3h),2.22-1.84(m,4h),1.29(d,j=6.3hz,3h),1.27-1.11(m,2h),1.02(s,1h)。lcms m/z 342.27[m+h]
+
.
[1680]
步骤2.[1-[(2's,6's,7s)-2-氯-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-2'-基]环丙基]甲醇(380)
[1681]
在小瓶中将1-[(2's,6's,7s)-2-氯-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-2'-基]环丙烷羧酸c129(15mg,0.044mmol)溶解于thf(200μl)中。将小瓶在氮气下冷却至0℃。缓慢添加硼烷四氢呋喃络合物(175μl 1m,0.1750mmol),并观察到大量气泡。使混合物缓慢升温至室温。4小时后,将反应物用饱和nahco3溶液淬灭并用dcm萃取(5x)。将合并的有机层经na2so4干燥,过滤,并在真空中浓缩。通过反相hplc纯化(方法:waters xselect csh c18 obd制备型柱;30x 150mm,5微米.梯度:具有0.2%甲酸的水中的乙腈),得到产物[1-[(2's,6's,7s)-2-氯-6'-甲基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,
4'-哌啶]-2'-基]环丙基]甲醇(甲酸盐)380(6.9mg,42%产率)。当制备c129时,由于对映异构体纯度的部分消除,380被分离为对映异构体的混合物,其中(2's,6's,7s)构型是主要对映异构体。1h nmr(400mhz,甲醇-d4)δ6.74(s,1h),3.93(t,j=5.5hz,2h),3.86(dd,j=11.7,1.5hz,1h),3.66-3.47(m,1h),3.24(d,j=11.7hz,1h),3.06(dd,j=12.4,2.6hz,1h),2.63(t,j=5.5hz,2h),2.27(ddt,j=16.7,14.2,2.8hz,2h),2.10(dd,j=14.6,12.5hz,1h),1.85(dd,j=14.7,12.2hz,1h),1.36(d,j=6.6hz,3h),0.93-0.79(m,1h),0.79-0.67(m,1h),0.56(ddt,j=17.2,9.5,5.4hz,2h)。lcms m/z 328.28[m+h]
+
.
[1682]
化合物381
[1683]
(2'r,6's,7s)-2-氯-2'-环丙基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](381)
[1684][1685]
步骤1.(2s,3r,6r)-6-环丙基-2-(1-甲基三唑-4-基)-4-氧代-哌啶-3-羧酸叔丁酯(c131)
[1686]
向(3r)-3-(叔丁氧基羰基氨基)-3-环丙基-丙酸c130(500mg,2.181mmol)于thf(4.5ml)中的溶液中添加cdi(390mg,2.405mmol)并将混合物在室温下搅拌3小时。添加双[(3-叔丁氧基-3-氧代-丙酰基)氧基]镁c109(449mg,1.310mmol)。20小时后,将反应物用mbte(10ml)和1n hcl(3ml)稀释。将有机层分离并用饱和碳酸氢钠水溶液(3ml)和盐水(3ml)洗涤,用硫酸镁干燥,过滤,并浓缩,产生(5r)-5-(叔丁氧基羰基氨基)-5-环丙基-3-氧代-戊酸叔丁酯。
[1687]
将粗材料溶解于dcm(4ml)并添加tfa(1ml,13mmol)。在1h 30min后,将溶液与dcm(4ml)共沸三次。
[1688]
将来自第二步骤的粗材料溶解于dcm(4ml)中并添加1-甲基三唑-4-甲醛(250mg,2.250mmol)。在搅拌超过2天后,将混合物直接装载到硅胶柱上以进行纯化(梯度:0-10%于dcm中的meoh)。将含有产物的级分合并并浓缩,产生(2s,3r,6r)-6-环丙基-2-(1-甲基三唑-4-基)-4-氧代-哌啶-3-羧酸叔丁酯c131(432mg,62%产率)。1h nmr(400mhz,氯仿-d)δ10.17(s,1h),8.11(s,1h),7.50(s,1h),4.22(s,3h),4.09-4.04(m,1h),2.67-2.64(m,1h),2.49-2.39(m,2h),1.39(s,9h),0.95(dq,j=8.1,2.7hz,1h),0.57(dd,j=8.2,1.5hz,2h),
0.32-0.24(m,2h)。
[1689]
步骤2.(2'r,6's,7s)-2-氯-2'-环丙基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](381)
[1690]
向(2s,3r,6r)-6-环丙基-2-(1-甲基三唑-4-基)-4-氧代-哌啶-3-羧酸叔丁酯c131(25mg,0.078mmol)于dcm(0.5ml)中的混合物中添加msoh(20μl,0.3082mmol)并将反应物回流。1小时后,添加2-(5-氯-3-噻吩基)乙醇s2(20mg,0.1230mmol)。将反应物搅拌过夜。将混合物冷却至室温并再用dcm(2ml)和饱和碳酸氢钠水溶液(2ml)稀释。将有机层分离并装载到硅胶柱上以进行纯化(梯度:0-10%于dcm中的meoh)。发现含有产物的级分仍处于所需纯度阈值下,因此将合并的级分浓缩并再稀释于dmso中。通过反相hplc纯化。方法:waters xselect csh c18 obd制备型柱;30x 150mm,5微米.梯度:具有0.1%三氟乙酸的水中的乙腈.将含有产物的级分用dcm(1ml)和饱和碳酸氢钠水溶液(1ml)稀释。将有机层干燥,产生(2'r,6's,7s)-2-氯-2'-环丙基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]381(5.3mg,18%产率)。1h nmr(400mhz,氯仿-d)δ7.45(s,1h),6.58(s,1h),4.35(dd,j=11.7,2.6hz,1h),4.06(s,3h),3.99-3.85(m,2h),2.68-2.50(m,2h),2.38-2.25(m,2h),2.20(dt,j=13.6,2.6hz,1h),1.80(dd,j=13.5,11.7hz,2h),1.65(dd,j=13.6,11.4hz,1h),0.79(qt,j=8.5,4.9hz,1h),0.46(tdd,j=10.3,8.3,5.4hz,2h),0.23-0.12(m,2h)。lcms m/z 365.3[m+h]
+
.
[1691]
化合物382
[1692]
(2'r,4s,6's,7s)-2-氯-2'-环丙基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(382)
[1693][1694]
步骤1.(2r,6s)-2-环丙基-6-(1-甲基三唑-4-基)哌啶-4-酮(c132)
[1695]
向(2s,3r,6r)-6-环丙基-2-(1-甲基三唑-4-基)-4-氧代-哌啶-3-羧酸叔丁酯c131(400mg,1.248mmol)于氯仿(8ml)中的混合物中添加2,2,2-三氟乙酸(500μl,6.490mmol)并将反应物回流。在搅拌过夜后,将混合物冷却至室温,用dcm(10ml)稀释,并用饱和碳酸氢钠水溶液(10ml)洗涤。将有机层用硫酸镁干燥,过滤,并浓缩并且然后少量稀释于dcm并装载到硅胶柱上以进行纯化(梯度:0-10%于dcm中的meoh)。将含有产物的级分合并并浓缩,产生(2r,6s)-2-环丙基-6-(1-甲基三唑-4-基)哌啶-4-酮c132(90mg,33%产率)。1h nmr(400mhz,氯仿-d)δ7.50(s,1h),4.22-4.15(m,1h),4.12(s,3h),2.67-2.64(m,2h),2.61-2.56(m,1h),2.47-2.40(m,1h),2.19(ddd,j=11.6,8.7,3.0hz,1h),0.95(ddt,j=13.2,8.4,4.1hz,1h),0.59-0.52(m,2h),0.33-0.21(m,2h)。
[1696]
步骤2.(2'r,6's,7s)-2-氯-2'-环丙基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](381)
[1697]
向(2r,6s)-2-环丙基-6-(1-甲基三唑-4-基)哌啶-4-酮c132(45mg,0.20mmol)和2-(5-氯-3-噻吩基)乙醇(43mg,0.26mmol)于dcm(1000μl)中的混合物中添加msoh(90μl,1.4mmol)并将混合物回流。在搅拌过夜后,将混合物冷却至室温并用饱和碳酸氢钠水溶液(1ml)淬灭。分离各层,并将有机层直接装载到硅胶柱上以进行纯化(梯度:0-10%于dcm中的meoh)。将含有产物的级分合并并浓缩,产生呈黄色油状物的(2'r,6's,7s)-2-氯-2'-环丙基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]381(74mg,90%产率)。lcms m/z 365.15[m+h]
+
.
[1698]
步骤3.1-[(2'r,6's,7s)-2-氯-2'-环丙基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(c133)
[1699]
向冷却至0℃的于dcm(1ml)中的(2'r,6's,7s)-2-氯-2'-环丙基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]381(74mg,0.203mmol)中添加dipea(50μl,0.2871mmol),随后添加tfaa(30μl,0.2158mmol)。35分钟后,将混合物用1n hcl(1ml)淬灭。将有机层分离并使其穿过相分离器。将有机层装载到硅胶柱上以进行纯化(梯度:0-50%于庚烷中的etoac)。将含有产物的级分合并并浓缩,产生呈白色固体的1-[(2'r,6's,7s)-2-氯-2'-环丙基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c133(51mg,50%产率)。lcms m/z 461.31[m+h]
+
.
[1700]
步骤4.(2r,4s,6s)-2'-氯-2-环丙基-6-(1-甲基三唑-4-基)-1-(2,2,2-三氟乙酰基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮(c134)
[1701]
将1-[(2'r,6's,7s)-2-氯-2'-环丙基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c133(51mg,0.102mmol)于氯苯(1ml)中的混合物用氮气真空吹扫5次。此时,添加5,5-二甲基-1,3-二溴乙内酰脲(20mg,0.07mmol)和2-[(e)-(1-氰基-1-甲基-乙基)偶氮基]-2-甲基-丙腈(1.5mg,0.009mmol),并将混合物再次真空吹扫5次。将混合物加热至75℃。15分钟后,将混合物冷却至室温并将其与饱和碳酸氢钠水溶液(1ml)混合。将有机层分离,将水层用乙酸乙酯(1ml)洗涤,并将合并的有机层用硫酸镁干燥,过滤,并浓缩。将材料干燥过夜,去除残余溶剂。
[1702]
在火焰干燥的烧瓶中将粗制泡沫用dmso(1ml)稀释并用氮气真空吹扫5次。将混合物加热至60℃,添加三乙胺(75μl,0.54mmol),并将深棕色溶液进一步加热至65℃(内部温度)。在75分钟后,将混合物冷却至室温并用水(5ml)和乙酸乙酯(5ml)稀释。将水层用乙酸乙酯(2x 5ml)洗涤,并且然后将合并的有机层用硫酸钠干燥,过滤,并浓缩。将粗材料用少量dcm稀释并装载到硅胶柱上以进行纯化(梯度:0-50%于庚烷中的etoac)。将含有产物的级分合并并浓缩,产生呈白色固体的(2r,4s,6s)-2'-氯-2-环丙基-6-(1-甲基三唑-4-基)-1-(2,2,2-三氟乙酰基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮c134(16mg,32%)。lcms m/z 475.21[m+h]
+
.
[1703]
步骤5.1-[(2'r,4s,6's,7s)-2-氯-2'-环丙基-4-羟基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(c135)
[1704]
向5:2二乙基乙胺/甲酸(20μl,0.04763mmol)于acn(500μl)中的混合物中添加n-[(1r,2r)-2-氨基-1,2-二苯基-乙基]-4-甲基-苯磺酰胺(0.12mg,3.274e-4mmol)和1,2,3,4,5-五甲基环戊烷四氯化铑(2+)(0.1mg,1.592e-4mmol)于acn(50μl)中的溶液。5分钟后,将溶液添加到(2r,4s,6s)-2'-氯-2-环丙基-6-(1-甲基三唑-4-基)-1-(2,2,2-三氟乙酰
基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮c134(16mg,0.03203mmol)并将反应物在-10℃下搅拌并缓慢升温过夜。此时,将混合物浓缩并直接装载到硅胶柱上以进行纯化(梯度:0-50%于庚烷中的etoac)。将含有产物的级分合并并浓缩,产生1-[(2'r,4s,6's,7s)-2-氯-2'-环丙基-4-羟基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c135(15mg,100%)。lcms m/z 477.24[m+h]
+
.
[1705]
步骤6.(2'r,4s,6's,7s)-2-氯-2'-环丙基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(382)
[1706]
将1-[(2'r,4s,6's,7s)-2-氯-2'-环丙基-4-羟基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c135(15mg,0.03mmol)溶解于meoh(0.2ml)中并加热至60℃,此时添加naoh(50μl 6m,0.30mmol)。在搅拌7小时后,观察到接近完全转化。将混合物用水(1ml)稀释。通过反相hplc纯化(方法:waters xselect csh c18 obd制备型柱;30x 150mm,5微米.梯度:于具有0.1%三氟乙酸的水中的乙腈),得到呈白色固体的(2'r,4s,6's,7s)-2-氯-2'-环丙基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(三氟乙酸盐)382(11.7mg,74%产率,在2个步骤内)。1hnmr(400mhz,甲醇-d4)δ8.03(s,1h),6.95(s,1h),4.49(t,j=3.4hz,1h),4.13(s,3h),4.04(dd,j=12.2,3.3hz,1h),3.86(dd,j=12.2,3.7hz,1h),3.32(d,j=1.8hz,1h),2.99(s,1h),2.64-2.50(m,2h),2.28(dd,j=14.8,12.6hz,1h),2.12-2.01(m,1h),1.02(dd,j=8.7,4.8hz,1h),0.74(d,j=8.1hz,2h),0.65-0.59(m,1h),0.42(s,1h)。lcms m/z 381.28[m+h]
+
.
[1707]
化合物383
[1708]
(2'r,4s,6's,7s)-2-氯-2'-环丙基-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(383)
[1709][1710]
向(2r,6s)-2-环丙基-6-(1-甲基三唑-4-基)哌啶-4-酮c132(45mg,0.20mmol)和2-[5-(三氟甲基)-3-噻吩基]乙醇s3(52mg,0.27mmol)于dcm(1000μl)中的混合物中添加msoh(90μl,1.387mmol)并将混合物回流。在搅拌过夜后,将混合物冷却至室温并用饱和碳酸氢钠水溶液(1ml)淬灭。分离各层,并将有机层直接装载到硅胶柱上以进行纯化(梯度:0-10%于dcm中的meoh)。将含有产物的级分合并并浓缩;然而,uplc分析指示存在中间体。将产物溶解于dmso中。通过反相hplc纯化(方法:waters xselect csh c18 obd制备型柱;30x 150mm,5微米.梯度:于具有5mm盐酸的水中的乙腈),得到呈白色固体的(2'r,6's,7s)-2'-环丙基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](盐酸盐)383(18.7mg,21%产率)。1hnmr(400mhz,甲醇-d4)δ8.05(s,1h),7.35(s,1h),
4.90-4.87(m,1h),4.13(s,3h),4.03(t,j=5.4hz,2h),2.99(s,1h),2.78(d,j=2.4hz,2h),2.54(d,j=17.3hz,2h),2.46-2.37(m,1h),2.15-2.05(m,1h),1.03(s,1h),0.74(d,j=8.2hz,2h),0.66-0.61(m,1h),0.40(s,1h)。lcms m/z 399.3[m+h]
+
.
[1711]
化合物384
[1712]
(2's,6's,7s)-2',4-二甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[5h-噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(384)
[1713][1714]
向冷却至0℃的(2s,4s,6s)-2-甲基-6-(1-甲基三唑-4-基)-1-(2,2,2-三氟乙酰基)-2'-(三氟甲基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮s32(53mg,0.1036mmol)于乙醚(1ml)中的混合物中添加溴(甲基)镁(35μl 3.4m,0.1190mmol)。将混合物在0℃下搅拌5分钟,然后用饱和氯化铵(2ml)淬灭并用tbme(3ml)稀释。将有机层用盐水洗涤并经mgso4干燥,过滤,并浓缩成粗残余物。向稀释于meoh(1ml)中的粗残余物中添加naoh(50μl 6m,0.3000mmol)。将混合物在60℃下搅拌2小时。通过反相hplc(方法:c18 waters sunfire柱(30x150 mm,5微米).梯度:于具有0.1%三氟乙酸的h2o中的mecn)直接纯化反应混合物,提供呈澄清玻璃状固体的产物(2's,6's,7s)-2',4-二甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[5h-噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(三氟乙酸盐)384(10.6mg,20%)。lcms m/z 403.27[m+1]
+
.
[1715]
化合物385
[1716]
(2s,4s,4's,6s)-2'-氯-4',5',5'-三氘化-2-甲基-6-(1-甲基三唑-4-基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-醇(385)
[1717][1718]
步骤1.(2s,4s,6s)-2'-氯-5',5'-二氘化-2-甲基-6-(1-甲基三唑-4-基)-1-(2,2,2-三氟乙酰基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮(c137)
[1719]
向40ml小瓶中添加(2s,4s,6s)-2'-氯-2-甲基-6-(1-甲基三唑-4-基)-1-(2,2,2-三氟乙酰基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮c136(404mg,0.817mmol),随后添加d2o(1.9ml)和mecn(5.6ml)。然后添加吡咯烷(7μl,0.08mmol)。添加另外的mecn(1ml)和二噁烷(2ml),并将反应混合物加热至75℃。18小时后,将混合的反应物用水洗涤并用etoac萃取(3x)。将合并的有机层用盐水洗涤,经na2so4干燥,过滤,并在真空中浓缩。通过硅胶色谱纯化(梯度:0至50%于庚烷中的etoac),得到(2s,4s,6s)-2'-氯-5',5'-二氘化-2-甲基-6-(1-甲基三唑-4-基)-1-(2,2,2-三氟乙酰基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮c137(366mg,97%)。lcms m/z 451.15[m+h]
+
.基于1h nmr,97.5% d。
[1720]
步骤2.1-[(2s,4s,4's,6s)-2'-氯-4',5',5'-三氘化-4'-羟基-2-甲基-6-(1-甲基三唑-4-基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-1-基]-2,2,2-三氟-乙酮(c138)
[1721]
向hcooh-d4(23μl,0.6095mmol)和三乙胺(32μl,0.23mmol)于dcm(500μl)中的混合物中添加1,2,3,4,5-五甲基环戊烷四氯化铑(2+)(0.195mg,3.104e-4mmol)和n-[(1r,
2r)-2-氨基-1,2-二苯基-乙基]-4-甲基-苯磺酰胺(0.222mg,6.058e-4mmol)于dcm(60μl)中的溶液。5分钟后,添加dcm(500μl)中的(2s,4s,6s)-2'-氯-5',5'-二氘化-2-甲基-6-(1-甲基三唑-4-基)-1-(2,2,2-三氟乙酰基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮c137(50mg,0.11mmol)。将所得橙色反应混合物在室温下搅拌过夜。将反应物用饱和水溶液碳酸氢钠、1n hcl和盐水洗涤,并将水层用etoac单独萃取。将合并的有机层经na2so4干燥,过滤,并在真空中浓缩。通过硅胶色谱纯化(梯度:0-50%于庚烷中的etoac),得到产物1-[(2s,4s,4's,6s)-2'-氯-4',5',5'-三氘化-4'-羟基-2-甲基-6-(1-甲基三唑-4-基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-1-基]-2,2,2-三氟-乙酮c138(45.8mg,93%)。1h nmr(300mhz,氯仿-d)δ7.59(s,1h),6.84(s,1h),5.53(s,1h),4.68-4.39(m,1h),4.13-4.03(m,3h),3.10(dd,j=15.1,7.4hz,1h),2.64(ddd,j=15.1,8.1,2.2hz,2h),2.12(s,1h),2.08(s,1h),1.44-1.25(m,3h)。lcms m/z454.17[m+h]
+
.
[1722]
步骤3.(2s,4s,4's,6s)-2'-氯-4',5',5'-三氘化-2-甲基-6-(1-甲基三唑-4-基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-醇(385)
[1723]
将1-[(2s,4s,4's,6s)-2'-氯-4',5',5'-三氘化-4'-羟基-2-甲基-6-(1-甲基三唑-4-基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-1-基]-2,2,2-三氟-乙酮c138(45.8mg,0.1008mmol)于meoh(800μl)中的溶液中用naoh水溶液(170μl 6m,1.020mmol)处理并加热至60℃。1小时后,将反应小瓶冷却至并用饱和nahco3水溶液淬灭并用etoac萃取(4x)。将合并的有机层经硫酸钠干燥并在真空中浓缩。通过硅胶色谱纯化(梯度:0-12%于dcm中的meoh),得到产物(2s,4s,4's,6s)-2'-氯-4',5',5'-三氘化-2-甲基-6-(1-甲基三唑-4-基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-醇385(24.3mg,61%产率)。1h nmr(400mhz,氯仿-d)δ7.45(s,1h),6.82(s,1h),4.38(dd,j=11.7,2.6hz,1h),4.05(s,3h),3.47(s,1h),3.37(dqd,j=12.7,6.4,2.5hz,1h),2.40(dt,j=13.9,2.6hz,1h),2.03(dt,j=13.5,2.6hz,1h),1.69(dd,j=13.9,11.8hz,1h),1.50(dd,j=13.5,11.3hz,1h),1.13(d,j=6.3hz,3h)。lcms m/z 358.13[m+h]
+
.
[1724]
化合物476-1的制备
[1725][1726]
向500ml三颈rbf中添加甲苯(125ml)和丙炔酸甲酯(10.09g,1.0当量)。在室温下缓慢添加tmsn3(27.65g,2.0当量)。将混合物加热至85-90℃持续24小时。将溶液冷却至0℃并用thf(200ml)稀释。将固体nano2(10.76g,1.3当量)添加到混合物中。向其中逐滴添加2.4m hcl(水溶液)(63ml,1.3当量),同时将内部温度保持在0-10℃下。添加后,将混合物在0-10℃下搅拌1小时。分离各层,并将水层用thf(2x 200ml)萃取。将合并的有机萃取物用3% nahco3(水溶液)(100ml)洗涤。将水层用thf(2x 100ml)萃取。将合并的有机萃取物经na2so4干燥,过滤,并在减压下浓缩。将粗残余物用己烷(200ml)洗涤并在室温下搅拌1小时。将混合物过滤,并将固体用己烷(2x 100ml)洗涤,得到白色粉末(12.3g,81%产率)。1h nmr(400mhz,dmso):δ8.53(s,1h),3.83(s,3h)。
[1727]
化合物477-1a的制备
[1728][1729]
向500ml三颈圆底烧瓶中添加thf(200ml)、476-1(14.8g,1.0当量)和na2co3(18.6g,1.5当量),随后缓慢分批添加
13
cd3i(20.0g,1.2当量)。将混合物在25℃下搅拌48小时。在反应完成后(tlc 1:1乙酸乙酯:己烷),通过na2so4垫过滤混合物。将滤饼用dcm和thf冲洗。将滤液在真空下浓缩,随后溶解于dcm(145ml)中并用5% na2so3(水溶液)(80ml)洗涤。将水相用dcm(4x 80ml)萃取。将合并的有机萃取物浓缩并用己烷(6x 300ml)洗涤。通过过滤收集固体并干燥,得到呈白色粉末的477-1a(8.4g,50.6%产率)。1h nmr(400mhz,dmso):δ8.70(d,j=0.8hz,lh),3.82(s,3h),3.32(s,3h)。
[1730]
化合物497a的制备
[1731][1732]
向圆底烧瓶中添加477-1a(10.0g,68.9mmol)和240ml无水dcm。将反应混合物冷却至-70℃。通过添加漏斗添加dibal(137ml,1m于dcm,137.8mmol)。将反应混合物在-70℃下搅拌2小时。在-70℃下通过添加漏斗添加甲醇(35ml)并搅拌1小时。缓慢添加酒石酸钠(40%,170ml)。混合物于室温搅拌过夜。将dcm层分离并将水层用dcm(4x 150ml)萃取。将合并的有机萃取物浓缩,并添加己烷(150ml,产生白色浆液,将其过滤并干燥,得到呈白色固体的497a(7.5g,94%产率)。1h nmr(400mhz,cdcl3)δ10.14(s,1h)8.09(s,1h)。
[1733]
化合物495a的制备
[1734][1735]
向圆底烧瓶中添加tbuoh(28.2g,380.7mmol)、[2-13
c]丙二酸(20.0g,190.4mmol)和mecn(300ml)。在5℃下在冰水浴中添加于mecn(300ml)中的dcc(43.2g,209.4mmol)。将白色浆液在室温下搅拌过夜。将反应混合物过滤并用mecn(2x 30ml)洗涤。将滤液浓缩,得到黄色油状物,通过硅胶快速色谱(100% dcm to 4%于dcm中的meoh)纯化,提供呈无色油状物的495a(20.7g,67%产率)。1h nmr(400mhz,cdcl3)δ3.35(d,j=132hz,2h),1.49(s,9h)。
[1736]
化合物496a的制备
[1737][1738]
向化合物495a(19.50g,121.0mmol)于thf(150ml)中的溶液中分批添加mg(otbu)2(11.46g,60.50mmol)。在每次添加后,观察到到少量放热,其中圆底烧瓶升温至触摸温度(~30℃)。混合物于室温搅拌过夜。3小时后,反应物是澄清的淡黄色溶液。将混合物经由旋转蒸发仪浓缩,并将残余物溶于乙醚(50ml)中并再次浓缩。将此重复3次。将所得固体在真空下干燥,得到灰白色固体(21.20g,102%产率)。在单个烧瓶中,将thf(230ml)中的化合物479(22.13g,108.9mmol)用cdi(19.04g,117.4mmol)分三批处理。将反应物在室温下在氮气下搅拌并观察到气体逸出。3小时后,添加先前制备的固体镁盐(21.20g)并将反应物在室温下搅拌过夜。将反应混合物倒入到1n hcl(270ml)中并用etoac(2x 200ml)萃取。将合并的有机萃取物用饱和碳酸氢钠(200ml)和盐水(200ml)洗涤,经硫酸钠干燥,并通过旋转蒸发仪浓缩,得到棕色油状物(粗制物,35.5g)。将该材料溶于thf(100ml)并用1nnaoh(100ml)处理并在室温下搅拌0.5小时。将该溶液用水(300ml)稀释并用etoac(3x 300ml)萃取。将合并的有机萃取物用0.5hcl(100ml)和盐水(200ml)洗涤,经硫酸钠干燥,并通过旋转蒸发仪浓缩,得到呈淡黄色油状物化合物496a(28.0g,85%产率)。1h nmr(400mhz,dmso-d6):δ6.75(d,j=7.2hz,1h),3.83(p,j=7.2hz,1h),3.43(d,j=132hz,2h),2.66-2.53(m,2h),1.39(s,9h),1.35(s,9h),1.00(d,j=6.8hz,3h)。
[1739]
化合物498a的制备
[1740][1741]
向在室温下化合物496a(28.0g,92.6mmol)于dcm(110ml)中的溶液中添加tfa(28.4ml,370.4mmol)。20小时后,tlc显示反应完成。将混合物经由旋转蒸发仪浓缩,并将残余物溶于dcm(100ml)并再次浓缩。将此重复3次。将所得固体在真空下干燥,得到粉色固体(35.0g)。将固体立即溶解于dcm(220ml(超声处理以溶解所有固体))并用化合物497a(10.13g,88.0mmol)处理。将反应物在室温下搅拌过夜。tlc显示497a完全消耗和产物形成。将反应物用饱和碳酸氢钠(270ml)淬灭并用dcm(3x 300ml)萃取。将有机萃取物用盐水(200ml)洗涤并经由旋转蒸发仪浓缩,得到粗制固体(23.0g)。将粗制固体在mtbe(70ml)中在室温下研磨1小时。经由真空过滤收集白色固体,用冷mtbe(2x 20ml)洗涤,并在高真空下干燥,得到呈白色固体的化合物498a(14.1g,53%产率)。1h nmr(400mhz,cdcl3):δ7.46(s,1h),4.50(d,j=10.8hz,1h),3.59(dd,j=11.2,132hz,1h),3.21-3.16(m,1h)2.55-2.50(m,1h),2.22-2.16(m,1h),2.00-1.90(br,1h),1.37(s,9h),1.27(d,j=6.4hz,3h)。
[1742]
制备化合物499a和500a
[1743][1744]
向化合物498a(14.1,47.1mmol)于dcm(180ml)中的溶液中添加msoh(15.3ml,235.5mmol)。将反应物加热至40℃过夜并通过tlc监测,直到反应完成,得到化合物499a。将反应混合物冷却至室温并用饱和碳酸氢钠溶液淬灭,分离各层,并将水层用dcm(5x 120ml)萃取。将合并的有机萃取物经由旋转蒸发仪浓缩,得到呈淡黄色固体的化合物499a(粗制物,9.15g)。将粗制固体在mtbe(60ml)中研磨3小时,然后过滤,得到白色固体(8.30g),通过硅胶快速色谱(2-3%于dcm中的meoh)进一步纯化所述固体,得到纯的化合物500a(6.80g 72%产率)。1h nmr(400mhz,cdcl3):δ7.45(s,1h),4.23(m,1h),3.18-3.10(m,1h),2.84-2.72(m,1h),2.49-2.41(m,1h+2x 0.5h),2.21-2.14(m,2x 0.5h),2.06-1.90(br s 1h),1.25(d,j=6.0hz,3h)。
[1745]
化合物386
[1746]
(2s,4s,4's,6s)-2-甲基-6-(1-(1
13
c)甲基三唑-4-基)-2'-(三氟甲基)螺[(5
13
c)嗪烷-4,7'-4,5-二氢噻吩并[2,3-c]吡喃]-4'-醇(386)
[1747][1748]
步骤1.(2s,4s,6s)-2-甲基-6-(1-(甲基-13
c-d3)-1h-1,2,3-三唑-4-基)-2'-(三氟甲基)-4',5'-二氢螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-5-13
c(c140)的合成
[1749]
向装备磁性搅拌器、加热套、温度探针、水冷却回流冷凝器和氮气入口/出口的500ml 3颈烧瓶中装入(2s,6s)-2-甲基-6-(1-(甲基-13
c-d3)-1h-1,2,3-三唑-4-基)哌啶-4-酮-5-13
c c139(10.4g,52.20mmol)和二氯甲烷(180ml),并搅拌5分钟。将2-[5-(三氟甲基)-3-噻吩基]乙醇(12.5g,63.71mmol)添加到混合物中,随后添加甲磺酸(24ml,369.8mmol)。将所得反应混合物升温至40℃,并在该温度下搅拌5天。将反应混合物用冰/水浴冷却至0℃,并用6n naoh溶液碱化,直到ph=11.5。将反应混合物用dcm(2x 100ml)萃取。将合并的有机级分经mgso4干燥,过滤,并在减压下浓缩。将粗残余物用mtbe(200ml)处理,老化1小时,并且然后通过中等烧结漏斗过滤,用mtbe(50ml)洗涤,并在真空下干燥,得到一批12g呈白色固体的所需产物。将母液在减压下浓缩。通过硅胶色谱纯化(梯度:0-20% meoh:dcm),得到第二批5.7g呈白色固体的所需产物。将两批分离的产物合并,得到呈白色固体的(2s,4s,6s)-2-甲基-6-(1-(甲基-13
c-d3)-1h-1,2,3-三唑-4-基)-2'-(三氟甲基)-4',5'-二氢螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-5-13
c(17.7g,90%)c140。1h nmr(400mhz,氯仿-d)δ7.44(t,j=0.7hz,1h),7.12(q,j=1.2hz,1h),4.44(dt,j=11.8,2.8hz,1h),4.04-3.89(m,2h),3.33(dtd,j=12.7,6.4,2.5hz,1h),2.79-2.61(m,2h),2.38(ddt,j=131.3,13.5,2.6hz,1h),2.14-2.04(m,1h),2.05-1.57(m,1h),1.49(dd,j=13.7,11.3hz,1h),1.13(d,j=6.3hz,3h)。
19
f nmr(376mhz,氯仿-d)δ-55.30。lcms m/z 378.07[m+1]
+
.
[1750]
步骤2.2,2,2-三氟-1-((2s,4s,6s)-2-甲基-6-(1-(甲基-13
c-d3)-1h-1,2,3-三唑-4-基)-2'-(三氟甲基)-4',5'-二氢螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-1-基-5-13
c)乙-1-酮(c141)的合成
[1751]
向装备磁性搅拌器、温度探针和氮气入口/出口的500ml 3颈烧瓶中装入(2s,4s,
6s)-2-甲基-6-(1-(1
13
c)甲基三唑-4-基)-2'-(三氟甲基)螺[(5
13
c)嗪烷-4,7'-4,5-二氢噻吩并[2,3-c]吡喃](17.5g,46.37mmol)和dcm(200ml),搅拌5分钟,并且然后用冰/水浴冷却至0℃。向混合物中添加n-乙基-n-异丙基-丙-2-胺(13ml,74.63mmol),随后添加(2,2,2-三氟乙酰基)2,2,2-三氟乙酸酯(7.5ml,53.96mmol)。将所得反应混合物在该温度下搅拌1小时。将反应混合物用饱和nahco3溶液(100ml)淬灭。分离各层,并将有机层用2m hcl水溶液(2x60ml)、水(100ml)、盐水(100ml)洗涤,然后经mgso4干燥,过滤并浓缩。通过硅胶色谱纯化(0-100% etoac:庚烷),提供呈白色泡沫的2,2,2-三氟-1-((2s,4s,6s)-2-甲基-6-(1-(甲基-13
c-d3)-1h-1,2,3-三唑-4-基)-2'-(三氟甲基)-4',5'-二氢螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-1-基-5-13
c)乙-1-酮c141(21g,96%)。1hnmr(400mhz,氯仿-d)δ7.59(s,1h),7.12(d,j=1.3hz,1h),5.60(s,1h),4.52-4.32(m,1h),3.91(t,j=5.5hz,2h),3.24(ddd,j=130.5,14.8,7.4hz,1h),2.88-2.59(m,2h),2.55-2.38(m,1h),2.07(d,j=21.9hz,1h),1.49-1.00(m,3h)。
19
f nmr(376mhz,氯仿-d)δ-55.41,-68.97。lcms m/z 474.02[m+1]
+
.
[1752]
步骤3.(2s,4s,6s)-2-甲基-6-(1-(甲基-13
c-d3)-1h-1,2,3-三唑-4-基)-1-(2,2,2-三氟乙酰基)-2'-(三氟甲基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'(5'h)-酮-5-13
c(c142)的合成
[1753]
第一步骤-光化学溴化:向装备磁性搅拌器、100w cfl光源和氮气入口/出口的1l烧瓶中装入2,2,2-三氟-1-((2s,4s,6s)-2-甲基-6-(1-(甲基-13
c-d3)-1h-1,2,3-三唑-4-基)-2'-(三氟甲基)-4',5'-二氢螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-1-基-5-13
c)乙-1-酮c141(21g,44.36mmol)和乙腈(400ml)。将所得反应混合物经由气体分配管用氮气流脱气15分钟。向混合物中添加n-溴琥珀酰亚胺(10.2g,57.31mmol),随后添加aibn(200mg,1.218mmol)。将所得反应混合物在100w cfl辐照下搅拌4小时。将反应混合物用10重量%亚硫酸氢钠水溶液(200ml)淬灭,搅拌10分钟,并且然后添加mtbe(300ml)。将有机相分离,用饱和碳酸氢钠水溶液(200ml)和盐水(200ml)洗涤,经mgso4干燥,过滤,并浓缩,得到呈褐色泡沫状的1-((2s,4s,6s)-4'-溴-2-甲基-6-(1-(甲基-13
c-d3)-1h-1,2,3-三唑-4-基)-2'-(三氟甲基)-4',5'-二氢螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-1-基-5-13
c)-2,2,2-三氟乙-1-酮(25g,102%)。该粗制材料不经进一步纯化即进入下一步骤。lcms m/z 553.94[m+1]
+
[1754]
第二步骤-kornblum氧化:将1-((2s,4s,6s)-4'-溴-2-甲基-6-(1-(甲基-13
c-d3)-1h-1,2,3-三唑-4-基)-2'-(三氟甲基)-4',5'-二氢螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-1-基-5-13
c)-2,2,2-三氟乙-1-酮(25g,102%)和二甲基亚砜(200ml)的混合物搅拌5分钟。向反应混合物中添加et3n(45ml,322.9mmol)。将所得反应混合物升温至75℃并搅拌3小时。将反应混合物冷却至室温,分配于mtbe(~400ml)、饱和nahco3水溶液(~200ml)与水(~400ml)之间,并搅拌10分钟。将有机相分离,并将水层用mtbe(2x 200ml)萃取。将合并的有机相用饱和nahco3溶液(~200ml)洗涤,经mgso4干燥,过滤,并浓缩,产生呈棕褐色泡沫的由以下的5:1比率的混合物组成的产物:酮(2s,4s,6s)-2-甲基-6-(1-(甲基-13
c-d3)-1h-1,2,3-三唑-4-基)-1-(2,2,2-三氟乙酰基)-2'-(三氟甲基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'(5'h)-酮-5-13
c c142和醇2,2,2-三氟-1-((2s,4s,6s)-4'-羟基-2-甲基-6-(1-(甲基-13
c-d3)-1h-1,2,3-三唑-4-基)-2'-(三氟甲基)-4',5'-二氢螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-1-基-5-13
c)乙-1-酮(21g,97%)于。酮和醇(5:1)的该粗制材料不经进一步纯化
即进入下一步骤。lcms m/z487.95[m+1]
+
.
[1755]
第三步骤
–
氧化:将来自第二步骤的粗材料溶液(21g,42.91mmol)(酮和醇的混合物,~5:1比率)和dcm(200ml)搅拌5分钟并且然后冷却至0℃。向反应混合物中添加固体nahco3(2.4g,28.57mmol),随后添加水(25ml)中的kbr(1.5g,12.60mmol)。向反应混合物中添加4-乙酰胺基-tempo(480mg,2.250mmol),随后在30分钟内非常缓慢地添加naocl(30ml 12%(w/w),58.32mmol),同时将内部温度保持低于7℃。将所得反应混合物搅拌1小时。将反应混合物用1m na2s2o3水溶液(100ml)淬灭。将有机相分离,用饱和nahco3水溶液(100ml)和盐水(100ml)洗涤,经mgso4干燥,并且然后过滤并浓缩。通过硅胶色谱纯化(梯度:0-100% etoac:庚烷),得到呈白色固体的(2s,4s,6s)-2-甲基-6-(1-(甲基-13
c-d3)-1h-1,2,3-三唑-4-基)-1-(2,2,2-三氟乙酰基)-2'-(三氟甲基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'(5'h)-酮-5-13
c c142(14g,66%)。1h nmr(400mhz,氯仿-d)δ7.73(q,j=1.2hz,1h),7.62(s,1h),5.65(s,1h),4.47(p,j=7.1hz,1h),4.36(d,j=4.4hz,2h),3.40(ddd,j=131.1,15.0,5.8hz,1h),2.99(dd,j=15.0,8.5hz,1h),2.65(dd,j=15.1,8.5hz,1h),2.23(d,j=23.2hz,1h),1.26(s,3h)。
19
f nmr(376mhz,氯仿-d)δ-55.96,-68.87。lcms m/z 488.01[m+1]
+
[1756]
步骤4.2,2,2-三氟-1-((2s,4s,4's,6s)-4'-羟基-2-甲基-6-(1-(甲基-13
c-d3)-1h-1,2,3-三唑-4-基)-2'-(三氟甲基)-4',5'-二氢螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-1-基-5-13
c)乙-1-酮(c143)的合成
[1757]
在mecn(120ml)中制备二氯(五甲基环戊二烯基)铑(iii)二聚体(72mg,0.1146mmol)和(r,r)-tsdpen(90mg,0.2456mmol)的溶液并且然后在室温下搅拌1小时。一次性添加甲酸-et3n(15.8ml,237.0mmol,5:2商用溶液),并然后将反应物冷却至-15℃。
[1758]
在单个烧瓶中,将acn(120ml)中的(2s,4s,6s)-2-甲基-6-(1-(甲基-13
c-d3)-1h-1,2,3-三唑-4-基)-1-(2,2,2-三氟乙酰基)-2'-(三氟甲基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'(5'h)-酮-5-13
c c142(14g,28.44mmol)冷却至-15℃,并且然后添加到先前制备的溶液中,同时将内部温度小心保持在-17℃与-20℃之间。将所得反应混合物升温至-10℃并在该温度下搅拌6小时。将反应混合物用饱和nahco3溶液(200ml)淬灭并在室温下搅拌6小时。然后将反应混合物用mtbe(2x 200ml)萃取。将合并的有机相用1n hcl(200ml)和盐水(200ml)洗涤,经硫酸钠干燥,过滤,并浓缩。将残余物溶解于dcm(~200ml)中,用3-巯基丙基乙基硫化物二氧化硅(spm32f金属清除树脂)(6g)处理,在室温下搅拌2小时,并且然后过滤并用dcm(60ml)洗涤。将合并的滤液浓缩。通过硅胶色谱纯化(梯度:0-100% etoac:庚烷),得到呈白色泡沫的2,2,2-三氟-1-((2s,4s,4's,6s)-4'-羟基-2-甲基-6-(1-(甲基-13
c-d3)-1h-1,2,3-三唑-4-基)-2'-(三氟甲基)-4',5'-二氢螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-1-基-5-13
c)乙-1-酮c143(13g,93%)。1h nmr(400mhz,氯仿-d)δ7.60(s,1h),7.38(q,j=1.2hz,1h),5.57(s,1h),4.58(dt,j=9.1,3.3hz,1h),4.50(s,1h),3.99(dd,j=12.4,3.2hz,1h),3.89(dd,j=12.4,3.4hz,1h),3.17(ddd,j=131.1,15.2,7.0hz,1h),2.68(ddd,j=132.9,15.2,8.3hz,2h),2.20(s,1h),2.10(d,j=9.1hz,1h),1.33(s,3h)。
19
f nmr(376mhz,氯仿-d)δ-55.56,-68.96。lcms m/z 490.05[m+1]
+
.
[1759]
步骤5.(2s,4s,4's,6s)-2-甲基-6-(1-(甲基-13
c-d3)-1h-1,2,3-三唑-4-基)-2'-(三氟甲基)-4',5'-二氢螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-5-13
c-4'-醇(386)的合成
[1760]
将2,2,2-三氟-1-((2s,4s,4's,6s)-4'-羟基-2-甲基-6-(1-(甲基-13
c-d3)-1h-1,2,3-三唑-4-基)-2'-(三氟甲基)-4',5'-二氢螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-1-基-5-13
c)乙-1-酮c143(13g,26.56mmol)和meoh(120ml)中的溶液搅拌5分钟。向反应混合物中一次性添加naoh水溶液(45ml 6m,270.0mmol)。将所得反应混合物升温至60℃并在该温度下搅拌1小时。将反应混合物冷却至室温,然后分配于冷水(100ml)与mtbe(200ml)之间并搅拌20分钟。然后将有机相分离。将水相用mtbe(2x 100ml)萃取。将合并的有机相用冷水(60ml)、盐水(100ml)洗涤,经mgso4干燥,过滤,并浓缩。将残余物在真空烘箱下在80℃下干燥14小时,得到呈白色固体的(2s,4s,4's,6s)-2-甲基-6-(1-(甲基-13
c-d3)-1h-1,2,3-三唑-4-基)-2'-(三氟甲基)-4',5'-二氢螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-5-13
c-4'-醇386(9.5g,90%)。1h nmr(400mhz,甲醇-d4)δ7.83(t,j=0.7hz,1h),7.46(q,j=1.2hz,1h),4.58(t,j=3.9hz,1h),4.35(dt,j=11.8,2.9hz,1h),4.08(dd,j=12.2,3.6hz,1h),3.86(dd,j=12.2,4.2hz,1h),3.35(ddd,j=11.5,6.3,3.2hz,1h),2.64-2.17(m,2h),1.78(ddd,j=127.7,13.8,11.8hz,1h),1.53(dd,j=13.6,11.4hz,1h),1.17(d,j=6.4hz,3h)。
19
f nmr(376mhz,甲醇-d4)δ-56.94.lcms m/z 394.1[m+1]
+
.
[1761]
s63的制备
[1762]
(2s,3r,6r)-2-(1-甲基-1h-1,2,3-三唑-4-基)-4-氧代-6-苯基哌啶-3-羧酸叔丁酯
[1763][1764]
步骤1.向(3r)-3-(叔丁氧基羰基氨基)-3-苯基-丙酸c144(500mg,1.885mmol)于thf(4.5ml)中的溶液中添加cdi(340mg,2.097mmol)并将混合物在室温下搅拌2.5小时。然后添加双[(3-叔丁氧基-3-氧代-丙酰基)氧基]镁c109(390mg,1.138mmol),并继续在室温下搅拌20小时。将反应物用tbme(10ml)和1n hcl(3ml)稀释。将有机层分离并用饱和碳酸氢钠(3ml)、盐水(3ml)洗涤。将有机层用硫酸镁干燥,过滤,并浓缩,产生呈白色结晶固体的(2s,3r,6r)-2-(1-甲基-1h-1,2,3-三唑-4-基)-4-氧代-6-苯基哌啶-3-羧酸叔丁酯s63,其立即用于步骤2。
[1765]
步骤2.向溶解于dcm(4ml)中的来自第一步骤的白色结晶固体中添加tfa(900μl,11.68mmol)。将混合物搅拌1小时,并且然后将混合物浓缩并通过dcm(3x 4ml)共沸,提供(5r)-5-氨基-3-氧代-5-苯基-戊酸叔丁酯的粗制混合物,其直接用于步骤3。
[1766]
步骤3.向来自步骤2的溶解于dcm(4ml)的粗制混合物中添加1-甲基三唑-4-甲醛s17(225mg,2.025mmol)并将混合物在室温下搅拌2小时。将混合物用饱和碳酸氢钠(2ml)淬灭。添加6n naoh以将ph调节至》9。将有机层分离并浓缩。通过硅胶色谱纯化(梯度:0-10% meoh:dcm),得到(2s,3r,6r)-2-(1-甲基三唑-4-基)-4-氧代-6-苯基哌啶-3-羧酸叔丁酯s63(347mg,52%)。观察到这种产物为酮和烯醇互变异构体的混合物。lcms m/z 357.23[m+
h]
+
[1767]
s64的制备
[1768]
1-[(2's,6'r,7s)-2-氯-2'-(1-甲基三唑-4-基)-6'-苯基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(s64)
[1769][1770]
步骤1.(2s,6r)-2-(1-甲基三唑-4-基)-6-苯基-哌啶-4-酮(c145)的合成
[1771]
向溶解于dcm(5ml)中的(2s,3r,6r)-2-(1-甲基三唑-4-基)-4-氧代-6-苯基-哌啶-3-羧酸叔丁酯s63(320mg,0.8978mmol)的混合物中添加msoh(300μl,4.623mmol)。将混合物回流1小时,然后冷却至室温并用饱和碳酸氢钠(5ml)淬灭。将有机层分离,并将水层用另外的dcm(3x 5ml)萃取。将合并的有机层用mgso4干燥,过滤,并浓缩。通过硅胶色谱纯化(0-10% meoh:dcm),得到呈黄色油状物的(2s,6r)-2-(1-甲基三唑-4-基)-6-苯基-哌啶-4-酮c145(98mg,43%)。1h nmr(400mhz,氯仿-d)δ7.53(s,1h),7.46-7.42(m,2h),7.40-7.30(m,3h),4.41(dd,j=9.8,5.2hz,1h),4.18-4.14(m,1h),4.13(s,3h),2.84-2.72(m,2h),2.65(d,j=7.6hz,2h)。
[1772]
步骤2.(2's,6'r,7s)-2-氯-2'-(1-甲基三唑-4-基)-6'-苯基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](c146)的合成
[1773]
向溶解于dcm(1000μl)中的(2s,6r)-2-(1-甲基三唑-4-基)-6-苯基-哌啶-4-酮c145(49mg,0.1912mmol)和2-(5-氯-3-噻吩基)乙醇s2(40mg,0.2459mmol)的混合物中添加msoh(90μl,1.387mmol)。将混合物回流过夜并且然后冷却至室温并用饱和碳酸氢钠(1ml)淬灭。通过硅胶色谱(梯度:0-10%meoh:dcm)直接纯化有机层,产生呈白色结晶固体的(2's,6'r,7s)-2-氯-2'-(1-甲基三唑-4-基)-6'-苯基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]c146(46mg,57%)。1h nmr(400mhz,氯仿-d)δ7.48(s,1h),7.45-7.38(m,2h),7.36-7.29(m,2h),7.24(td,j=5.3,4.8,2.4hz,1h),6.58(s,1h),4.58(dd,j=11.7,2.6hz,1h),4.32(dd,j=11.6,2.5hz,1h),4.06(s,3h),4.02(t,j=5.5hz,2h),2.72-2.54(m,2h),2.44(dt,j=13.6,2.6hz,1h),2.22(dt,j=13.6,2.6hz,1h),1.86(ddd,j=21.7,
13.6,11.7hz,3h)。lcms m/z 401.15[m+1]
+
[1774]
步骤3.1-[(2's,6'r,7s)-2-氯-2'-(1-甲基三唑-4-基)-6'-苯基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(s64)的合成
[1775]
将溶解于dcm(1ml)中的(2's,6'r,7s)-2-氯-2'-(1-甲基三唑-4-基)-6'-苯基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]c146的溶液冷却至0℃。添加dipea(25μl,0.1435mmol),随后添加tfaa(20μl,0.1439mmol)。将混合物搅拌20min,并且然后用1n hcl(1ml)淬灭。使有机层穿过相分离器。通过硅胶色谱纯化(0-50% etoac:庚烷),得到呈澄清油状物的1-[(2's,6'r,7s)-2-氯-2'-(1-甲基三唑-4-基)-6'-苯基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮s64(52mg,52%)。lcms m/z 497.26[m+1]
+
[1776]
化合物387
[1777]
(2's,4s,6'r,7s)-2-氯-2'-(1-甲基三唑-4-基)-6'-苯基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(387)
[1778][1779]
步骤1.(2s,4s,6r)-2'-氯-2-(1-甲基三唑-4-基)-6-苯基-1-(2,2,2-三氟乙酰基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮(c147)的合成
[1780]
在氮气气氛下向1-[(2's,6'r,7s)-2-氯-2'-(1-甲基三唑-4-基)-6'-苯基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮s64(52mg,0.09901mmol)于氯苯(1ml)中的溶液中添加5,5-二甲基-1,3-二溴乙内酰脲(20mg,0.06995mmol)和2-[(e)-(1-氰基-1-甲基-乙基)偶氮基]-2-甲基-丙腈(1.5mg,0.009135mmol)。将混合物加热至75℃持续15分钟。将混合物冷却至室温并用饱和碳酸氢钠(1ml)淬灭。将有机层分离,用mgso4干燥,过滤,并浓缩,提供粗制泡沫。
[1781]
在氮气气氛下将粗制泡沫溶解于dmso(1ml)中。将混合物加热至60℃并添加三乙胺(75μl,0.5381mmol)。将深棕色溶液加热至65℃并搅拌75分钟。将混合物冷却至室温,并用乙酸乙酯(5ml)和水(5ml)稀释。将水层用另外的乙酸乙酯(2x 5ml)洗涤。将合并的有机层用na2so4干燥,过滤,并浓缩。通过硅胶色谱纯化(梯度:0-50% etoac:庚烷),得到呈淡黄色固体的(2s,4s,6r)-2'-氯-2-(1-甲基三唑-4-基)-6-苯基-1-(2,2,2-三氟乙酰基)螺[哌
啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮c147(21mg,39%)。lcms m/z 511.21[m+1]
+
[1782]
步骤2.1-[(2's,4s,6'r,7s)-2-氯-4-羟基-2'-(1-甲基三唑-4-基)-6'-苯基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮(c148)的合成
[1783]
向5:2甲酸-三乙胺络合物(20μl,0.04763mmol)于mecn(500μl)中的混合物中添加溶解于mecn(50μl)中的n-[(1r,2r)-2-氨基-1,2-二苯基-乙基]-4-甲基-苯磺酰胺(0.12mg,3.274e-4
mmol)和二氯(五甲基环戊二烯基)铑(iii)二聚体(0.1mg,1.592e-4
mmol)的溶液。5分钟后,将所得溶液添加到-10℃的(2s,4s,6r)-2'-氯-2-(1-甲基三唑-4-基)-6-苯基-1-(2,2,2-三氟乙酰基)螺[哌啶-4,7'-噻吩并[2,3-c]吡喃]-4'-酮c147(21mg,0.03908mmol)于mecn(500μl)中的冷却溶液中。将反应混合物缓慢升温至室温并搅拌过夜。将混合物浓缩并通过硅胶色谱(梯度:0-50% etoac:庚烷)纯化,产生呈白色固体的1-[(2's,4s,6'r,7s)-2-氯-4-羟基-2'-(1-甲基三唑-4-基)-6'-苯基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c148。lcms m/z 514.94[m+1]
+
[1784]
步骤3.(2's,4s,6'r,7s)-2-氯-2'-(1-甲基三唑-4-基)-6'-苯基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇(387)的合成
[1785]
将溶解于meoh(250μl)中的1-[(2's,4s,6'r,7s)-2-氯-4-羟基-2'-(1-甲基三唑-4-基)-6'-苯基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-1'-基]-2,2,2-三氟-乙酮c148的溶液加热至60℃。然后添加naoh(75μl 6m,0.4500mmol),并继续回流3小时。将混合物用mtbe(3ml)和饱和氯化铵(3ml)稀释。将有机层分离,并将水层用另外的mtbe(2x 3ml)萃取。使合并的有机层穿过相分离器并浓缩,产生呈白色固体的(2's,4s,6'r,7s)-2-氯-2'-(1-甲基三唑-4-基)-6'-苯基-螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]-4-醇387(13.8mg,82%)。1hnmr(400mhz,甲醇-d4)δ7.88(d,j=3.2hz,1h),7.48-7.41(m,2h),7.37-7.30(m,2h),7.30-7.21(m,1h),6.88(s,1h),4.60-4.47(m,2h),4.37(dd,j=11.8,2.6hz,1h),4.12(dt,j=11.5,3.4hz,1h),4.07(s,3h),3.93-3.84(m,1h),2.49(dt,j=13.9,2.6hz,1h),2.35-2.22(m,1h),1.89(ddd,j=13.8,11.8,1.5hz,2h)。lcms m/z 417.26[m+1]
+
.
[1786]
s65的制备
[1787]
2-(二氟甲基)-6-(1-甲基三唑-4-基)哌啶-4-酮(s65)
[1788][1789]
向冷却至0℃的4-氨基-5,5-二氟-戊-2-酮(盐酸盐)c149(250mg,1.440mmol)于乙醇(15ml)中的溶液中添加1-甲基三唑-4-甲醛s17(175mg,1.512mmol)、l-脯氨酸(35mg,0.3040mmol)和et3n(210μl,1.507mmol)。将混合物升温至室温并72小时。将混合物浓缩,并溶解于dcm(10ml)和饱和碳酸氢钠(5ml)中。将水层用另外的dcm(2x 10ml)萃取,并将合并
的有机层浓缩。通过硅胶色谱纯化(梯度:0-10% meoh:dcm),得到呈黄色油状物的2-(二氟甲基)-6-(1-甲基三唑-4-基)哌啶-4-酮s65(303mg,91%)。1h nmr(400mhz,氯仿-d)δ7.51(s,1h),5.78(tdd,j=56.0,16.1,4.3hz,1h),4.30(dt,j=9.8,4.7hz,1h),4.13(s,3h),3.51(d,j=5.0hz,1h),3.41(d,j=18.7hz,1h),2.79-2.69(m,1h),2.60(s,1h),2.43(dd,j=14.4,11.9hz,1h),2.30(s,1h)。lcms m/z 231.18[m+h]
+
.分离作为4:1dr混合物的产物。
[1790]
化合物388
[1791]
2-氯-2'-(二氟甲基)-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](388)
[1792][1793]
向2-(二氟甲基)-6-(1-甲基三唑-4-基)哌啶-4-酮s65(140mg,0.6081mmol)于dcm(3ml)中的溶液中添加2-(5-氯-3-噻吩基)乙醇s2(100μl,0.8085mmol),随后添加msoh(200μl,3.082mmol)。将混合物回流过夜。将混合物冷却至室温并用饱和碳酸氢钠淬灭。将有机层分离并浓缩。通过硅胶色谱纯化(梯度:0-10% meoh:dcm),得到呈黄色油状物的2-氯-2'-(二氟甲基)-6'-(1-甲基三唑-4-基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]388(139mg,53%)。1hnmr(400mhz,氯仿-d)δ7.47(s,1h),6.62(s,1h),5.66(td,j=56.5,5.2hz,1h),4.48(dd,j=11.8,2.7hz,1h),4.10(d,j=1.5hz,3h),3.98(t,j=5.5hz,2h),3.64-3.51(m,1h),2.65(td,j=5.5,3.1hz,2h),2.41-2.36(m,1h),2.24-2.16(m,1h),2.13(dd,j=14.8,6.6hz,1h),1.84(dd,j=13.6,11.8hz,1h)。lcms m/z 375.14[m+1]
+
.
[1794]
化合物389
[1795]
2'-(二氟甲基)-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶](389)
[1796][1797]
向2-(二氟甲基)-6-(1-甲基三唑-4-基)哌啶-4-酮s65(140mg,0.6081mmol)于dcm
(3ml)中的溶液中添加2-[5-(三氟甲基)-3-噻吩基]乙醇s3(160mg,0.8155mmol),随后添加msoh(200μl,3.082mmol)。将所得混合物回流48小时。将混合物冷却至室温并用饱和碳酸氢钠淬灭。将有机层分离并浓缩。通过反相hplc纯化(方法:c18 waters sunfire柱(30x150 mm,5微米).梯度:于具有0.1%三氟乙酸的h2o中的mecn),提供产物,通过用dcm稀释,随后进行饱和碳酸氢钠洗涤来中和所述产物。使溶液穿过相分离器,并将有机层干燥,产生呈澄清油状物的2'-(二氟甲基)-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[4,5-二氢噻吩并[2,3-c]吡喃-7,4'-哌啶]389(12mg,5%)。1h nmr(400mhz,氯仿-d)δ7.49(s,1h),7.16(q,j=1.1hz,1h),5.68(td,j=56.4,5.1hz,1h),4.51(dd,j=11.7,2.7hz,1h),4.10(s,3h),4.02(t,j=5.5hz,2h),3.64-3.54(m,1h),2.75(td,j=5.4,2.8hz,2h),2.43(dt,j=13.7,2.7hz,1h),2.25(dd,j=13.4,2.7hz,1h),1.91(dd,j=13.6,11.7hz,1h),1.72-1.66(m,1h)。lcms m/z 409.21[m+1]
+
[1798]
化合物390
[1799]
(2's,4s,6's,7r)-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[6,7-二氢噻吩并[3,2-c]吡喃-4,4'-哌啶]-7-醇(390)
[1800]
[1801]
步骤1.2,2,2-三氟-1-[(2's,4s,6's)-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[6,7-二氢噻吩并[3,2-c]吡喃-4,4'-哌啶]-1'-基]乙酮(c150)的合成在0℃下向(2's,4s,6's)-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[6,7-二氢噻吩并[3,2-c]吡喃-4,4'-哌啶]8(900mg,2.365mmol)(经由s26中间体使用“方法b”制备)和dipea(550μl,3.158mmol于dcm(15ml)中的溶液中逐滴添加tfaa(400μl,2.878mmol)。三十分钟后,将反应物用饱和碳酸氢钠溶液(10ml)稀释,并使混合物穿过相分离器,将其用dcm(2x 10ml)萃取。将有机物在真空中浓缩。通过硅胶色谱纯化(梯度:0-50%于庚烷中的etoac),得到呈白色泡沫的2,2,2-三氟-1-[(2's,4s,6's)-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[6,7-二氢噻吩并[3,2-c]吡喃-4,4'-哌啶]-1'-基]乙酮c150(1.03g,91%)。1h nmr(300mhz,氯仿-d)δ7.59(s,1h),7.39(s,1h),5.59(s,1h),4.42(q,j=7.0hz,1h),4.11(s,3h),3.92(t,j=5.5hz,2h),3.24(dd,j=14.8,6.5hz,1h),2.96-2.73(m,2h),2.48(dd,j=14.9,8.4hz,1h),2.40-1.93(m,2h),1.52-0.94(m,3h)。lcms m/z 469.07[m+h]
+
。
[1802]
步骤2.(2s,4s,6s)-2-甲基-6-(1-甲基三唑-4-基)-1-(2,2,2-三氟乙酰基)-2'-(三氟甲基)螺[哌啶-4,4'-噻吩并[3,2-c]吡喃]-7'-酮(c151)的合成
[1803]
向2,2,2-三氟-1-[(2's,4s,6's)-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[6,7-二氢噻吩并[3,2-c]吡喃-4,4'-哌啶]-1'-基]乙酮c150(1.03g,2.153mmol)于乙腈(18ml)中的混合物中添加n-羟基苯邻二甲酰亚胺(260mg,1.594mmol)和二乙酸钴四水合物(120mg,0.4818mmol)并且然后将混合物用氧气气球真空吹扫三次。将反应物加热至45℃并在氧气气球气氛下搅拌过夜。将混合物冷却至室温,用氮气真空吹扫三次,并且然后用水(10ml)和饱和碳酸氢盐水溶液(20ml)稀释。将混合物用dcm(3x 20ml)萃取,并将有机物经硫酸钠干燥并在真空中浓缩。通过硅胶色谱纯化(梯度:0-40%于庚烷中的etoac),得到呈白色泡沫的(2s,4s,6s)-2-甲基-6-(1-甲基三唑-4-基)-1-(2,2,2-三氟乙酰基)-2'-(三氟甲基)螺[哌啶-4,4'-噻吩并[3,2-c]吡喃]-7'-酮c151(440mg,42%)。1h nmr(300mhz,氯仿-d)δ8.03-7.38(m,2h),5.64(s,1h),4.51-4.30(m,3h),4.13(s,3h),3.48-3.35(m,1h),2.67(dd,j=15.2,8.5hz,1h),2.24(s,2h),1.47-0.91(m,3h)。lcms m/z 483.11[m+h]
+
。
[1804]
步骤3.2,2,2-三氟-1-[(2's,4s,6's,7r)-7-羟基-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[6,7-二氢噻吩并[3,2-c]吡喃-4,4'-哌啶]-1'-基]乙酮(c152)的合成
[1805]
向n-[(1r,2r)-2-氨基-1,2-二苯基-乙基]-4-甲基-苯磺酰胺(1.5mg,0.004093mmol)和1,2,3,4,5-五甲基环戊烷;四氯化铑(2+)(1mg,0.001592mmol)于acn(200μl)中的预混合溶液中添加甲酸(40μl,1.060mmol)和tea(50μl,0.3587mmol)的溶液。10分钟后,将混合物冷却至0℃并添加(2s,4s,6s)-2-甲基-6-(1-甲基三唑-4-基)-1-(2,2,2-三氟乙酰基)-2'-(三氟甲基)螺[哌啶-4,4'-噻吩并[3,2-c]吡喃]-7'-酮c151(80mg,0.1653mmol)于acn(1.5ml)中的溶液。将反应物在0℃下搅拌,并在一小时后,将其用饱和碳酸氢盐水溶液淬灭并通过相分离器用dcm(2x 3ml)萃取。将有机物在真空中浓缩。通过硅胶色谱纯化(梯度:0-30% etoac于dcm),得到呈无色膜状物的2,2,2-三氟-1-[(2's,4s,6's,7r)-7-羟基-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[6,7-二氢噻吩并[3,2-c]吡喃-4,4'-哌啶]-1'-基]乙酮c152(65mg,81%)。1h nmr(300mhz,氯仿-d)δ7.77-7.26(m,2h),5.57(s,1h),4.78-4.67(m,1h),4.48(d,j=7.9hz,1h),4.11(s,3h),3.94(ddd,j=
41.9,12.2,3.7hz,2h),3.16(dd,j=15.0,6.1hz,1h),2.52(dd,j=15.1,8.5hz,1h),2.44-1.98(m,3h),1.55-0.83(m,3h)。lcms m/z 485.09[m+h]
+
。
[1806]
步骤4.(2's,4s,6's,7r)-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[6,7-二氢噻吩并[3,2-c]吡喃-4,4'-哌啶]-7-醇(390)的合成
[1807]
向2,2,2-三氟-1-[(2's,4s,6's,7r)-7-羟基-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[6,7-二氢噻吩并[3,2-c]吡喃-4,4'-哌啶]-1'-基]乙酮c152(65mg,0.1331mmol)于meoh(1.3ml)中的溶液中添加naoh(900μl 2m,1.800mmol)并将混合物加热至50℃。50分钟后,将混合物冷却至室温,用水(2ml)稀释,并通过相分离器用dcm(2x 3ml)萃取。将有机物在真空中浓缩,得到透明膜状物。将残余物溶于dcm和庚烷中并汽提,得到呈白色泡沫的(2's,4s,6's,7r)-2'-甲基-6'-(1-甲基三唑-4-基)-2-(三氟甲基)螺[6,7-二氢噻吩并[3,2-c]吡喃-4,4'-哌啶]-7-醇390(48.3mg,91%)。1h nmr(300mhz,氯仿-d)δ7.45(s,1h),7.15(s,1h),4.69(s,1h),4.40(dt,j=11.8,1.9hz,1h),4.10-3.93(m,6h),3.45-3.33(m,1h),2.25(d,j=13.9hz,1h),1.91(d,j=13.6hz,1h),1.81-1.70(m,1h),1.58(dd,j=13.3,11.5hz,1h),1.14(d,j=6.3hz,3h)。lcms m/z 389.1[m+h]
+
。
[1808]
化合物391
[1809][1810]
步骤1.向化合物500a(8.84g,44.4mmol)和489a(9.50g,57.7mmol)于dcm(250ml)中的混合物中添加msoh(34.6ml,532.8mmol)。将混合物加热至40℃并搅拌过夜。tlc显示反
脱气5分钟。经由滴液漏斗将所得溶液逐滴添加到上述催化剂溶液中,在添加化合物504b期间将内部温度保持在约0℃下。在添加化合物504b后,将反应混合物在0℃至2℃(内部温度)下搅拌4小时。tlc显示反应几乎完成。添加饱和nahco3溶液(50ml),并移除冷却浴。将混合物搅拌2小时并用etoac(3x 50ml)萃取。将合并的有机萃取物用盐水(100ml)洗涤,经na2so4干燥,过滤,并浓缩至干燥。通过硅胶快速柱色谱纯化产物,用etoac/dcm(1/10至1/3)洗脱。将产物级分合并并在真空中浓缩,得到呈白色泡沫的产物505b(2.90g,93%产率)。hplc:98.3%。lcms:458.21(m+1)。1h nmr(400mhz,cdcl3):δ7.59(s,1h),6.83(s,1h),5.53(br s,1h),4.64(d,j=8.4hz,0.5h),4.47(m,1h),4.25(d,j=8.0hz 0.5h),4.16-4.00(m,2h),3.80(m,0.5h),3.65(m,0.5h),3.26(m,0.5h),2.93(m,0.5h),2.81(m,0.5h),2.51-2.45(m,1h),2.16-2.03(m,1.5h),1.29(br s,3h)。
[1815]
步骤6.向250ml烧瓶中装入溶解于meoh(100ml)和水(25ml)中的化合物505b(6.44g,14.07mmol)。添加固体naoh(6.5g,162.5mmol),并将搅拌的反应混合物升温至60℃(油浴温度)持续3小时。tlc指示反应已经完成。将反应混合物冷却至室温并然后浓缩至干燥。通过硅胶快速柱色谱使用meoh/dcm/nh4oh(7/93/0.5)纯化残余物,得到呈灰白色固体的化合物391(4.35g,86%产率)。hplc:99.4%。lcms:362.21(m+1)。1h nmr(400mhz,dmso-d6):δ7.88(s,1h),6.95(s,1h),5.37(t,j=3.2hz,1h),4.57(d,j=5.2hz,0.5h),4.20(d,j=5.2hz,0.5h),4.12-4.05(m,2h),3.82-3.71(m,1h),3.47-3.41(m,0.5h),3.15(d,j=4.8hz,1h),3.11-3.06(m,1h),2.33-1.97(m,1.5h),1.75-1.68(m,0.5h),1.43-1.36(m,0.5h),1.27-1.21(m,1h),1.01(d,j=6.4hz,3h)。
[1816]
固态nmr实验(化合物174和181形式)
[1817]
使用配备有bruker-biospin 4mm hfx探头的bruker-biospin 400mhz宽口径谱仪。将样品装填至4-mm zro2转子中并在魔角旋转(mas)条件下以通常设定为12.5khz的旋转速度旋转。使用1h mas t1饱和恢复弛豫实验测量质子弛豫时间以设定
13
c和
31
p交叉极化(cp)mas实验的适当循环延迟。使用
19
f mas t1饱和恢复弛豫实验测量氟弛豫时间以设定
19
f mas实验的适当循环延迟。将碳以及磷cpmas实验的cp接触时间设定为2ms。采用具有线性斜坡(50%至100%)的cp质子脉冲。在外部参考样品(甘氨酸)上优化碳hartmann-hahn匹配,同时在实际样品上优化磷hartmann-hahn匹配。使用tppm15去耦序列在大约100khz的场强下利用质子去耦记录所有碳、磷和氟谱。
[1818]
k2的制备
[1819][1820]
将thf(3720ml,6.2体积)装入到5l玻璃烧瓶中,然后在20℃下添加k1(600g,3.47mol,576.92ml,通过q-nmr得到92.6%纯度,1当量)。将混合物冷却至0℃并将mg(oet)2(198.46g,1.73mol,0.5当量)装入反应器中。将所得混合物在0-5下搅拌10分钟,然后升温至20℃并搅拌18小时,得到乳白色悬浮液。在40℃下在减压下蒸馏雾状溶液,以去除thf
koh溶液。装入156g 45% koh,导致ph为11-12。将混合物用dcm(6x 900ml)萃取。将合并的有机层经硫酸钠(300g)干燥并在真空下在25℃下浓缩,直到获得产物的雾状浆液。添加正庚烷(200ml),并将混合物在25℃下进一步浓缩,以去除溶剂(200ml)。将此过程重复三次。过滤所得溶液,并将滤饼用正庚烷(200ml)洗涤。将固体在真空下在40℃下干燥10小时,得到186g k7(92.9%产率)。
[1826]
k8的制备
[1827][1828]
步骤1(j2):在1000l反应器中在氮气下搅拌,将j1(85.0kg,663.1mol,1.0当量)溶解于dmf(162.3kg)中,并且然后冷却至-10℃-0℃。在单个500l反应器中,在氮气下搅拌,将nbs(122.7kg,689.6mol,1.04当量)溶解于dmf(241.0kg)中。在5小时内将nbs溶液缓慢添加到1000l反应器中,同时将温度保持在-10-0℃之间。在添加后,将反应混合物在-10-0℃下保持1-2小时。将饱和nacl水溶液(480kg)添加到反应混合物,随后添加etoac(460.7kg),并将反应混合物搅拌30分钟。将有机层分离并将水层用etoac(230.4kg)萃取。将有机层合并并用0.5n hcl(420.0kg)洗涤。分离后,添加饱和nacl溶液(300kg),并将混合物搅拌30分钟。分离各相并在40-50℃下浓缩有机层,得到呈棕色液体的j2(147.95kg,92.3%纯度,75% qnmr,80.78%产率)。
[1829]
步骤2(j3):在1000l反应器中将j2(147.95kg,qnmr 75%,535.8mol,1.0当量)用acoh(349.65kg)和ac2o(82.05kg,803.7mol,1.5当量)处理,同时在氮气下搅拌。将混合物加热至90-100℃持续5-10小时,并且直到通过gc保留少于0.5% j2。将混合物冷却至35-40℃。将nis(138.6kg,616.2mol,1.15当量)添加到1000l反应器中,并将混合物在35-40℃下搅拌6-10小时。当保留少于0.5%的中间体时,将混合物冷却至20℃-30℃并转移到3000l反应器中。添加mtbe/庚烷(250kg/226.4kg)和水(333kg)的混合物。将混合物搅拌30分钟并且然后分离。将水层用mtbe/庚烷(250kg/226.4kg)的混合物萃取。将有机层合并,并添加13% nahso3水溶液(510.6kg)。在将混合物搅拌30分钟后,分离各层,并将有机层用1m naoh
(461.8kg)和水(333kg)洗涤。将有机层在40-60℃下浓缩,得到呈棕色液体的j3(220.75kg,92.3%纯度,85.57%qnmr,94%产率)。
[1830]
步骤3(j4):在3000l反应器中将j3(111kg,85.57% qnmr,252.2mol,1.0当量)、cui(12.06kg,63.3mol,0.25当量)和2,6-二甲基砒啶(6.78kg,63.3mol,0.25当量)溶解于dmac(356.25kg)中,在氮气下搅拌并且然后加热至85-100℃。将氟磺酰基二氟乙酸甲酯(mfsda,194.65kg,1013.2mol,4.0当量)添加到3000l反应器中,同时将温度保持在85-100℃之间。在将反应混合物在90-95℃下保持1-4小时后,保留少于5.0% j3并将反应混合物冷却至5-15℃。在另一个3000l反应器中,装入水(1140kg)和正庚烷(439.3kg),并将混合物冷却至10-20℃。将反应物在10-20℃下淬灭到此反应器中,并将所得混合物搅拌30分钟。将各层过滤并且然后分离。将水相用正庚烷(220kg)萃取,并将合并的有机物用20% nacl(570kg)洗涤并用mgso4(9.5kg,10%w/w)干燥。将混合物过滤并在35-45℃下浓缩,得到粗制j4。对另外三批j4(109.8kg,qnmr 85.57%)+(110.2kg,qnmr 85.1%)+(108.15kg,qnmr 85.1%)重复该相同程序。将总共四批粗制j4合并并蒸馏,得到呈黄色液体的j4(246.5kg,89.6%纯度,87% qnmr,67.7%产率)。
[1831]
步骤4(j5):在3000l反应器中在搅拌下将naoh(61.63kg,1540.8mol,2.28当量)溶解于水(493kg)中。装入j4(246.5kg,87% qnmr,676.2mol,1.0当量)和四丁基溴化铵(tbab,12.33kg,38.25mol,0.057当量),随后装入2-methf(1059.95kg)。将反应混合物加热至65-75℃并在该温度下保持1-4小时,此时通过hplc分析确定保留少于1.0% j4。将反应混合物冷却至30℃,并分离各相。将有机层用水(739.5kg)洗涤两次并经mgso4(36.98kg)干燥。将混合物过滤并在40-50℃下浓缩至干燥。添加正庚烷(167.6kg),并将混合物再次浓缩,以去除残余水。将该过程重复一次,得到呈黄色液体的j5(203.2kg,89.46% qnmr,94.57%纯度,97.72%产率)。
[1832]
步骤5(j6/k8):在2000l反应器中将j5(203.2kg,89.46% qnmr,660.8mol,1.0当量)溶解于thf(817.2kg)中,在氮气下搅拌。将溶液冷却至-50至-30℃并装入n-buli(377.5kg,1387.7mol,2.1当量),同时将温度保持在-50至-30℃之间。在将反应混合物在-50至-30℃下保持1-2小时后,保留少于1.0% j5。在15℃下将混合物淬灭到20% nh4cl水溶液(671.9kg),并将所得混合物搅拌30分钟并分离。将水相用etoac(817kg)萃取。将合并的有机相用20%nh4cl水溶液(671.9kg)洗涤两次,随后用20% nacl水溶液(408.6kg)洗涤并且然后在40-55℃下浓缩至干燥。添加thf(100kg),并将混合物浓缩,以去除残余水。将该过程重复一次,得到呈黄色液体的j6/k8(147.8kg,89.71%纯度,83.62% qnmr,95.41%产率)。
[1833]
步骤6(j7):在3000l反应器中将j6/k8(147.8kg,83.62% qnmr,627.0mol,1.0当量)和三乙胺(95.2kg,940.5mol,1.5当量)溶解于thf(587.0kg)中,在氮气下搅拌。将混合物冷却至-10-0℃。在单个1000l反应器中将3,5-二硝基苯甲酰氯(173.5kg,752.4mol,1.2当量)溶解于thf(587.0kg),并将所得溶液在-10-5℃下转移到3000l反应器中。在将反应混合物升温至10-20℃并搅拌1.5-2小时后,保留1.0% j6/k8。将8% nahco3水溶液(667.4kg)和etoac(500kg)添加到3000l反应器中。将混合物搅拌30分钟并且然后分离。将有机层用8% nahco
3水溶液
(667.4kg)洗涤,随后用10% nacl水溶液(680kg)洗涤,并且然后在40-55℃下浓缩。添加正庚烷(168kg),并将混合物在40-55℃下浓缩。添加etoac(300kg)
和正庚烷(420kg),并将混合物加热至65-75℃,同时搅拌1-2小时。将浆液冷却至15-25℃,搅拌1-2小时,并且然后过滤。将固体用etoac(450kg)和etoh(352kg)的组合处理,并将所得混合物加热至65-75℃,同时搅拌1-2小时。将混合物冷却至5-10℃,搅拌1-2小时,并过滤。将滤饼用etoh(50kg)洗涤并在40-50℃下干燥,得到呈淡黄色固体的j7(206.4kg,99.04%纯度,83.59%产率)。
[1834]
步骤7(k8):在3000l反应器中在搅拌下将lioh
·
h2o(66.57kg,1586.5mol,3.0当量)溶解于水(619.2kg)中。装入j7(206.4kg,528.8mol,1.0当量)和thf(928.8kg)。在将混合物在30-40℃下搅拌3小时后,保留少于1% j7。分离各层,并将thf层在40-55℃下浓缩。添加mtbe(1548kg),并将所得混合物用8% nahco3水溶液(668.7kg)洗涤两次并且然后用20% nacl水溶液(743kg)洗涤。将混合物经mgso4(20.64kg,10%w/w)干燥1-2小时并过滤。将有机相在40-50℃下浓缩。添加正庚烷(138kg),并将混合物浓缩,以去除残余mtbe。将该过程重复一次,并将所得溶液浓缩,产生呈淡黄色、棕色液体的k8(89.9kg,98.61% qnmr,99.24%纯度,86.72%产率)。
[1835]
化合物181磷酸盐水合物
[1836]
[1837][1838]
步骤1.将k7(70g,0.360mol,1.0当量)和2-[5-三氟甲基)-3-噻吩基]乙醇k8(74.2g,0.378mol,1.05当量)于二氯甲烷(210ml,3体积)中的溶液冷却至5℃。将甲磺酸(210.6ml,3.24mol,9当量)装入反应器中,同时将内部温度保持低于30℃。将所得反应混合物加热至39℃。18小时后,hplc分析指示大于99%转化为k9。将反应混合物冷却至30℃,装入二氯甲烷(280ml,4体积),并进一步冷却至0℃。将ph用4n氢氧化钠(830ml)调节至约ph 10。将有机层分离,并将水相用dcm(350ml,5体积)反萃取。将合并的有机物用水(350ml,5体积)洗涤并在减压下浓缩至3.5总体积。将所述批次装入mtbe(5体积)并在减压下浓缩至3.5总体积。将该放入/取出循环再重复三次,并将所得3.5体积混合物用mtbe(6.5体积)稀释,提供10体积混合物。将浆液加热至50℃并搅拌5小时,然后在2小时内装入正庚烷(700ml,10体积)。将所得悬浮液在5小时内冷却至20℃并搅拌18小时。将悬浮液过滤,用1:2mtbe/正庚烷(2x 140ml,2x 2体积)洗涤,并在真空下干燥,同时在50℃下用氮气冲洗18小时,得到103g k9(77%产率)。
[1839]
步骤2.将k9(50g,0.134mol,1.0当量)and三乙胺(22.5ml,0.161mol,1.2当量)于二氯甲烷(380ml,7.6体积)中的溶液冷却至5℃。在5℃下,将三氟乙酸酸酐(20.5ml,0.148mol,1.1当量)装入反应器中,同时将内部温度保持低于15℃。将所得反应混合物在5℃下搅拌1小时,此时hplc显示99.8%转化为k10。在5℃下向反应混合物中装入水(200ml,4体积)。将有机层分离并依次用5% nahco3(200ml,4体积)、2n hcl(2x 200ml,2x 4体积)和水(2x 200ml,2x 4体积)洗涤。将有机层在减压下浓缩至3.5总体积。装入mtbe(400ml,8体积),并将所述批次在减压下浓缩至3.5体积.将该放入/取出循环再重复两次,并在最后循环后将混合物浓缩至3体积。将溶液加热至40℃并在1小时内装入正庚烷(190ml,2体积)。在2小时内将所述批次冷却至20℃,产生悬浮液。在2小时内装入正庚烷(500ml,10体积),并将所得悬浮液搅拌18小时。将悬浮液过滤,用5% mtbe/正庚烷(2x 125ml,2x 2.5体积)洗涤,并在真空下干燥,同时在50℃下用氮气冲洗18小时,得到53gk10(84%产率)。
[1840]
步骤3.将k10(70g,149.4mmol,1.0当量)和1,3-二溴-5,5
’‑
二甲基乙内酰脲
(29.9g,104.6mmol,0.7当量)于无水氯苯(280l,4体积)中的悬浮液用次表面氮气气泡鼓泡60分钟。将混合物加热至75℃并在60分钟内在该温度下装入制备的偶氮二异丁腈(0.49g,3mmol,0.02当量)于无水氯苯(70ml,1体积)中的溶液。在搅拌2小时后,在75℃下,hplc分析显示转化为k11。将反应混合物冷却至60℃并在30分钟内装入无水脱气的dmso(350ml,5体积),随后在30分钟内添加无水脱气三乙胺(104ml,747mmol,5当量)。将反应器顶部空间用氮气充分吹扫,并将所述批次加热至75℃。15小时后,hplc分析显示》99% k11转化为k12。将所述批次冷却至20℃并用二氯甲烷(210ml,3体积)稀释。将所述批次进一步冷却至5℃并装入水(350ml,5体积),同时将溶液温度保持低于30℃。将有机层分离,并将水层用二氯甲烷(210ml,3体积)反萃取。将有机相合并并依次用2n hcl(350ml,5体积)和水(2x 350ml,2x 5体积)洗涤。将有机相在减压下浓缩至3总体积。向溶液中装入ipa(560ml,8体积)并在减压下浓缩至3体积。将该放入/取出循环再重复两次,得到3体积溶液,将其进一步用ipa(70ml,1体积)稀释。将所得4体积混合物加热至75℃,提供均匀溶液并且然后冷却至50℃。在50℃下接种溶液(0.1重量%),搅拌1小时,并在2小时内进一步冷却至20℃。在20℃下再搅拌18小时后,在1小时内向浆液中装入正庚烷(70ml,1体积)。将浆液在20℃下搅拌4小时,过滤,用1:1ipa/正庚烷(2x 70ml,2x 2体积)洗涤,并在真空下干燥,同时在50℃下用氮气冲洗18小时,得到31.2g k12(43%产率,来自k10)。将干燥的k12悬浮于ipa(93ml,3体积)中,加热至80℃,并在该温度下搅拌2小时。在1小时内将溶液冷却至70℃并搅拌1小时。在5小时内将悬浮液冷却至20℃并在该温度下搅拌18小时。将悬浮液过滤,用1:1ipa/正庚烷(2x 35ml,2x 0.5体积)洗涤,并在真空下干燥,同时在50℃下用氮气冲洗18小时,得到28.8g k12(40%产率,来自k10)。
[1841]
步骤5.将五甲基环戊二烯基氯化铑(iii)二聚体(154mg,0.002当量)和(r,r)-tsdpen(182mg,0.004当量)合并于乙腈(240ml,4体积)中,并将混合物用氮气鼓泡,同时在20℃下搅拌1小时。将混合物冷却至-15℃并在30分钟内添加制备的甲酸(27.0ml,5.5当量)和三乙胺(38.1ml,2.2当量)的混合物并且将所得红色/橙色溶液在-15℃下搅拌15分钟。单独制备k12(60g,1.0当量)于乙腈(240ml,4体积)中的溶液并在45分钟内将其添加到冷的催化剂溶液中。将混合物用此次表面氮气气泡鼓泡15分钟,在-15℃下搅拌20小时,升温至0℃,并再搅拌20小时。将温度调节至约20℃并将混合物装入mtbe(360ml,6体积)和18% nacl(水溶液)(360ml,6体积)中。将各相混合,并分离各相。将有机相依次用18% nacl(水溶液)(2x 360ml,6体积)、4% nahco3(水溶液)(360ml,6体积)和18% nacl(水溶液)(180ml,3体积)洗涤。将反应溶液在减压下浓缩至3总体积,然后通过添加mtbe(360ml,6体积)并在减压下浓缩至3体积来将溶剂转换为mtbe。将该放入/取出循环再重复3次。将所得溶液用mtbe稀释至4总体积并装入dcm(240ml,4体积)和mtbe预洗涤的siliamets dmt树脂(30g,50重量%)。将混合物在20℃下剧烈搅拌2小时。在真空下过滤树脂浆液。将反应烧瓶用2:1dcm:mtbe(120ml,2体积)溶液冲洗并将冲洗液转移到树脂。将所得浆液混合,然后在真空下过滤。通过将2:1dcm:mtbe(120ml,2体积)的溶液添加到树脂、混合、然后在真空下过滤来将树脂用所述溶液再冲洗一次。将冲洗液和初始滤液合并并转移回反应烧瓶中,在转移后使用2:1dcm:mtbe(30ml,0.5体积)作为最终冲洗液。将滤液与mtbe预洗涤的siliamets dmt树脂(30g,50重量%)合并并在20℃下剧烈搅拌2小时。树脂浆液是在真空下。将2:1dcm:mtbe(120ml,2体积)的溶液用于冲洗反应烧瓶,并将冲洗液转移到玻璃料的树脂中。将浆液
混合并在真空下过滤。将2:1dcm:mtbe(120ml,2体积)的溶液装入玻璃料的树脂中并将浆液混合,然后在真空下过滤。将合并的滤液转移回反应烧瓶,使用2:1dcm:mtbe(30ml,0.5体积)作为冲洗液。将滤液与mtbe预洗涤的siliamets dmt树脂(30g,50重量%装载)合并并剧烈搅拌18小时。将所得树脂浆液在真空下过滤。。将反应烧瓶用2:1dcm:mtbe(120ml,2体积)溶液冲洗。将冲洗液添加到玻璃料的树脂,并将浆液混合,然后在真空下过滤。将2:1dcm:mtbe(120ml,2体积)的溶液添加到玻璃料的树脂,并将浆液混合,然后在真空下过滤。将合并的滤液转移到烧瓶,然后浓缩至3总体积(180ml)的溶液。添加mtbe(480ml,8体积),并将溶液浓缩至3总体积(180ml)。将该放入/取出循环再重复两次。将所得溶液用mtbe稀释至5体积(300ml),加热至50℃并搅拌3小时。在60分钟内添加正庚烷(240ml,4体积),并将浆液在50℃下再保持1小时。在3小时内将浆液冷却至20℃并搅拌过夜。将浆液在真空下过滤。将滤饼用1:1mtbe:庚烷(2x 60ml,2x 1体积)冲洗,并将固体在真空下在50℃下干燥18小时,产生58.5gk13(83%产率)。
[1842]
步骤6.将k13(43.5g,89mmol,1当量)和甲醇(150.0ml,3体积)合并并搅动,直到观察到完全消失。在30分钟内逐滴添加6n naoh(89ml,6当量),并将混合物加热至60℃并搅拌1小时,此时实现完全转化为化合物181。将反应溶液冷却至15℃并用乙酸异丙酯(250ml,5.75体积)处理。然后添加水(100ml,2.3体积),并将混合物搅动30分钟。将各相分离,并将水相用乙酸异丙酯(250ml,5.75体积)反萃取。将有机物合并并用10% nacl(水溶液)(2x250ml,2x 5.75体积)和水(250ml,5.75体积)洗涤。将有机物浓缩至4.0总体积(174ml)。向溶液中装入mtbe(11.5体积,500ml)并再次浓缩至4.0体积。将该放入/取出循环再重复三次。添加mtbe(75ml,1.75体积),得到5.75体积(250ml)溶液。在20℃下搅拌时,在2小时内添加水(3.2ml,180mmol,2当量),从而诱导结晶。将浆液在20℃下搅拌1小时,然后加热至50℃并在该温度下搅拌3小时。将悬浮液冷却至20℃并搅拌18小时。将浆液在真空下过滤,并将滤饼用mtbe(100ml,2.3体积)洗涤。将固体在50℃下在真空下干燥18小时,提供29g化合物181游离形式单水合物(化合物181.h2o)(81%产率)。
[1843]
步骤7.方法a.将1当量的化合物181游离形式单水合物装入反应器中,随后装入6体积的mek。在20℃下开始搅动。一旦获得澄清溶液,就精良过滤溶液并将其装回反应器中。将水(0.2体积)添加到澄清溶液并继续搅动。添加1重量%的化合物181磷酸盐作为晶种。在单个容器中,将1.02当量85重量%的磷酸用3.8体积的mek稀释。然后在3小时内将该磷酸溶液缓慢添加到反应器中。将最终浆液在20℃下搅动2小时,然后在真空下过滤。将所得湿饼用3体积的mek洗涤。将湿饼在真空下在氮气流下在80℃下干燥,产生化合物181磷酸盐水合物(化合物181.h3po4)(约90%产率)。
[1844]
方法b.将1当量的化合物181游离形式单水合物装入反应器中,随后装入6体积的mek。在20℃下开始搅动,并且一旦获得澄清溶液,就精良过滤溶液并将其装回反应器中。将水(0.2体积.)添加到澄清溶液中并继续搅动。在单个容器中,将1.02当量85重量%的磷酸用3.8体积的mek稀释。然后在3小时内将该磷酸溶液缓慢添加到反应器中。将最终浆液在20℃下搅动2小时,然后在真空下过滤。将所得湿饼用3体积的mek洗涤。将湿饼在真空下在氮气流下在80℃下干燥,产生化合物181磷酸盐水合物(化合物181.h3po4)(约90%产率)。
[1845]
注意:化合物181磷酸盐水合物是结晶水合物.
[1846]
xrpd和vh-xrpd
[1847]
,使用配备有密封管源和pixcel 1d medipix-3检测器的panalytical empyrean系统(malvern panalytical inc,westborough,massachusetts),在室温(25
±
2℃)下以透射模式获得化合物181磷酸盐水合物的粉末x射线粉末衍射(xrpd)谱(图1)。利用铜辐射在45kv的电压和40ma的电流下运行x射线发生器。将粉末置于样品台ap chc台和chc室中。将chc室连接至水泵,所述水泵收集相对湿度。以一定增量逐步改变室内的相对湿度,以5%开始,持续1小时,然后增加至10%并保持一小时,随后将10%相对湿度(rh)逐步增加至60%并且每次保持一小时,从60%升高至90%并保持1小时。然后将chc室在90%下再保持一小时,然后然后从90%减小至80%并保持3小时,然后从80%减小至70%并保持3小时,然后从70%减小至60%并保持3小时,然后从60%以10% rh逐步减小至10%并且每一步保持一小时,并且最后从10%减小至5%并保持一小时。在该小时时间点,在约3
°
2θ至约40
°
2θ的范围内运行xrpd收集,其中步长为0.0131303
°
并且每步49s。可变湿度xrpd(vh-xrpd):观察到化合物181磷酸盐水合物具有连续峰位移,其全部是(在
±
0.2
°
2θ内)5-90%相对湿度(图2,表14)。
[1848]
表14.化合物181磷酸盐水合物的xrpd衍射图的峰列表
[1849]
[1850]
tga
[1851]
使用ta instruments q5000 tga对化合物181磷酸盐水合物进行热重分析。从环境温度到250℃以10℃/min的加热速率在氮气吹扫下对重量为大约1-10mg的样品进行扫描。tga热分析图显示从环境温度开始升温直至150℃的约0.5%重量损失(图3)。
[1852]
dsc
[1853]
使用ta instruments q2000 dsc对化合物181磷酸盐水合物进行差示扫描量热法(dsc)分析。将重量为1-10mg之间的样品称量到带有针孔的铝卷边密封盘中。将该盘置于量热计单元的样品位置中。将空盘置于参考位置。关闭量热计单元,并使氮气流穿过所述单元。设定程序以调节每60秒0.32
°
,然后以2℃/min的加热速率将温度升高至300℃。热分析图显示约226℃和251℃的两个吸热峰(图4)。
[1854]
ssnmr
[1855]
在275k和43%相对湿度(rh)下使用12.5khz旋转并使用金刚烷作为参考来获得化合物181磷酸盐水合物的
13
c cpmas(图5,表15)。
[1856]
表15.化合物181磷酸盐水合物的
13
c cpmas的峰列表
[1857]
峰#化学位移[ppm]强度[相对]1146.342.12145.845.63144.042.54141.758.35139.351.86129.446.47128.652.38126.646.8973.687.51073.283.21166.138.91264.343.71362.755.11462.162.31548.944.61647.370.81745.450.61843.039.61941.648.82038.4100.02136.748.32216.094.4
[1858]
在275k以及0%、6%、22%、43%、53%、75%和100%相对湿度(rh)下使用12.5khz旋转并使用金刚烷作为参考来获得化合物181磷酸盐水合物的
19
fmas(图6、7;表16、17)。
[1859]
表16.化合物181磷酸盐水合物在43% rh下的
19
f mas的峰列表
[1860]
峰#化学位移[ppm]强度[相对]1-53.811.02-57.412.5
[1861]
表17.相对湿度对化合物181磷酸盐水合物的
19
f mas的作用
[1862][1863][1864]
在275k以及0%、6%、22%、43%、53%、75%和100%相对湿度(rh)下使用12.5khz旋转并使用金刚烷作为参考来获得化合物181磷酸盐水合物的
31
pcpmas(图8、9;表18、19)。
[1865]
表18.化合物181磷酸盐水合物在43% rh下的
31
p cpmas的峰列表
[1866]
峰#化学位移[ppm]强度[相对]14.246.422.6100.0
[1867]
表19.相对湿度对化合物181磷酸盐水合物的
31
p cpmas的作用
[1868]
rh[%]峰1[ppm]峰2[ppm]06.12.665.12.6224.42.6334.22.6434.22.6534.12.5754.02.51003.82.5
[1869]
化合物181游离形式单水合物的替代性制备
[1870]
将无定形化合物181(30mg)添加到盐水(1ml)中。在轻度涡旋以查看材料是否溶解后,形成白色乳状沉淀。将样品置于环境温度下过夜。使用0.22μm pvdf eppendorf过滤管过滤固体材料,用冰冷水冲洗。在真空烘箱中在45℃下干燥样品过夜。湿饼和干燥的材料二者都是结晶化合物181游离形式单水合物。
[1871]
xrpd
[1872]
使用配备有密封管源和pixcel 1d medipix-3检测器的panalytical empyrean系统(malvern panalytical inc,westborough,massachusetts),在室温下以透射模式获得化合物181游离形式单水合物的粉末x射线粉末衍射(xrpd)谱(图10,表20)。利用铜辐射
在45kv的电压和40ma的电流下运行x射线发生器。将粉末样品置于具有麦拉膜的96孔样品架上并装载至仪器中。在约3
°
2θ至约40
°
2θ的范围内扫描样品,其中步长为0.0131303
°
并且每步为49s。
[1873]
表20.化合物181游离形式单水合物的xrpd衍射图的峰列表
[1874][1875][1876]
tga
[1877]
使用ta5500 discovery tga对化合物181游离形式单水合物进行热重分析。从环境温度到250℃以10℃/min的加热速率在氮气吹扫下对重量为大约1-10mg的样品进行扫描。tga热分析图显示从环境温度开始升温直至100℃的约~3-4%重量损失(图11)。
[1878]
dsc
[1879]
使用ta instruments q2000 dsc对化合物181游离形式单水合物进行差示扫描量热法(dsc)分析。将重量为1-10mg之间的样品称量到带有针孔的铝卷边密封盘中。将该盘置于量热计单元的样品位置中。将空盘置于参考位置。关闭量热计单元,并使氮气流穿过所述单元。设定程序以调节每60秒0.32
°
,然后以2℃/min的速率将温度加热至300℃。热分析图显示约61℃、94℃和111℃的三个吸热峰(图12)。
[1880]
ssnmr
[1881]
在275k和43%相对湿度(rh)下使用12.5khz旋转并使用金刚烷作为参考来获得化合物181游离形式单水合物的
13
c cpmas(图13,表21)。另外,在275k下使用12.5khz旋转并使用金刚烷作为参考来获得化合物181游离形式单水合物在脱水后(在转子中在80℃下过夜(2x),80℃周末与p2o5孵育)的
13
c cpmas(图14,表22)。
[1882]
表21.化合物181游离形式单水合物的
13
c cpmas的峰列表
[1883][1884][1885]
表22.脱水化合物181游离形式单水合物的
13
c cpmas的峰列表
[1886]
峰#化学位移[ppm]强度[相对]1150.933.42150.053.93135.364.64129.630.15127.229.26126.632.6774.7100.0868.414.9961.538.61050.748.51148.822.21248.341.71347.523.41447.241.71536.845.21635.842.51725.656.2
[1887]
在275k和43%相对湿度(rh)下使用12.5khz旋转并使用金刚烷作为参考来获得化合物181游离形式单水合物的
19
f mas(图15,表23),其中减去
19
f背景。另外,在275k下使用12.5khz旋转并使用金刚烷作为参考来获得化合物181游离形式单水合物在脱水后(在转子中在80℃下过夜(2x),80℃周末与p2o5孵育)的
19
f mas(图16,表24),其中减去
19
f背景。
[1888]
表23.化合物181游离形式单水合物的
19
f mas的峰列表
[1889]
峰#化学位移[ppm]强度[相对]
1-55.812.5
[1890]
表24.脱水化合物181游离形式单水合物的
19
f mas的峰列表
[1891]
峰#化学位移[ppm]强度[相对]1-55.512.5
[1892]
化合物181磷酸盐甲醇溶剂化物
[1893]
将无定形化合物181(50mg)添加到mek(0.3ml)中。向其中添加0.27ml0.5m h3po4于meoh中的储备溶液。将样品在环境温度下搅拌过夜。使用0.22μm pvdf eppendorf过滤管过滤固体并用在冰上冷冻的4:1正庚烷/mek(v/v)洗涤。用正庚烷进行后续洗涤,从而产生固体白色粉末。湿材料的xrpd显示产物是化合物181磷酸盐甲醇溶剂化物。
[1894]
xrpd
[1895]
使用配备有密封管源和pixcel 3d medipix-3检测器的panalytical empyrean系统(malvern panalytical inc,westborough,massachusetts),在室温下以透射模式获得化合物181磷酸盐甲醇水合物的粉末x射线粉末衍射(xrpd)谱(图17,表25)。利用铜辐射在45kv的电压和40ma的电流下运行x射线发生器。将粉末样品置于具有麦拉膜的96孔样品架上并装载至仪器中。在约3
°
2θ至约40
°
2θ的范围内扫描样品,其中步长为0.0131303
°
并且每步为49s。
[1896]
表25.化合物181磷酸盐甲醇溶剂化物的xrpd衍射图的峰列表
[1897]
xrd峰角度(
°
2θ
±
0.2)强度%115.8100.0220.789.2312.759.548.554.2519.545.5618.736.8713.935.6810.230.3922.529.51021.527.4113.926.41220.024.91319.224.51424.924.01519.623.31621.821.51727.421.31812.921.01925.220.82014.820.7
2117.318.0229.617.82320.417.02417.615.92516.015.52611.413.92718.413.72825.512.52927.912.23027.611.13112.510.83223.510.7
[1898]
ssnmr
[1899]
在275k下使用12.5khz旋转并使用金刚烷作为参考来获得化合物181磷酸盐甲醇溶剂化物的
13
c cpmas(图18,表26)。
[1900]
表26.化合物181磷酸盐甲醇溶剂化物的
13
c cpmas的峰列表
[1901]
峰#化学位移[ppm]强度[相对]1146.854.02145.850.83143.947.84140.682.35139.566.06129.471.67128.556.28127.958.29126.746.51073.894.91172.295.21266.366.81364.261.71462.869.11561.677.91649.756.91748.580.31847.1100.01945.557.92043.051.02140.573.12240.165.6
2338.966.22437.762.12536.858.62617.778.32715.778.5
[1902]
在275k下使用12.5khz旋转并使用金刚烷作为参考来获得化合物181磷酸盐甲醇溶剂化物的
19
f mas(图19,表27),其中减去
19
f背景。
[1903]
表27.化合物181磷酸盐甲醇溶剂化物的
19
f mas的峰列表
[1904]
峰#化学位移[ppm]强度[相对]1-54.711.82-57.712.5
[1905]
在275k下使用12.5khz旋转并使用金刚烷作为参考来获得化合物181磷酸盐甲醇溶剂化物的
31
p cpmas(图20,表28)。
[1906]
表28.化合物181磷酸盐甲醇溶剂化物的
31
p cpmas的峰列表
[1907]
峰#化学位移[ppm]强度[相对]12.593.921.8100.0
[1908]
化合物181磷酸盐mek溶剂化物
[1909]
在hplc小瓶中将化合物181磷酸盐水合物(25mg)添加到2-丁酮(mek)(1ml)中。将样品混合并形成浆液。将浆液置于5℃冷室中,用小搅拌棒搅拌11天。将固体材料离心并在室温下使用0.22μm pvdf eppendorf过滤管过滤。湿饼样品的xrd显示它是化合物181磷酸盐mek溶剂化物。
[1910]
xrpd
[1911]
使用配备有密封管源和pixcel 1d medipix-3检测器的panalytical empyrean系统(malvern panalytical inc,westborough,massachusetts),在室温(25
±
2℃)下以透射模式获得化合物181磷酸盐mek溶剂化物的粉末x射线粉末衍射(xrpd)谱(图21,表29)。利用铜辐射在45kv的电压和40ma的电流下运行x射线发生器。将粉末样品置于具有麦拉膜以及样品上的聚酰亚胺胶带的96孔样品架上并装载至仪器中。在约3
°
2θ至约40
°
2θ的范围内扫描样品,其中步长为0.0131303
°
并且每步为49s。
[1912]
表29.化合物181磷酸盐mek溶剂化物的xrpd衍射图的峰列表
[1913]
xrd峰角度(
°
2θ
±
0.2)强度%120.1100.0215.485.738.680.8415.736.5519.432.1618.232.0721.730.8
821.929.0913.228.61023.825.91110.825.11210.524.11321.023.01422.821.71517.518.81618.418.21726.716.81822.414.4193.812.4208.311.02116.510.6
[1914]
ssnmr
[1915]
在275k下使用12.5khz旋转并使用金刚烷作为参考来获得化合物181磷酸盐mek溶剂化物的
13
c cpmas(图22,表30)。
[1916]
表30.化合物181磷酸盐mek溶剂化物的
13
c cpmas的峰列表
[1917]
[1918][1919]
在275k下使用12.5khz旋转并使用金刚烷作为参考来获得化合物181磷酸盐mek溶剂化物的
19
f mas(图23,表31),其中减去
19
f背景。
[1920]
表31.化合物181磷酸盐mek溶剂化物的
19
f mas的峰列表
[1921]
峰#化学位移[ppm]强度[相对]1-53.610.02-55.25.23-57.212.5
[1922]
在275k下使用12.5khz旋转并使用金刚烷作为参考来获得化合物181磷酸盐mek溶剂化物的
31
p cpmas(图24,表32)。
[1923]
表32.化合物181磷酸盐mek溶剂化物的
31
p cpmas的峰列表
[1924][1925][1926]
化合物181磷酸盐水合物的替代性制备在n2吹扫下将2.05g化合物181磷酸盐甲醇溶剂化物在50℃下干燥21小时。所得固体是化合物181磷酸盐水合物。
[1927]
化合物174半水合物
[1928][1929]
步骤1.在0℃下在1小时内将k7(4153g,1当量,81.11%纯度通过q-nmr得到,21.53mmol,1当量)和k8(3651g,22.45mmol,1.05当量)于二氯甲烷(33.2l,8体积)中的溶液用甲磺酸(14384g,149.7mol,7当量)处理。将所得混合物在40℃下加热。在14小时后,分析显示k7的》99%消耗。将反应混合物冷却至10℃并用4n氢氧化钠(40l)调节至约ph 10。将有机层分离,经硫酸钠(1.5kg)干燥,并在减压下蒸发在25℃下,得到呈灰白色固体的粗制k14(8.1kg)。将该固体悬浮于甲基叔丁基醚(22l),在10℃下搅拌2.5小时,并且然后过滤。将滤
饼用甲基叔丁基醚(4l)洗涤并在真空下干燥,同时在20℃下用氮气冲洗18小时,得到纯化的5950g k14(96.8%产率)。
[1930]
步骤2.将k14(5937g,17.52mol,1当量)和n,n-二异丙基乙基胺(3967ml,22.78mol,1.3当量)于二氯甲烷(59l,10体积)中的溶液冷却至0-5℃并在40分钟内用三氟乙酸酸酐(2680ml,19.27mol,1.1当量)处理,同时将反应温度保持低于14℃。将所得反应混合物在0-10℃下搅拌。2小时后,hplc分析指示》99.5%转化。将反应混合物冷却至5℃并用饱和盐水(27l)稀释。将所得混合物用6n氢氧化钠溶液(5l)调节至约ph 10,同时将温度保持低于12℃。将所得混合物20分钟搅拌,然后分离各层。将有机层依次用2n hcl(3x 22l)、水(3x 22l)和盐水(22l)洗涤,然后经硫酸钠(1kg)干燥并在减压下在30℃下蒸发,得到粗制k15(7519g)。在50℃下将粗材料悬浮于甲基叔丁基醚(16l)和正庚烷(8l)的混合物中持续5小时,然后在5小时内冷却至20℃。在20℃下搅拌18小时后,将悬浮液过滤。将滤饼用甲基叔丁基醚(8l)和正庚烷(4l)的混合物洗涤,然后并在真空下干燥,同时在20℃下用氮气冲洗18小时,得到6824g k15(94.1%产率)。
[1931]
步骤3和4.在100l夹套玻璃反应器中将k15(5879g,13.52mol,1当量)、偶氮二异丁腈(178g,1.082mol,0.08当量)和1,3-二溴-5,5-二甲基乙内酰脲(2900g,10.14mol,0.75当量)于氯苯(41.2l,7体积)中的悬浮液用氮气鼓泡20分钟。将反应混合物然后加热至70℃。30分钟后,hplc分析指示》99%转化为k16。将反应物冷却至45℃,用无水二甲基亚砜(41.2l,7体积)和三乙胺(9.42l,67.55mol,5当量)处理,并在65℃下加热。12小时后,hplc分析指示k16完全消耗。将反应混合物冷却至0℃并分成两个相等的半部分。将每个半部分用冰冷水(22l)处理,将温度保持低于15℃,然后用乙酸乙酯(2x20l)萃取。将水层用乙酸乙酯(18l)萃取。将合并的有机层用水(2x 24l)、盐水(24l)洗涤,经无水硫酸钠(2kg)干燥,然后在减压下在50℃下蒸发,得到半固体残余物,将其与甲醇(2x 4l)在50℃下共蒸发,得到呈深棕色固体的粗产物(6.65kg)。将残余物在65℃下用甲醇(32l)研磨5小时,在5小时内冷却至15℃,然后过滤,得到3643g k17。将固体在65℃下用丙酮和甲醇的1:2混合物(18l)研磨5小时,在5小时内冷却至20℃,然后过滤。将滤饼用丙酮和甲醇的1:2混合物(2x 3l)冲洗,随后在20℃下用甲醇(3l)冲洗。将产物在氮气对流下在20℃下干燥18小时,得到2780g k17(45.8%产率)。
[1932]
步骤5.将n-[(1r,2r)-2-氨基-1,2-二苯基-乙基]-4-甲基-苯磺酰胺(22.1g,0.06mol,0.01当量)和二氯-(1,2,3,4,5-五甲基环戊-2,4-二烯-1-基)铑二聚体(18.26g,0.03mol,0.005当量)于乙腈(12l)中的溶液在20℃下搅拌30分钟,然后冷却至-5℃。在0℃下将该溶液添加到k17(2704g,6.024mol,1当量)于乙腈(16l)以及甲酸(1.25l,33.13mol,5.5当量)和三乙胺(1.85l,13.25mol,2.2当量)的混合物(预混合并预冷却至0℃下)中的悬浮液中。将所得混合物在0℃下搅拌并通过hplc监测反应进程。31小时后,hplc分析指示》99.9%转化为k18。将反应混合物用碳酸氢钠(2.1kg)于水(30l)中的溶液稀释。将所得混合物在10℃下搅拌15分钟,然后升温至15℃并用甲基叔丁基醚(12l)稀释。将所得混合物在15℃下搅拌15分钟。将各层分离并将水层用甲基叔丁基醚(12l)萃取。将合并的有机层依次用1n hcl(2x 11l)和盐水(2x 11l)洗涤。在第二次盐水洗涤期间,将盐水层的ph用固体碳酸氢钠(288g)调节至约~8。将有机层经无水硫酸钠(2kg)干燥并在减压下蒸发,得到3.4kg粗制k18。将粗制k18于甲基叔丁基醚(35l)中的溶液在20℃下用siliamets dmt(1.7kg)处理
18小时,然后过滤。将滤饼用甲基叔丁基醚(5l)冲洗。在3次连续运行中在50℃下将合并的滤液用siliamets dmt(1.7kg,0.5体积)处理5小时。在各处理之间将混合物冷却至20℃并过滤。在45℃下在减压下蒸发在最终研磨后的滤液,得到2.4kg k18(68.9%产率)。
[1933]
步骤6.在20℃下将k18(1785.8g(对nmr纯度进行校正),3.96mol)于甲醇(12.5l,7体积)中的溶液用6n氢氧化钠(5.0l,29.71mol,7.5当量,预冷却至5℃)处理,在100l夹套玻璃反应器中在20分钟内分4等份添加。将所得溶液在40℃下搅拌。1.5小时后,lc-ms分析指示》99.9%转化。将反应混合物冷却至5-10℃并用6n hcl(4l)调节至约ph 10至11。将反应混合物在减压下在37℃下部分蒸发,以去除甲醇。将混合物用乙酸异丙酯(18l)和水(2l)稀释。将所得悬浮液加热至46℃,得到澄清相。在46℃下搅拌15分钟后,分离各层。将水层在40℃下用乙酸异丙酯(10l)萃取。将合并的有机层用半饱和盐水(10l)洗涤,随后在40℃下用水(5l)洗涤。将有机层在减压下在40℃下蒸发至干燥,得到1298g粗制化合物174(化合物174)(~92%产率)。
[1934]
步骤7.在40℃下在减压下将化合物174(1207g,3.4mol(对于~90%1hnmr纯度进行校正),1当量)与甲基乙基酮(4l)共蒸发。将残余物溶解于甲基乙基酮(6l)并过滤(~8μm多孔性)。将滤液连同水(40ml,2.2mol,0.65当量)装入反应器中。将所得溶液加热至60-62℃。在一小时内将正庚烷(6l,5体积)加入热溶液中,将温度保持在60-62℃下。将所得混合物接种(1g,约0.1%wt)并在62℃下加热一小时。在5小时内将所得溶液冷却至20℃。在20℃下搅拌18小时后,在20℃下通过whatman#113滤纸过滤悬浮液。将滤饼用正庚烷和甲基乙基酮(3l)的4:1混合物以2等份洗涤。将产物在氮气对流下在20℃下干燥3小时,得到1091.5g呈白色粉末的化合物174半水合物(化合物174.0.5h2o)(86.1%产率)。
[1935]
化合物174磷酸半水合物的制备1
[1936]
在10ml小瓶中称入628mg化合物174半水合物,然后添加约7.6ml 2-methf。约3.7ml 0.5m h3po4,通过与约0.42ml 6m h3po4(水溶液)混合来预配制并将约4.6ml甲醇逐滴添加到小瓶中。将混合物在环境温度下用磁力搅拌棒搅拌两天。然后通过离心收集固体,并在40℃真空烘箱中干燥过夜。回收的固体总量为670mg。
[1937]
化合物174磷酸半水合物的制备2
[1938]
将1当量化合物174半水合物装入反应器中,随后装入8体积的2-methf。将混合物在40℃下搅动。用1重量%的化合物174磷酸盐半水合物在40℃下接种澄清溶液。在另一个容器中,用0.35体积的水、3体积的2-methf和0.6体积的丙酮稀释1.02当量的85重量%的磷酸。然后在2小时内将该磷酸溶液缓慢添加到反应器中。然后在5小时内将所得浆液冷却至20℃。将最终浆液在20℃下搅动不少于2小时,然后在真空下过滤。用3体积的2-methf洗涤所得湿滤饼。在真空下在50℃下在氮气流下干燥湿饼,得到约94%的化合物174磷酸盐半水合物。
[1939]
xrpd
[1940]
使用配备有密封管源和pixcel 3d medipix-3检测器的panalytical empyrean系统(malvern panalytical inc,westborough,massachusetts),在室温(25
±
2℃)下以透射模式记录x射线粉末衍射(xrpd)谱(图24,表33)。利用铜辐射在45kv的电压和40ma的电流下运行x射线发生器。将粉末样品置于具有麦拉膜的96孔样品架上并装载至仪器中。在约3
°
2θ至约40
°
2θ的范围内扫描样品,其中步长为0.0131303
°
并且每步为49s。
[1941]
表33.化合物174磷酸酯半水合物的xrpd衍射图的峰列表
[1942][1943][1944]
tga
[1945]
使用来自ta instrument的ta discovery 550tga对化合物174磷酸盐半水合物进行热重分析。从25℃至350℃以10℃/min的加热速率在氮气吹扫下对重量为1-10mg的样品
进行扫描。通过thermal advantage q series
tm
软件收集数据,并通过trios和/或通用分析软件(ta instruments,new castle,de)进行分析。热分析图显示从环境温度到150℃的2.4%重量损失(图25)。
[1946]
dsc
[1947]
使用ta discovery 550dsc对化合物174磷酸盐半水合物进行dsc。将重量为1-10mg之间的样品称量到带有针孔的铝卷边密封盘中。将该盘置于量热计单元的样品位置中。将空盘置于参考位置。关闭量热计单元并使氮气流穿过所述单元。加热程序设定为以10℃/min的加热速率将样品加热至250℃的温度。当运行完成时,通过trios和/或通用分析软件(ta instruments,new castle,de)分析数据。热分析图显示在123℃和224℃附近的两个吸热峰(图26)。
[1948]
ssnmr
[1949]
在275k下使用12.5khz旋转并使用金刚烷作为参照来获得化合物174磷酸半水合物的
13
c cpmas(图27,表34)。另外,在275k下使用12.5khz旋转并使用金刚烷作为参照来获得化合物174磷酸半水合物在脱水后的
13
ccpmas(图28,表35)。
[1950]
表34.化合物174磷酸盐半水合物的
13
c cpmas的峰列表
[1951]
[1952][1953]
表35.脱水化合物174磷酸盐半水合物的
13
c cpmas的峰列表
[1954][1955][1956]
在275k下使用12.5khz旋转并使用金刚烷作为参照来获得化合物174磷酸盐半水合物的
13
c cpmas(图29a,表36a)。此外,在275k使用12.5khz旋转并使用金刚烷作为参照来获得化合物174磷酸半水合物在脱水后的
31
pcpmas(图29b,表36b)。
[1957]
表36a.化合物174磷酸盐半水合物的
31
p cpmas的峰列表
[1958]
峰#化学位移[ppm]强度[相对]13.1100.02-1.170.63-1.828.0
[1959]
表36b.脱水化合物174磷酸盐半水合物的
31
p cpmas的峰列表
[1960]
峰#化学位移[ppm]强度[相对]15.681.924.4100.033.285.043.096.7
[1961]
化合物174半水合物的替代性制备
[1962]
将100mg无定形化合物174添加到玻璃小瓶中。向其中添加0.4ml mek,并且所有固体溶解。然后添加3l水以帮助半水合物的形成。向该混合物中直接添加0.25ml正庚烷。在室温下搅拌18小时后,过滤固体,用1:4mek/正庚烷(v/v)冲洗,然后用100%正庚烷冲洗。收集固体,在真空烘箱(60℃)中干燥过夜,并表征。
[1963]
xrpd
[1964]
使用配备有密封管源和pixcel 1d medipix-3检测器的panalytical empyrean系统(malvern panalytical inc,westborough,massachusetts),在室温(25
±
2℃)下以透射模式记录x射线粉末衍射(xrpd)谱(图30,表37)。利用铜辐射在45kv的电压和40ma的电流下运行x射线发生器。将粉末样品置于具有麦拉膜的96孔样品架上并装载至仪器中。在约3
°
2θ至约40
°
2θ的范围内扫描样品,其中步长为0.0131303
°
并且每步为49s。
[1965]
表37.化合物174半水合物的xrpd衍射图的峰列表(室温)
[1966]
号.位置[
±
0.2,
°
2θ]相对强度[%]117.1100.0220.487.3319.174.146.566.255.746.8614.432.6712.125.6811.422.3925.522.01012.320.51118.919.9129.419.91322.419.71421.818.81515.817.71622.714.51722.414.21820.812.91925.012.4
2026.112.12129.012.02226.111.82327.011.82419.311.52525.111.22625.311.1276.210.92827.910.92928.410.73015.710.0
[1967]
此外,在原位变温xrpd(vt-xrpd)的一项测试中,观察到化合物174半水合物在升高的温度下显示峰位移。使用配备有密封管源和pixcel 1d medipix-2检测器的panalytical empyrean系统(malvern panalytical inc,westborough,massachusetts),在30-90℃下以反射模式记录变温x射线粉末衍射(vt-xrpd)谱。从30℃至90℃以10℃的增量逐步改变温度,在每个温度下保持1小时,随后进行xrd收集。将样品室用室内氮气吹扫。利用铜辐射在45kv的电压和40ma的电流下运行x射线发生器。在约3
°
2θ至约40
°
2θ的范围内扫描样品,其中步长为0.0131303
°
并且每步为49.725s。
[1968]
发现三种不同的xrpd模式,分别在:(1)环境温度到30℃;(2)40-50℃;和(3)60-90℃。在环境温度和湿度下重新平衡后,样品恢复到其初始形态。从环境温度到30℃的xrpd光谱与在室温(25
±
2℃)下收集的xrpd光谱相同(在0.2
±
2θ内)。表38列出了40-50℃之间观察到的峰,并且表39列出了60-90℃之间观察到的峰。
[1969]
表38.化合物174半水合物的xrpd衍射图的峰列表(40-50℃)
[1970][1971][1972]
表39.化合物174半水合物(60-90℃)的xrpd衍射图的峰列表
[1973][1974][1975]
tga
[1976]
使用来自ta instrument的ta discovery 550tga对化合物174半水合物进行热重分析。从25℃至300℃以10℃/min的加热速率在氮气吹扫下对重量大约为1-10mg的样品进行扫描。通过thermal advantage q seriestm软件收集数据,并通过trios和/或通用分析软件(ta instruments,new castle,de)进行分析。热分析图显示从环境温度到150℃的2.4%重量损失(图31)。
[1977]
dsc
[1978]
使用ta instruments q2000 dsc对化合物174半水合物进行dsc。将重量为1-10mg
之间的样品称量到带有针孔的铝卷边密封盘中。将该盘置于量热计单元的样品位置中。将空盘置于参考位置。关闭量热计单元并使氮气流穿过所述单元。加热程序设定为以10℃/min的加热速率将样品加热至300℃的温度。当运行完成时,通过trios和/或通用分析软件(ta instruments,new castle,de)分析数据。热分析图显示在77℃、107℃和125℃附近的吸热峰(图32)。
[1979]
ssnmr
[1980]
在275k下使用12.5khz旋转并使用金刚烷作为参照来获得化合物174半水合物的
13
c cpmas(图33,表40)。此外,在275k下使用12.5khz旋转并使用金刚烷作为参照来获得脱水后化合物174半水合物的
13
c cpmas(在转子中在环境温度下周末并在80℃下过夜)(图34,表41)。
[1981]
表40.化合物174半水合物的
13
c cpmas的峰列表
[1982]
[1983][1984]
表41.脱水化合物174半水合物的
13
c cpmas的峰列表
[1985]
峰#化学位移[ppm]强度[相对]1151.920.52147.217.33142.223.64141.525.65139.434.16132.929.67130.714.48125.521.19124.424.010121.117.51174.6100.01267.639.41364.158.31461.830.91549.924.81649.225.81747.742.91847.339.91946.630.32045.226.82144.333.92239.826.2
2338.526.32435.322.22522.641.12622.443.6
[1986]
实施例3:用于检测和测量化合物的apol1抑制剂性质的测定
[1987]
multitox-fluor多重细胞毒性测定
[1988]
multitox-fluor多重细胞毒性检测是一种添加单一试剂的同质荧光检测,其同时测量培养孔中活细胞和死细胞的数量。所述测定通过检测两种不同的蛋白酶活性来测量细胞活力和细胞毒性。活细胞蛋白酶活性限于完整的活细胞,并使用荧光、细胞渗透肽甘氨酰基-苯丙氨酰基氨基氟香豆素(gf-afc)底物进行测量。底物进入完整的细胞,在那里它被裂解产生与活细胞数量成比例的荧光信号。这种活细胞蛋白酶活性标记在膜完整性丧失和泄漏到周围培养基中时变得失活。使用第二种不渗透细胞的荧光肽底物(双aaf-r110底物)测量从失去膜完整性的细胞中释放的死细胞蛋白酶。使用死细胞与活细胞的比率来使数据标准化。
[1989]
简言之,在湿润37℃培养箱中在10.03、3.24、1.13、0.356、0.129、0.042、0.129、0.0045、0.0015、0.0005μm的3-(2-(4-氟苯基)-1h-吲哚-3-基)-n-((3s,4r)-4-羟基-2-氧代吡咯烷-3-基)丙烯酰胺的存在下,一式两份地将tet诱导的转基因apol1 t-rex-hek293细胞系与50ng/ml tet一起孵育24小时以诱导apol1。将multitox试剂添加到每个孔中,并放回培养箱中再放置30分钟。在envision板读取器上读取板。使用死细胞与活细胞的比率来进行标准化,并使用genedata screener(basel,switzerland)软件导入、分析和拟合数据。使用对照、无tet(100%存活力)和50ng/ml tet处理(0%存活力)的百分比对数据进行标准化,并使用smart fit进行拟合。用于multitox测定的试剂、方法和完整方案如下所述。
[1990]
表42.multi-tox测定中使用的试剂
[1991]
[1992]
表43.multi-tox测定中使用的设备
[1993][1994]
multi-tox测定方案
[1995]
将含有tet诱导型表达系统(t-rex
tm
;invitrogen,carlsbad,ca)和腺相关病毒位点1paavs1-puro-apol1 g0或paavs1-puro-apol1 g1或paavs1-puro-apol1 g2克隆g0 dc2.13、g1 dc3.25和g2 dc4.44的人胚肾(hek293)细胞系在t-225烧瓶中细胞生长培养基(dmem、10%无tet fbs、2mm l-谷氨酰胺、100单位/ml青霉素-链霉素、5μg/ml杀稻瘟素s hcl、1μg/ml嘌呤霉素而盐酸盐)中在~90%融合下生长。用dpbs洗涤细胞,然后用胰蛋白酶消化以从烧瓶中解离。用培养基淬灭胰蛋白酶,然后以200g使细胞沉淀,并重悬于新鲜细胞测定培养基(dmem、2%无tet fbs、2mm l-谷氨酰胺、100单位/ml青霉素-链霉素)中。对细胞进行计数,并稀释至1.17
×
106个细胞/ml。使用multidrop分配器将20μl细胞(23,400/孔)分配到384孔聚-d-赖氨酸包被板的每个孔中。然后将板在室温下孵育一小时。
[1996]
四环素是诱导apol1表达所必需的。在细胞测定培养基中将1mg/ml于水中的tet储备液稀释至250ng/ml(5x)。用multidrop分配器将60μl细胞测定培养基(无tet对照)分配到384-pp圆底板的第1和24列中,并且将60μl5x tet分配到第2至23列中。
[1997]
使用模板384_apol1cell_dr10n2_50um_v3订购全球化合物档案库中的测定就绪板。分配200nl于dmso中的化合物。在multitox测定中,最终最高浓度为10μm,10点3倍稀释,一式两份。
[1998]
将20μl从5x tet板转移到arp并混合,然后将5μl 5x tet和化合物转移到细胞板并使用bravo混合。将细胞板置于湿润的37℃5% co2培养箱中24小时。
[1999]
根据制造商的方案进行multitox-fluor多重细胞毒性测定。在将细胞与tet和化合物一起孵育24小时后,使用multidrop分配器将25μl 1x multitox试剂添加到每个孔中;将板置于板振荡器(600rpm)上2分钟,然后短暂离心并放回37℃培养箱中30分钟。使用envision板读取器读取细胞活力(激发:400nm,发射:486nm)和细胞毒性(激发:485nm,发射:535nm)。报道了死细胞(细胞毒性)与活细胞(生存力)的比率。在genedata中导出并分析数据。使用对照、无tet(100%生存力)和50ng/ml tet处理(0%生存力)的百分比对数据进行标准化,并使用genedata中的智能拟合设置进行拟合。
[2000]
使用apol1重组蛋白进行布氏布氏锥虫溶解测定
[2001]
布氏布氏锥虫是一种血流寄生虫,人类、大猩猩和狒狒由于在其hdl颗粒中存在apol1蛋白而对该寄生虫免疫。该蛋白经由位于寄生虫鞭毛袋中的tbhphb受体被寄生虫摄取并被hdl颗粒中包含的hpr蛋白结合,从而触发寄生虫的受体内吞作用。
[2002]
在内吞作用后,形成的含有hdl颗粒的囊泡从早期内体成熟为晚期内体,并随后成熟为溶酶体。伴随而来的囊泡内腔中的ph变化触发了apol1蛋白插入到晚期内体/溶酶体的膜中,从而触发了溶酶体膜透化,并作为进一步的下游事件触发了锥虫溶解。布氏布氏锥虫对所有三种apol1变体(g0、g1和g2)造成的溶解都敏感。
[2003]
布氏布氏锥虫溶解测定法是使用重组apol1蛋白变体进行的寄生虫溶解测定法,随后是通过向测定孔添加alamarblue试剂进行的存活的荧光检测法,该试剂为通用的代谢
氧化还原指示剂(alamarblue测定)。
[2004]
简言之,alamarblue活性化合物刃天青是一种蓝色、水溶性、无毒且细胞可渗透的分子,可由吸光度跟踪,其通过各种代谢途径被还原成试卤灵,一种可由吸光度或荧光跟踪的红色化合物。该测定允许通过在标准曲线上用已知量的接种锥虫/孔插值求取荧光值(flu)来计算溶解结束时相对于未处理条件的存活百分比(每个孔中剩余的活锥虫的百分比)。
[2005]
试剂和材料布氏布氏锥虫(atcc,目录号pra-382)
[2006]
o lister 427vsg 221血流形式。
[2007]
解冻/扩增培养基(atcc培养基2834改良hmi-9培养基)
[2008][2009]
测定培养基(无酚红/无fbs):使用当天制备
[2010][2011]
hmi-9(10x)
[2012][2013]
次黄嘌呤储备液(100x)-9(10x)
[2014][2015]
表44.用于裂解测定的布氏布氏锥虫中的培养基试剂
[2016][2017]
表45.布氏布氏锥虫裂解测定中使用的材料
[2018][2019]
[2020]
设备
[2021]
o e1-clip tip移液器12通道可调2-125μl,目录号4672070bt
[2022]
o thermofisher multidrop 384,目录号5840300
[2023]
o multidrop
[2024]
o agilent bravo,目录号g5409a
[2025]
o bravo
[2026]
o spectramax m5
[2027]
即用型测定板(arp)
[2028]
o arp有两种格式:
[2029]
10mm最终最高浓度,稀释2.5倍。
[2030]
5mm最终最高浓度,稀释3倍。
[2031]
■
二者均有10点剂量反应。
[2032]
■
黑色测定板中最终0.1%的dmso。
[2033]
■
化合物在黑色测定板中稀释1000倍。
[2034]
■
每个板设计用于14种化合物,两份平行。
[2035]
o在最终黑色测定板中:
[2036]
·
第1列:仅培养基(无apol1)(100%活)
[2037]
·
第2-23列:0.05μg/ml apol1(~ec
90
)(10%活,有apol1)
[2038]
·
第24列:0.1μg/ml apol1(ec
100
)(大约0%活)
[2039]
测定程序
[2040]
布氏布氏锥虫培养
[2041]
方案a
[2042]
步骤1,第1天
[2043]
o在35℃下解冻细胞不超过2分钟。
[2044]
o将一个小瓶轻轻重新悬浮在20ml预热的培养基中,并且在37℃和5%co2下在t75烧瓶中孵育。
[2045]
o不要去除冷冻保护剂。
[2046]
步骤2,第4天
[2047]
o在室温下以800xg离心5分钟。
[2048]
o在1ml培养基中再悬浮。
[2049]
o进行1:25倍稀释(10μl/240μl培养基)。
[2050]
o在血细胞计数器上计数(加入寄生虫后)。
[2051]
·
让静置1-2分钟以便寄生虫沉降。
[2052]
·
计数应为大约100个活的活动性寄生虫/16个网格或大约25
×
106个寄生虫/烧瓶。
[2053]
o通过在20ml培养基中加入1
×
106个寄生虫/t75烧瓶来使寄生虫传代。
[2054]
o通过在46.6ml培养基中加入2.33
×
106个寄生虫/t175烧瓶来使寄生虫传代。
[2055]
·
每个t75烧瓶对于大约1.5
×
384孔测定板应足够。
[2056]
·
每个t175烧瓶对于大约3.8
×
384孔测定板应足够。
[2057]
步骤3,第6天
[2058]
o以800xg离心5分钟。
[2059]
·
每个75起始烧瓶再悬浮于3ml测定培养基中(无酚红、无fbs)。
[2060]
·
每个175烧瓶再悬浮于7ml测定培养基中(无酚红、无fbs)。
[2061]
o进行1:25倍稀释。
[2062]
o通过血细胞计数器计数。
[2063]
·
每个t75烧瓶设置应具有大约75x 106个寄生虫/烧瓶(验证倍增时间=8.7小时
±
1小时)。
[2064]
·
每个t175烧瓶设置应具有大约175x 106个寄生虫/烧瓶(验证倍增时间=8.7小时
±
1小时)。
[2065]
·
每个384孔板需要46
×
106个寄生虫(每孔120,000个寄生虫)。
[2066]
方案b
[2067]
步骤1,第1天
[2068]
o在35℃下解冻细胞不超过2分钟。
[2069]
o将一个小瓶轻轻重新悬浮在20ml预热的培养基中,并且在37℃和5%co2下在t75烧瓶中孵育。
[2070]
o不要去除冷冻保护剂。
[2071]
步骤2,第2天
[2072]
o在室温下以800xg离心5分钟。
[2073]
o在1ml培养基中再悬浮。
[2074]
o进行1:25倍稀释(10μl/240μl培养基)。
[2075]
·
让静置1-2分钟以便寄生虫沉降。
[2076]
·
计数应为大约100个活的活动性寄生虫/16个网格或大约8
×
106个寄生虫/烧瓶。
[2077]
o通过在20ml培养基中加入1.25
×
106个寄生虫/t75烧瓶来使寄生虫传代。
[2078]
·
每个t75烧瓶设置应有大约1.5
×
384孔测定板。
[2079]
·
每个t175烧瓶设置应有大约3.8
×
384孔测定板。
[2080]
步骤3,第5天
[2081]
o以800xg离心5分钟。
[2082]
·
每个t75起始烧瓶再悬浮于3ml测定培养基中(无酚红、无fbs)。
[2083]
·
每个t175起始烧瓶再悬浮于7ml测定培养基中(无酚红、无fbs)。
[2084]
o进行1:25倍稀释。
[2085]
o通过血细胞计数器计数。
[2086]
·
每个t75烧瓶应具有大约75x 106个寄生虫/烧瓶(验证倍增时间:7.7小时+1小时)。
[2087]
·
每个t175烧瓶应具有大约175x 106个寄生虫/烧瓶(验证倍增时间:7.7小时+1小时)。
[2088]
溶解测定设置
[2089]
apol1 g1蛋白
[2090]
o从-70℃中取出一份1.2mg/ml apol1蛋白储备液。
[2091]
o确定实验所需的量:
[2092]
·
每个384孔板需要11.5ml 0.1μg/ml的apol1。
[2093]
·
对于对照,每个384孔板需要0.5ml 0.2μg/ml的apol1。
[2094]
o在测定培养基中进行初始的1:10稀释(10μl/90μl)(现为120μg/ml)。
[2095]
·
使用最终浓度为0.05μg/ml的apol1获得~ec
50
。需要为每批新使用的蛋白质确定此值。
[2096]
·
添加30ml/孔浓度为0.1μg/ml的2x apol1。
[2097]
溶液a:对于0.1μg/ml的2x储备液,在10ml中测量8.33μl(120μg/ml)。
[2098]
溶液b:对于0.2μg/ml的2x储备液对照,在10ml中测量16.67μl(120μg/ml)。
[2099]
multidrop
[2100]
o黑色测定板(384孔黑色孔透明底,目录号3762)。
[2101]
第1列:分配30μl/孔的测定培养基(无apol1)。
[2102]
第2-23列:分配30μl/孔的溶液a(0.1μg/ml apol1)。
[2103]
第24列:分配30μl/孔的溶液b(0.2μg/ml apol1)。
[2104]
o储存板(聚丙烯储存板,目录号3656)。
[2105]
第1-24列:每孔分配80μl测定培养基(无apol1)(30ml培养基/板)。
[2106]
bravo:化合物转移
[2107]
o将储存板、即用型测定板(arp)和黑色测定板置于台面上。
[2108]
·
从储存板向arp转移20μl并混合。
[2109]
·
从arp向黑色测定板转移6μl并混合。
[2110]
■
[2111]
·
黑色测定板现已准备好加入锥虫了。
[2112]
锥虫加入:
[2113]
一旦黑色测定板加入了化合物,就开始收获锥虫,如布氏布氏锥虫培养章节的步骤3中所描述。
[2114]
o计数锥虫并以5
×
106/ml在测定培养基(无酚红且无fbs)中制备。
[2115]
·
每个384孔板需要9.2ml 5
×
106个锥虫/ml(46
×
106/板)。
[2116]
o使用e1-clip multichannel 12通道2-125μl可调移液器向384孔板的每个孔中加入24μl 5
×
106锥虫混合物。
[2117]
o加入完成后,在表面上轻击板以确保每个孔内都有液体。
[2118]
o将板置于板振荡器上大约10秒,振荡以确保均匀分布并且没有液滴留在任何边缘上。
[2119]
o在37℃和5% co2条件下,在孵育期中放置过夜(16小时)。
[2120]
o每个孔应包含60μl:
[2121]
30μl 2x apol1培养基、6μl 10x化合物和24μl锥虫溶液。
[2122]
alamarblue添加
[2123]
o在孵育器中过夜16小时后,从储存在冰箱中的瓶中取出所需量的alamarblue(2.3ml/板),并且在37℃水浴中短暂预热。
[2124]
o使用e1-clip multichannel 12通道2-125μl可调移液器添加6μl/孔。
[2125]
o避光,并且将板在37℃和5% co2下孵育2.5小时。
[2126]
o在spectramax(softmax pro 6.4软件,激发:555nm,发射:585nm)上读取。
[2127]
化合物1至390的效力数据
[2128]
式i化合物可用作apol1活性的抑制剂。下表46示出了使用上述程序的化合物1至390的ic
50
。在下表46中,下列含义适用:对于ip
50
(即,细胞增殖的ic
50
)和ic
90
:“+++”意指≤50nm;“++”意指在50nm与500nm之间;“+”意指≥500nm。n.d.=未测定。
[2129]
表46.化合物1至390的效力数据
[2130]
[2131]
[2132]
[2133]
[2134]
[2135]
[2136]
[2137][2138]
其他实施方案
[2139]
本公开仅提供所公开主题的示例性实施方案。本领域技术人员将从本公开和实施
方案中容易地认识到,在不脱离如以下权利要求书中限定的本公开的精神和范围的情况下,可在其中进行各种改变、修改和变化。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1