作为多巴胺D1受体正变构调节剂的苯基-3,4-二氢异喹啉-2(1H)-基-乙-1-酮衍生物的制作方法
作为多巴胺d1受体正变构调节剂的苯基-3,4-二氢异喹啉-2(1h)-基-乙-1-酮衍生物
1.本发明提供了某些苯基-3,4-二氢异喹啉-2(1h)-基-乙-1-酮相关的化合物,其药物组合物,以及它们用于治疗多巴胺能cns障碍的方法,多巴胺能cns障碍包括帕金森病、阿尔茨海默病、精神分裂症和注意力缺陷多动障碍(adhd)。
2.许多目前使用的药物通过多巴胺受体直接或间接起作用。这些药物包括用于帕金森病的多巴胺激动剂和多巴胺前体l-dopa、用于注意力缺陷障碍和发作性睡病的多巴胺释放剂和用于抑郁症的多巴胺再摄取抑制剂。d1受体在运动活动和奖励中具有重要作用,并且在维持工作记忆、注意力和执行功能的较高认知功能中具有特殊作用(arnsten af,cereb.cortex(2013)123,2269-2281)。迄今为止,开发用于临床应用的d1激动剂的尝试还没有成功,这促使寻找增加d1受体活性的替代方法。
3.一种这样的方法是鉴定多巴胺d1受体的变构增效剂,也称为正变构调节剂或pam。(svensson k等人,j.pharmacol.exp.ther.(2017)360:117
–
128)。变构调节剂是通过结合至与受体上的正构结合位点不同的位点(变构结合位点)来增强(正变构调节剂或pam)或抑制(负变构调节剂或nam)天然配体的作用的药剂。通过增加多巴胺对d1受体的亲和力,d1增效剂可以放大对内源性多巴胺的响应,增加多巴胺释放时和释放处的d1张力。这种活性模式与d1激动剂相反,只要d1激动剂存在,就可以激活它所能接触到的所有d1受体。在认知和运动活性的动物模型中,d1激动剂显示钟形的剂量-反应关系,这可能是由于在较高剂量下发生过度刺激。由于d1受体的恒定活化,一些d1激动剂也显示出耐受性快速发展。相反,因为d1增效剂将依赖于内源性张力并受到正常反馈控制,所以它可能具有低得多的过度刺激倾向。考虑到多巴胺和d1受体信号传导参与这些中枢神经系统功能,可增强d1受体活性的d1增效剂可提供用于治疗某些多巴胺相关疾病的替代和/或改善剂。
4.帕金森病是以脑中多巴胺能神经元损失为特征的慢性进行性神经变性病症。帕金森病表现为静止性震颤以及其它运动症状(例如运动徐缓和姿势不稳定)和非运动症状(例如认知缺损、睡眠障碍和抑郁)。目前用于治疗帕金森病的疗法包括施用非选择性多巴胺前体如左旋多巴和多巴胺受体激动剂。直接作用的多巴胺受体激动剂疗法还可能与冲动控制障碍、精神病和认知恶化有关,因为它们对d2受体的亲和力相对较大。精神分裂症是一种具有复杂病理机制的衰弱性疾病。精神分裂症的一个组成部分是认知缺损,其可能与d1受体活化或d1受体下调的缺乏有关。已经假设相对于d2调节更具有选择性的d1活化可有效治疗与精神分裂症相关的认知缺损。阿尔茨海默病是一种慢性进行性神经变性病症,其特征在于大脑皮质和某些皮质下区域中神经元和突触的损失。疾病进展包括认知缺损,其被假设为至少部分由于d1受体活化降低;因此,d1活化可在治疗与阿尔茨海默病相关的认知缺损中提供治疗益处。adhd是一种神经发育障碍,其特征在于难以集中注意力、过度活动或难以根据人的年龄控制其行为。假设d1活化可在adhd的治疗中提供治疗益处。因此,对于安全和有效治疗与精神分裂症、帕金森病、阿尔茨海默病和/或adhd相关的认知或其它神经功能缺损(例如替代的和/或改进的多巴胺d1受体正变构调节剂(d1 pam)),仍然显著存在未满足的需求。
5.wo 2014/193781记载了某些3,4-二氢异喹啉-2(1h)-基化合物作为d1 pam用于治疗与帕金森病、阿尔茨海默病、精神分裂症、抑郁症或adhd相关的认知缺损。
6.本发明提供某些新颖化合物,其为多巴胺1受体(d1)的选择性pam,并表现出药理学特性的有利组合,如响应于多巴胺的人类d1受体信号传导增强、高口服体内可用性和在对习惯环境的动物的运动活化的体内功效。因此,认为本发明的化合物可用于治疗帕金森病、阿尔茨海默病、精神分裂症和/或adhd。本发明的化合物可提供此类病症的替代治疗。
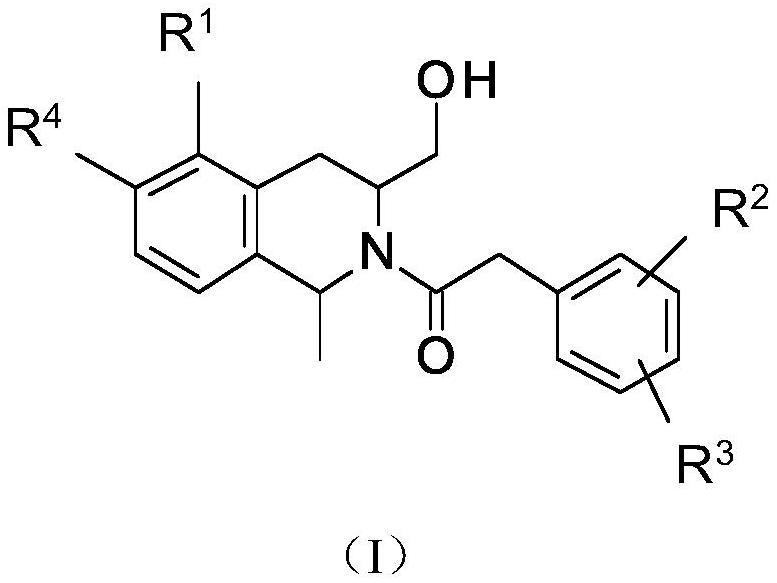
7.本发明提供一种具有式i的化合物:
[0008][0009]
其中:
[0010]
r1是
[0011]
r2是-f或
–
cl;
[0012]
r3是-f或
–
cl;且
[0013]
r4是-h或-f;
[0014]
条件是当r1为时,则r4为-f。
[0015]
具有式i的化合物在本发明的治疗方法中特别有用,但某些构型是优选的。以下段落描述了这样的优选构型。尽管如在式i中体现的本发明考虑了所述化合物的所有单独的对映异构体和非对映异构体,以及对映异构体和/或非对映异构体的混合物,包括外消旋体,但是具有如下所述的绝对构型的化合物是优选的。应当理解,这些优选方案适用于本发明的治疗方法和新化合物。
[0016]
式i的一个特别的实施例是一种具有式ia的化合物:
[0017][0018]
其中:
[0019]
r1是是
[0020]
r2是-f或
–
cl;
[0021]
r3是-f或
–
cl;且
[0022]
r4是-h或-f;
[0023]
条件是当r1为时,则r4为-f。
[0024]
式i的一个特别的实施例是一种具有式ib的化合物:
[0025][0026]
其游离碱形式也可命名为2-(2-氯-6-氟-苯基)-1-[(1s,3r)-6-氟-3-(羟基甲基)-5-(3-羟基-3-甲基-丁基)-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙酮。
[0027]
此外,本发明提供一种药物组合物,其包含具有式i、ia和/或ib的化合物,以及药学上可接受的载体、稀释剂或赋形剂。
[0028]
以下具体实施例为具有式i或ia的化合物。
[0029]
本发明提供了一种化合物,其为
[0030][0031]
本发明提供了一种化合物,其为
[0032][0033]
本发明提供了一种化合物,其为
[0034][0035]
本发明提供了一种化合物,其为
[0036]
[0037]
本发明提供了一种化合物,其为
[0038][0039]
本发明提供了一种化合物,其为
[0040][0041]
本发明提供了一种化合物,其为
[0042][0043]
本发明提供了一种化合物,其为
[0044][0045]
本发明提供了一种化合物,其为
[0046][0047]
本发明提供了一种化合物,其为
[0048][0049]
此外,本发明提供上文刚刚列出的特定实施例之一的化合物,和药学上可接受的载体、稀释剂或赋形剂。例如,本文所述的化合物实施例考虑共结晶。
[0050]
本发明的化合物是对多巴胺2(d2)受体具有最小活性的、多巴胺1(d1)受体的选择性pam。本发明的化合物可进一步提供其治疗益处,同时避免药物-药物相互作用的风险。因此,本发明的化合物被认为可用于治疗其中降低d1活性起作用且不期望d2活化的病症,例如帕金森病和精神分裂症,包括某些相关症状的缓解,例如与帕金森病相关的运动症状和认知缺损以及与精神分裂症相关的认知缺损和阴性症状,例如轻度认知缺损或痴呆。还认为本发明的化合物作为单一疗法或与其它疗法组合可用于改善帕金森病的运动症状。还认为本发明的化合物可用于治疗阿尔茨海默病的某些症状,例如认知缺损,例如轻度认知缺损。此外,本发明的化合物被认为可用于治疗adhd的某些症状。还认为本发明的化合物可用于治疗多巴胺能cns障碍,其包括帕金森病、阿尔茨海默病、路易体痴呆(lbd)、血管性痴呆、精神分裂症、adhd、抑郁、孤独症、慢性肌肉骨骼痛、纤维肌痛、认知缺损障碍、睡眠障碍、白天睡眠过多、发作性睡病、轮班工作障碍、创伤性脑损伤、慢性创伤性脑病、肥胖症和食欲调节、心境障碍、嗜睡(lethargy)、情感淡漠(apathy)和成瘾性障碍。
[0051]
此外,本发明提供了一种具有式i的化合物,其用于治疗。此外,本发明提供了一种具有式ia的化合物,其用于治疗。此外,本发明提供了一种具有式ib的化合物,其用于治疗。
[0052]
另一方面,本发明提供一种药物组合物,其包含具有式i、ia或ib的化合物,以及一种或多种药学上可接受的载体、稀释剂或赋形剂。此外,本发明的该方面提供了一种用于治疗帕金森病,例如与帕金森病相关的认知缺损的药物组合物,其包含具有式i、ia或ib的化合物,以及一种或多种药学上可接受的赋形剂、载体或稀释剂。在本发明该方面的另一个实施例中,提供了一种用于减轻与帕金森病相关的运动损害的药物组合物,其包含具有式i、ia或ib的化合物,以及一种或多种药学上可接受的赋形剂、载体或稀释剂。
[0053]
在本发明该方面的另一个实施例中,提供了一种用于治疗阿尔茨海默病,例如减
轻与阿尔茨海默病相关的认知缺损的药物组合物,其包含具有式i、ia或ib的化合物,以及一种或多种药学上可接受的赋形剂、载体或稀释剂。
[0054]
本发明该方面的另一个实施例提供了一种用于治疗精神分裂症,例如减轻与精神分裂症有关的认知缺损的药物组合物,其包含具有式i、ia或ib的化合物,以及一种或多种药学上可接受的赋形剂、载体或稀释剂。
[0055]
本发明的另一个实施例提供了一种用于治疗adhd的药物组合物,其包含具有式i、ia或ib的化合物,以及一种或多种药学上可接受的赋形剂、载体或稀释剂。
[0056]
此外,本发明提供了一种用于治疗帕金森病,例如与帕金森病有关的认知缺损,或例如减轻与帕金森病有关的运动损害的方法,其包含给予有需要的患者有效量的具有式i、ia或ib的化合物。
[0057]
此外,本发明提供了一种用于治疗阿尔茨海默病,例如与阿尔茨海默病相关的认知缺损的方法,其包含给予有需要的患者有效量的具有式i、ia或ib的化合物。
[0058]
此外,本发明提供了一种用于治疗精神分裂症,例如与精神分裂症相关的认知缺损的方法,其包含给予有需要的患者有效量的具有式i、ia或ib的化合物。
[0059]
此外,本发明提供了一种用于治疗adhd的方法,其包含给予有需要的患者有效量的具有式i、ia或ib的化合物。
[0060]
在该方面的一个实施例中,本发明提供了一种具有式i、ia或ib的化合物,其用于治疗帕金森病。在一个具体的实施例中,本发明提供了一种具有式i、ia或ib的化合物,其用于治疗与帕金森病相关的认知缺损。在另一个具体的实施例中,本发明提供了一种具有式i、ia或ib的化合物,其用于减轻与帕金森病相关的运动损害。
[0061]
此外,本发明提供了一种具有式i、ia或ib的化合物,其用于治疗精神分裂症,例如用于治疗与精神分裂症相关的认知缺损。
[0062]
此外,本发明提供了一种具有式i、ia或ib的化合物,其用于治疗adhd。
[0063]
此外,本发明提供了一种具有式i、ia或ib的化合物,其用于治疗阿尔茨海默病,例如用于治疗与阿尔茨海默病相关的认知缺损。
[0064]
在另一方面,本发明提供了具有式i、ia或ib的化合物在制备用于治疗帕金森病,例如治疗与帕金森病有关的认知缺损或减轻与帕金森病有关的运动损害的药物中的用途。
[0065]
此外,本发明提供了具有式i、ia或ib的化合物在制备用于治疗精神分裂症,例如治疗与精神分裂症有关的认知缺损的药物中的用途。
[0066]
此外,本发明提供了具有式i、ia或ib的化合物在制备用于治疗阿尔茨海默病,例如治疗与阿尔茨海默病有关的认知缺损的药物中的用途。
[0067]
此外,本发明提供了具有式i、ia或ib的化合物在制备用于治疗adhd的药物中的用途。
[0068]
尽管可以直接施用本发明方法中使用的化合物而无需任何制剂,但所述化合物通常以药物组合物的形式施用,所述药物组合物包含作为活性成分的具有式i、ia或ib的化合物,以及至少一种药学上可接受的载体、稀释剂和/或赋形剂。这些组合物可以通过多种途径施用,包括口服、舌下、鼻、皮下、静脉内和肌内。这样的药物组合物及其制备方法是本领域熟知的。参见例如remington:the science and practice of pharmacy(university of the sciences in philadelphia,ed.,21st ed.,lippincott williams&wilkins co.,
2005)。
[0069]
组合物优选配制成单位剂型,每个剂量含有约0.5mg至约800mg活性成分。术语“单位剂型”是指适合作为用于人类受试者和其它哺乳动物的单位剂量的物理离散单位,每个单位含有经计算以产生所需治疗效果的预定量的活性物质以及至少一种合适的药学上可接受的载体、稀释剂和/或赋形剂。应当理解,实际施用的化合物的量将由医师根据相关情况来确定,所述相关情况包括待治疗的病症、所选择的施用途径、所施用的实际化合物、个体患者的年龄、体重和反应以及患者症状的严重程度。经考虑,本发明的化合物(例如在本发明的药物组合物中)将通过长期施用治疗用于治疗阿尔茨海默病、帕金森病和/或精神分裂症,例如与这些疾病相关的轻度认知缺损。
[0070]
本发明的化合物可以与用于治疗或改善多巴胺能cns障碍的其它药物组合使用。这样的一种或多种其它药物可以通过通常使用的途径和量与本发明的化合物同时或顺序施用。例如,可与本发明化合物组合的有效治疗帕金森病的其它活性成分包括但不限于:(a)多巴胺前体如左旋多巴;美左旋多巴(melevodopa)和依替左旋多巴(etilevodopa);和(b)多巴胺激动剂,包括普拉克索(pramipexole)、罗匹尼罗(ropinirole)、阿扑吗啡(apomorphine)、罗替戈汀(rotigotine)、溴隐亭(bromocriptine)、卡麦角林(cabergoline)和培高利特(pergolide),(c)单胺氧化酶抑制剂,包括司来吉兰(selegiline)和雷沙吉兰(rasagiline),(d)comt抑制剂,包括托卡朋(tolcapone)和恩他卡朋(entacapone),(e)乙酰胆碱酯酶抑制剂,包括利凡斯的明(rivastigmine)和多奈哌齐(donepezil),(f)抗抑郁药,包括舍曲林(sertraline)、西酞普兰(citalopram)、米氮平(mirtazapine)和曲唑酮(trazodone)。此外,例如,可与本发明化合物组合的有效治疗阿尔茨海默病的其它活性成分包括但不限于:乙酰胆碱酯酶抑制剂,包括利凡斯的明和多奈哌齐,nmda拮抗剂,包括美金刚(memantine),某些抗精神病药,包括利培酮(risperidone)、喹硫平(quetiapine)、阿立哌唑(aripiprazole)、奥氮平(olanzapine)和匹莫范色林(pimavanserin),以及抗抑郁药,包括舍曲林、西酞普兰、米氮平和曲唑酮。
[0071]
如本文所用,术语“患者”是指需要治疗病症或疾病的哺乳动物,例如人。人是优选的患者。如本文所用,用本发明的化合物治疗的患者患有多巴胺能cns障碍,并且照此共享病因学方面,因为已知多巴胺信号传导障碍促成这些疾病。本文所用的多巴胺能cns障碍包括帕金森病、阿尔茨海默病、路易体痴呆(lbd)、血管性痴呆、精神分裂症、adhd、抑郁、孤独症、慢性肌肉骨骼痛、纤维肌痛、认知缺损障碍、睡眠障碍、白天睡眠过多、发作性睡病、轮班工作障碍、创伤性脑损伤、慢性创伤性脑病、肥胖症和食欲调节、心境障碍、嗜睡、情感淡漠和成瘾性障碍。可以通过本领域技术人员已知的已建立的方法来实现识别患有这些多巴胺能cns障碍的患者。
[0072]
本发明提供了一种将具有式i、ia或ib的化合物与多巴胺前体同时、分开或顺序组合用于治疗患者多巴胺能疾病的方法。本发明提供了一种将具有式i、ia或ib的化合物与多巴胺前体同时、分开或顺序组合用于治疗患者帕金森病的方法。
[0073]
本发明提供了一种将具有式i、ia或ib的化合物与多巴胺前体同时、分开或顺序组合用于治疗患者阿尔茨海默病的方法。
[0074]
本发明提供了一种将具有式i、ia或ib的化合物与多巴胺激动剂同时、分开或顺序组合用于治疗患者多巴胺能障碍的方法。本发明提供了一种将具有式i、ia或ib的化合物与
多巴胺激动剂同时、分开或顺序组合用于治疗患者帕金森病的方法。
[0075]
本发明提供了一种将具有式i、ia或ib的化合物与多巴胺激动剂同时、分开或顺序组合用于治疗患者阿尔茨海默病的方法。
[0076]
在本发明的实施例中,患者是已被诊断为具有医学风险、病状或病症,如多巴胺能cns障碍,需要用本文所描述的给药方案进行治疗的人。在可以通过本发明的方法治疗的病症通过已建立和接受的分类如ad、pd、lbd已知的那些情况下,它们的分类可以在各种众所周知的医学文本中找到。例如,目前,《精神病症诊断和统计手册(diagnostic and statistical manual of mental disorders)》(dsm-5)的第5版提供了用于识别本文所描述的病症中的许多病症的诊断工具。此外,《国际疾病分类(international classification of diseases)》第十次修订(icd-10)为本文所描述的病症中的许多病症提供了分类。技术人员将认识到,对于本文所描述的病症存在替代性命名法、疾病分类学和分类系统,包含如在dsm-5和icd-10中描述的那些,并且术语和分类系统随着医学科学进步而发展。患有帕金森病的受试者的认知障碍通常被称为神经认知病症。dsm-5(《精神病症诊断和统计手册》第5版)中的诊断标准描述了从先前的表现水平的显著认知降低的证据(神经心理学测试记录的个体或通知者的担忧),并且认知缺陷可能或可能不干扰日常活动的独立性。这被认定为重度或轻度神经认知病症。如本文所用,“体重减轻”是指体重减少和/或慢性体重管理,其中治疗促进将体重维持在期望的范围内。
[0077]
如本文所用,术语“治疗(treatment/treating)”或“减轻(mitigating)”旨在指其中可减缓、中断、阻止、控制或停止现有病症的进展和/或其症状的减少,但不一定指示所有症状的完全消除的所有过程。
[0078]
如本文所用,术语具有式i、ia或ib的化合物的“有效量”是指在患者体内有效加强多巴胺介导的反应的量,即剂量。优选的“有效量”可确定为与未治疗的患者相比可促进患者觉醒或警觉状态的量。在确定具有式i、ia或ib的化合物的有效量或剂量时,考虑许多因素,包括但不限于待施用的化合物及其特定制剂;患者体型、年龄、一般健康状况;疾病的牵涉程度或严重性;所述个体患者的响应;施用的方式;以及其它相关情况。
[0079]
本文使用的缩写定义如下:
[0080]“bn”是指苄基。
[0081]“cas#”是指化学文摘登记号。
[0082]“巴豆基”是指丁-2-烯-1-基。
[0083]“cns”是指中枢神经系统。
[0084]“1d-noesy”是指一维核极化效应(nuclear overhauser effect)nmr谱。
[0085]“dcm”是指二氯甲烷或二氯甲烷。
[0086]“dipea”是指n,n-二异丙基乙胺。
[0087]“dmso-d
6”是指完全氘化的二甲亚砜。
[0088]“ee”是指对映体过量。
[0089]“esms”是指电喷雾质谱。
[0090]“etoac”是指乙酸乙酯。
[0091]“h”是指小时或小时数。
[0092]“hatu”是指1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化
物六氟磷酸盐或n-[(二甲基氨基)-1h-1,2,3-三唑并-[4,5-b]吡啶-1-基亚甲基]-n-甲基甲铵六氟磷酸盐n-氧化物。
[0093]“hplc”是指高效液相色谱。
[0094]“lah”是指氢化铝锂。
[0095]“meoh”是指甲醇或甲基醇。
[0096]“min”是指分钟或分钟数。
[0097]“nmr”是指核磁共振波谱。
[0098]“pg”是指保护基。
[0099]“ph”是指苯基。
[0100]“oac”是指乙酸根。
[0101]“rac
‑”
是指外消旋或外消旋物。
[0102]“rt”是指室温或环境温度。
[0103]“sfc”是指超临界流体色谱法。
[0104]“tbdms”是指叔丁基二甲基甲硅烷基。
[0105]“tbdps”是指叔丁基二苯基甲硅烷基。
[0106]“thf”是指四氢呋喃。
[0107]“t
r”是指保留时间。
[0108]“w/w”是指重量对重量或重量比重量作为质量单位。
[0109]“wt%”是指重量百分比。
[0110]“z”是指苄氧基羰基,-c(o)och2ph,作为保护基。
[0111]
一般化学
[0112]
本发明的化合物可以通过本领域已知和理解的一般方法或通过本文所述的方法制备。用于这些方案的步骤的合适的反应条件是本领域公知的,并且溶剂和共试剂的适当取代是本领域技术人员已知的。同样地,本领域技术人员将理解的是,合成中间体可以根据需要或期望通过各种熟知的技术分离和/或纯化,并且经常地,可以在随后的合成步骤中直接使用各种中间体而几乎不纯化或不纯化。此外,本领域技术人员将理解,在一些情况下,引入部分的顺序不是关键的。
[0113]
方案1
[0114][0115]
方案1描述了化合物8的制备。本领域技术人员将认识到溶解在极性质子溶剂中的适当二取代的苯丙氨酸1(例如,x=br,cl,i;r4=h,f),可以用合适的强酸酯化以获得酯化的盐2。随后通过用碱水溶液洗涤盐以获得游离碱,溶解在非质子溶剂中,并加入适当的酰基氯进行酰化以获得3。2-取代的n-酰化苯丙氨酸甲酯3通过用多聚甲醛在合适的强酸中处理并搅拌环化得到四氢异喹啉4是本领域公知的。脱甲基化和脱羧可以通过用酸的水溶液处理并在回流下搅拌来实现,以获得作为相应胺盐的5。本领域技术人员将认识到,n-保护的四氢异喹啉6可以通过将胺盐5溶解在合适的极性非质子溶剂中,加入碱和合适的酸酐或氯甲酸烷基酯以获得氨基甲酸叔丁酯6来形成。随后还原为甲醇衍生物7可以使用一系列还原剂,例如用金属氢化物、硼氢化物盐或乙硼烷在极性非质子溶剂中进行。o-保护的四氢异喹啉8可以通过首先用适当的强酸处理n-保护的四氢异喹啉7并在真空下浓缩来实现。此后,可将胺盐溶于合适的非质子溶剂中,用碱和合适的保护基(pg)(例如,pg=osi,obn,omom等)处理,得到8。例如,用酸稳定的甲硅烷基如tbdms或tbdps保护伯醇是本领域公知的。
[0116]
方案2
[0117][0118]
方案2示出了制备必需的取代的中间体3(例如,x=br,cl,i;r4=f)的替代方法。本领域技术人员将认识到,适当二取代的苯甲醛13(r4=f)和n-保护的氨基-2-二甲氧基磷
酰基乙酸酯14之间的霍纳尔-沃兹沃思-埃蒙斯(horner-wadsworth-emmons)缩合可用于制备烯-氨基甲酸酯15(例如,pg=z)。随后在h2下用合适的手性助剂,例如用rh(i)过渡金属催化剂进行手性还原,如本领域充分描述的,得到n-保护的16。交换出n-保护基可以在本领域技术人员可识别的多种条件下完成,以获得3。化合物3的后续转化可以如方案1中进行以提供化合物8(例如,x=br,cl,i;r4=f)。
[0119]
方案3
[0120][0121]
方案3描述了i’型化合物的合成。由四氢异喹啉8形成亚胺可以在本领域技术人员可识别的各种氧化条件下完成,特别是仲胺的卤化和随后用合适的强碱消除以提供二氢异喹啉9。可以使用立体选择性格氏反应,通过用合适的烷基卤化镁处理亚胺9,以获得反式四氢异喹啉10。四氢异喹啉10的相对构型可以用适当的nmr谱实验,特别是1d-noesy测定。随后的n-酰化可用本领域熟知的酰胺偶联技术实现,例如苯并三唑-1-基-氧三吡咯烷鏻六氟磷酸盐,在温和的非亲核碱存在下,以制备化合物11。使用适当取代的化合物11(例如,x=br,cl,i等)形成碳-碳键可以在过渡金属催化下实现,例如使用pd、pt、ni或cu,与适当的硼酸酯或三氟硼烷盐,如本领域中众所周知的。或者,在heck-或sonogashira-型条件下分别与适当取代的烯烃或炔烃的偶联可以如本领域公知的那样实现。例如,可以实现钯介导的11与适当取代的环丙基三氟硼烷(trifluoroboranuide)的交叉偶联,以获得类型12的取代的环丙基化合物。另外,可以实现钯介导的与11适当取代的烯烃的交叉偶联,以获得类型12的烷基取代的化合物。技术人员将认识到,保护的醇12(其中pg=osi,obn,omom等)的脱保护可以在多种条件下进行。例如,可以用四丁基氟化铵除去甲硅烷基保护基,得到i’型手性化合物。
[0122]
在以下示例性制备和实例中,溶剂通常在减压下除去(蒸发)。在一些方法中,指示的产率是通过蒸发或过滤分离并且不经进一步纯化直接使用的产物的代表性粗产率。通过适当的nmr谱,特别是1d-noesy,确定了所述四氢异喹啉框架周围的相对立体化学。在侧链取代中取代的环丙基周围的构型的情况下,描述相对构型而不确定绝对立体化学。在侧链取代中的其它额外手性中心的情况下,尚未确定绝对构型。在这两种情况下,当通过手性色谱技术分离时,非对映异构体对被命名为“异构体1”或“异构体2”。
[0123]
制备1
[0124]
2-溴-d-苯基丙氨酸甲酯盐酸盐
[0125][0126]
将2-溴-d-苯丙氨酸(22.4g,91.8mmol)溶于meoh(459ml)中。在室温下加入乙酰氯(65.3ml,917.7mmol),并将所得反应混合物搅拌36小时。在减压下浓缩反应混合物,得到标题化合物(27.2g,》99%产率)。esms(m/z)258/260[m-cl,
79
br/
81
br].
[0127]
制备2
[0128]
2-溴-n-(甲氧基羰基)-d-苯基丙氨酸甲酯
[0129][0130]
将2-溴-d-苯基丙氨酸甲酯盐酸盐(27.2g,92.3mmol)溶于dcm(923ml)和水(185ml)的两相混合物中。在室温下加入nahco3(31g,369.4mmol)和氯甲酸甲酯(7.9ml,101.6mmol),并将所得混合物搅拌2.5小时。将混合物用水稀释并用dcm萃取。用na2so4干燥有机萃取物,过滤,减压浓缩滤液。将所得残余物通过快速硅胶色谱法纯化,用10-75%etoac的己烷溶液梯度洗脱,将所需色谱级分的溶剂蒸发后得到标题化合物(29.1g,》99%产率)。esms(m/z):316/318[m+h,
79
br/
81
br].
[0131]
制备3
[0132]
(3r)-5-溴-3,4-二氢-1h-异喹啉-2,3-二甲酸二甲酯
[0133][0134]
在室温下,将2-溴-n-(甲氧基羰基)-d-苯基丙氨酸甲酯(29.1g,92.1mmol)和多聚甲醛(9.13g,101.3mmol)的混合物在含有浓h2so4(38.4ml,719.9mmol)的冰醋酸(115ml,2mol)中搅拌7小时。将反应混合物在水和etoac之间分配,分离各层,并用etoac萃取水层。用na2so4干燥合并的有机萃取物,过滤,减压浓缩滤液。所得残余物通过快速硅胶色谱来纯化,用5-40%etoac的己烷溶液梯度洗脱,所需色谱级分的溶剂蒸发后得到标题化合物(27.6g,91%产率)。esms(m/z):328/330[m+h,
79
br/
81
br].
[0135]
制备4
[0136]
(3r)-5-溴-1,2,3,4-四氢异喹啉-3-羧酸甲酯盐酸盐
[0137][0138]
将(3r)-5-溴-3,4-二氢-1h-异喹啉-2,3-二甲酸二甲酯(27.6g,84mmol)溶解于5n hcl(330.6ml,1.7mol)中,并将所得混合物加热回流三天。将混合物减压浓缩,得到白色固体。通过过滤收集固体,用乙醚洗涤,并在40℃真空干燥过夜,得到(3r)-5-溴-1,2,3,4-四氢异喹啉-3-甲酸盐酸盐(1:1)(20.8g,71.1mmol)。将乙酰氯(50.6ml,711.0mmol)添加至(3r)-5-溴-1,2,3,4-四氢异喹啉-3-甲酸盐酸盐(1:1)(20.8g,71.1mmol)于meoh(474ml)中的0℃混合物中。将混合物温热至室温并搅拌36小时。将混合物减压浓缩并干燥,得到标题化合物(21.9g,85%产率)。esms(m/z):270/272[m-cl,
79
br/
81
br].
[0139]
制备5
[0140]
2-叔丁基-3-甲基-(3r)-5-溴-3,4-二氢-1h-异喹啉-2,3-二羧酸酯
[0141][0142]
将(3r)-5-溴-1,2,3,4-四氢异喹啉-3-羧酸甲酯盐酸盐(21.0g,68.5mmol)溶解于1,4-二噁烷(685ml)中。在室温加入饱和nahco3水溶液(685ml,17.5mol)和二碳酸二叔丁酯(29.9g,137.0mmol),并将两相混合物在室温搅拌90分钟。混合物用etoac萃取,有机萃取物用na2so4干燥,过滤,滤液减压浓缩。所得残余物通过快速硅胶色谱来纯化,用5-50%etoac的己烷溶液梯度洗脱,所需色谱级分的溶剂蒸发后得到标题化合物(19.5g,77%产率)。ms(m/z):270/272[m-t
boc+h,
79
br/
81
br].
[0143]
制备6
[0144]
(3r)-5-溴-3-(羟基甲基)-3,4-二氢-1h-异喹啉-2-羧酸叔丁酯
[0145][0146]
将2m硼氢化锂(99.4ml,198.8mmol)于thf和meoh(10.1ml,248.5mmol)中的溶液添加至2-叔丁基-3-甲基-(3r)-5-溴-3,4-二氢-1h-异喹啉-2,3-二羧酸酯(18.4g,49.7mmol)于thf(497ml)中的溶液中,并将所得混合物在室温下搅拌40分钟。将反应用水淬灭并用etoac萃取。分离有机萃取物,用na2so4干燥,过滤,减压浓缩滤液。将所得残余物通过快速硅胶色谱纯化,用5-80%etoac的己烷溶液梯度洗脱。将溶剂从所需色谱级分中蒸发,并将所得残余物在高真空下干燥过夜,得到呈白色固体标题化合物(19.1g,》99%产率)。esms(m/z):286/288[m-t
bu+h,
79
br/
81
br].
[0147]
制备7
[0148]
[(3r)-5-溴-1,2,3,4-四氢异喹啉-3-基]甲氧基-叔丁基-二甲基-硅烷
[0149][0150]
在室温下将三氟乙酸(75.5ml,998.3mmol)添加至(3r)-5-溴-3-(羟基甲基)-3,4-二氢-1h-异喹啉-2-羧酸叔丁酯(15.5g,45.3mmol)于dcm(226ml)中的溶液中。将反应混合物在室温搅拌30分钟并减压浓缩。将得到的残余物真空干燥,得到[(3r)-5-溴-1,2,3,4-四氢异喹啉-3-基]甲醇2,2,2-三氟乙酸,为湿固体。将[(3r)-5-溴-1,2,3,4-四氢异喹啉-3-基]甲醇2,2,2-三氟乙酸溶解于dcm(753ml)中。加入1h-咪唑(51.3g,753mmol)、n,n-二甲基-4-吡啶胺(460mg,3.77mmol)和叔丁基二甲基氯硅烷(13.6g,90.4mmol),并将所得混合物在室温搅拌过夜。加入饱和nh4cl溶液,混合物用dcm萃取。分离dcm层,用na2so4干燥,过滤,减压浓缩滤液。从与(3r)-5-溴-3-(羟基甲基)-3,4-二氢-1h-异喹啉-2-羧酸叔丁酯(6.6g,19.4mmol)基本上相同的反应运行合并粗产物。将合并的粗产物通过快速硅胶色谱纯化,用5-40%etoac的己烷溶液梯度洗脱,在所需色谱级分的溶剂蒸发后得到标题化合物(14.3g,89%产率)。esms(m/z):356/358[m+h,
79
br/
81
br].
[0151]
制备8
[0152]
[(3r)-5-溴-3,4-二氢异喹啉-3-基]甲氧基-叔丁基-二甲基-硅烷
[0153][0154]
将[(3r)-5-溴-1,2,3,4-四氢异喹啉-3-基]甲氧基-叔丁基-二甲基-硅烷(4.2g,11.8mmol)溶解于乙醚(118ml)中,并添加n-氯琥珀酰亚胺(2.36g,17.7mmol)。将所得混合物在室温下搅拌30分钟并减压浓缩。将所得残余物溶解于5%koh于meoh中的溶液(42.0ml,30.3mmol)中,并在室温下搅拌30分钟。将混合物倒入水中并用dcm萃取。dcm萃取物经na2so4干燥并过滤,滤液在减压下浓缩。所得残余物通过快速硅胶色谱来纯化,用5-100%etoac的己烷溶液梯度洗脱,得到标题化合物(3.40g,82%产率)。esms(m/z):354/356[m+h,
79
br/
81
br].
[0155]
可替代地,将n-氯琥珀酰亚胺(106.7g,790mmol)添加至[(3r)-5-溴-3,4-二氢异喹啉-3-基]甲氧基-叔丁基-二甲基-硅烷(220g,520mmol)于thf(3.85l)中的溶液中,在室温下在适当容器中搅拌30分钟。将混合物减压浓缩,并将残余物溶于5%w/w koh的甲醇溶液(2.2l,1.7摩尔)中,同时在室温下搅拌30分钟。将混合物加入水(3l)中,并用dcm(3
×
1l)萃取三次。将合并的有机萃取物用无水mgso4干燥并过滤,并将滤液减压浓缩,得到标题化合物(210g,》99%产率)。esms(m/z):354/356[m+h,
79
br/
81
br].
[0156]
制备9
[0157]
[(1s,3r)-5-溴-1-甲基-1,2,3,4-四氢异喹啉-3-基]甲氧基-叔丁基-二甲基-硅烷
[0158][0159]
将[(3r)-5-溴-3,4-二氢异喹啉-3-基]甲氧基-叔丁基-二甲基-硅烷(3.4g,9.6mmol)溶解于乙醚(160ml)中,并在干冰-丙酮浴中冷却至-78℃。滴加3m甲基氯化镁于thf中的溶液(26.9ml,80.6mmol)。将混合物缓慢温热至室温并搅拌过夜。通过加入饱和nh4cl溶液缓慢淬灭反应混合物。所得混合物用dcm萃取,用naso4干燥,过滤,滤液减压浓缩。将粗产物与来自用1.7mmol[(3r)-5-溴-3,4-二氢异喹啉-3-基]甲氧基-叔丁基-二甲基-硅烷基本上相同的反应运行的物质合并。将合并的残余物通过快速硅胶色谱纯化,用5-65% etoac的己烷溶液梯度洗脱,将所需色谱级分的溶剂蒸发后得到标题化合物(3.78g,》99%产率)。esms(m/z):370/372[m+h,
79
br/
81
br].
[0160]
化合物[(1s,3r)-5-溴-1-甲基-1,2,3,4-四氢异喹啉-3-基]甲氧基-叔丁基-二甲基-硅烷的相对构型通过nmr谱、使用1d-noesy测定。在1.30ppm处选择性激发甲基,在3.11ppm处产生对于ha的noe。这种noe增强仅与其中甲基和ha在环的同侧的构型(反式异构体)一致,因为在顺式异构体中,甲基质子离ha太远而不能表现出noe。由于已知3位的绝对化学为r,则1位的绝对化学推断为s。
[0161][0162]
制备10
[0163]
1-[(1s,3r)-5-溴-3-[[叔丁基(二甲基)甲硅烷基]氧基甲基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2,6-二氯苯基)乙酮
[0164][0165]
将2,6-二氯苯乙酸(3.7g,18mmol)与[(1s,3r)-5-溴-1-甲基-1,2,3,4-四氢异喹啉-3-基]甲氧基-叔丁基-二甲基-硅烷(5g,13.5mmol)、hatu(7.7g,20mmol)和dipea(7.1ml,41mmol)在dcm(70ml)中合并。将混合物在氮气层下在室温下搅拌2小时。将反应溶液在减压下浓缩,并将所得残余物通过快速硅胶色谱法纯化,用0-25% etoac的己烷溶液
梯度洗脱,在所需色谱级分的溶剂蒸发后得到标题化合物(6.2g,81%产率)。esms(m/z):558(m+1)。
[0166]
制备11
[0167]
反式-2-[(1s,3r)-3-[[叔丁基(二甲基)甲硅烷基]氧基甲基]-2-[2-(2,6-二氯苯基)乙酰基]-1-甲基-3,4-二氢-1h-异喹啉-5-基]环丙烷羧酸乙基酯
[0168][0169]
在微波小瓶中将1-[(1s,3r)-5-溴-3-[[叔丁基(二甲基)甲硅烷基]氧基甲基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2,6-二氯苯基)乙酮(500mg,0.9mmol)与[反式-2-乙氧基羰基环丙基]-三氟-硼氢化钾(300mg,1.3mmol;参见pct/fr2013/053057),kh2po4(370mg,2.7mmol)和1,1'-双(二苯基膦基)二茂铁-二氯化钯(ii)二氯甲烷络合物(115mg,0.1mmol)在1,4-二噁烷(8ml,93.2mmol)和水(2ml)中合并。将小瓶加盖,用氮气吹扫,并在120℃下在微波中照射2小时。将反应溶液在减压下浓缩,并将所得残余物通过快速硅胶色谱法纯化,用0-25% etoac的己烷溶液梯度洗脱,在所需色谱级分的溶剂蒸发后得到反式异构体混合物形式的标题化合物(245mg,46%产率)。esms(m/z):590(m+1)。
[0170]
制备12
[0171]
1-[(1s,3r)-3-[[叔丁基(二甲基)甲硅烷基]氧基甲基]-5-[反式-2-(2-羟基-2-甲基-丙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2,6-二氯苯基)乙酮
[0172][0173]
将反式-2-[(1s,3r)-3-[[叔丁基(二甲基)甲硅烷基]氧基甲基]-2-[2-(2,6-二氯苯基)乙酰基]-1-甲基-3,4-二氢-1h-异喹啉-5-基]环丙烷羧酸乙基酯(240mg,0.4mmol)在thf(4ml)中搅拌并冷却至0℃。在0℃下经5分钟经由注射器逐滴添加3m甲基氯化镁于thf中的溶液(1.75ml,5.25mmol)。将所得混合物在氮气下搅拌1小时,用饱和nh4cl水溶液淬灭,并用etoac萃取。分离有机层,用na2so4干燥,过滤,减压浓缩得到的滤液,得到标题化合物,为环丙基取代基周围的反式异构体混合物,不经进一步纯化即适用。esms(m/z):576(m+1)。
[0174]
制备13
[0175]
顺式-2-[(1s,3r)-3-[[叔丁基(二甲基)甲硅烷基]氧基甲基]-2-[2-(2,6-二氯苯基)乙酰基]-1-甲基-3,4-二氢-1h-异喹啉-5-基]环丙烷羧酸乙基酯
[0176][0177]
基本上使用制备2中所述的方法,将1-[(1s,3r)-5-溴-3-[[叔丁基(二甲基)甲硅烷基]氧基甲基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2,6-二氯苯基)乙酮(500mg,0.9mmol)与[顺式-2-乙氧基羰基环丙基]-三氟-硼氢化钾(wz5-e16784-076-b,300mg,1.3mmol;参见pct/fr2013/053057)合并,得到标题化合物,为顺式异构体的混合物(280mg,53%产率)。esms(m/z):590(m+1)。
[0178]
制备14
[0179]
1-[(1s,3r)-3-[[叔丁基(二甲基)甲硅烷基]氧基甲基]-5-[顺式-2-(2-羟基-2-甲基-丙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2,6-二氯苯基)乙酮异构体1
[0180]
以及
[0181]
1-[(1s,3r)-3-[[叔丁基(二甲基)甲硅烷基]氧基甲基]-5-[顺式-2-(2-羟基-2-甲基-丙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2,6-二氯苯基)乙酮异构体2
[0182][0183]
基本上使用在制备3中描述的方法,顺式-2-[(1s,3r)-3-[[叔丁基(二甲基)甲硅烷基]氧基甲基]-2-[2-(2,6-二氯苯基)乙酰基]-1-甲基-3,4-二氢-1h-异喹啉-5-基]环丙烷羧酸乙基酯(280mg,0.5mmol)和3m甲基溴化镁于thf中的溶液(2ml,6mmol)的混合物,随后通过快速色谱法在硅胶上纯化,使用0-25%etoac的己烷溶液梯度,在所需色谱级分的溶剂蒸发后得到标题化合物,为分离的顺式异构体1-[(1s,3r)-3-[[叔丁基(二甲基)甲硅烷基]氧基甲基]-5-[顺式-2-(2-羟基-2-甲基-丙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2,6-二氯苯基)乙酮异构体1(110mg,38%产率)和1-[(1s,3r)-3-[[叔丁基(二甲
基)甲硅烷基]氧基甲基]-5-[顺式-2-(2-羟基-2-甲基-丙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2,6-二氯苯基)乙酮异构体2(65mg,23%产率)。每个的esms(m/z):576(m+1)。
[0184]
制备15
[0185]
4-[(1s,3r)-3-[[叔丁基(二甲基)甲硅烷基]氧基甲基]-2-[2-(2-氯-6-氟-苯基)乙酰基]-1-甲基-3,4-二氢-1h-异喹啉-5-基]丁-2-酮
[0186][0187]
将1-[(1s,3r)-5-溴-3-[[叔丁基(二甲基)甲硅烷基]氧基甲基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2-氯-6-氟-苯基)乙酮(2g,3.7mmol)、三(二亚苄基丙酮)二钯(0)(169mg,0.2mmol)和二叔丁基-(1-苯基吲哚-2-基)磷烷(187mg,0.55mmol)悬浮于dmf(12.3ml)中。加入n,n-甲基二环己胺(0.9ml,4mmol)和丁-3-烯-2-醇(0.45ml,5.2mmol),并通过用氮气鼓泡使混合物脱气10分钟。将反应容器密封,并在搅拌下加热至100℃持续1.5小时。将反应混合物冷却至室温并用etoac稀释。分离有机层,用饱和氯化钠水溶液洗涤,用na2so4干燥,过滤,减压浓缩。所得残余物通过硅胶快速色谱来纯化,使用5-30% etoac的己烷溶液梯度,在所需色谱级分的溶剂蒸发后得到呈白色泡沫状的标题化合物(1.5g,75%产率)。esms(m/z):532(m+1)。
[0188]
制备16
[0189]
1-((1s,3r)-3-(((叔丁基二甲基甲硅烷基)氧基)甲基)-5-((r)-4,4-二氟-3-甲基-3-((三甲基甲硅烷基)氧基)丁基)-1-甲基-3,4-二氢异喹啉-2(1h)-基)-2-(2-氯-6-氟苯基)乙-1-酮
[0190]
以及
[0191]
1-((1s,3r)-3-(((叔丁基二甲基甲硅烷基)氧基)甲基)-5-((s)-4,4-二氟-3-甲基-3-((三甲基甲硅烷基)氧基)丁基)-1-甲基-3,4-二氢异喹啉-2(1h)-基)-2-(2-氯-6-氟苯基)乙-1-酮
[0192][0193]
将4-[(1s,3r)-3-[[叔丁基(二甲基)甲硅烷基]氧基甲基]-2-[2-(2-氯-6-氟-苯基)乙酰基]-1-甲基-3,4-二氢-1h-异喹啉-5-基]丁-2-酮(580mg,1.1mmol)和18-冠-6(29mg,0.1mmol)溶解于1,2-二甲氧基乙烷(5.5ml)中。加入(二氟甲基)三甲基硅烷(0.30ml,2mmol),然后加入csf(17mg,0.1mmol)。将混合物加热至60℃并搅拌过夜。加入另外的(二氟甲基)三甲基硅烷(0.30ml,2mmol)、18-冠-6(29mg,0.1mmol)和csf(17mg,0.1mmol),并将反应混合物在60℃搅拌4小时。将混合物冷却至室温,用etoac稀释,并将分离的有机萃取物用饱和nacl水溶液洗涤。用na2so4干燥有机层,过滤,减压浓缩滤液。所得残余物通过快速硅胶色谱来纯化,使用0-10% etoac的己烷溶液梯度,在所需色谱级分的溶剂蒸发后得到呈白色泡沫状的标题化合物的混合物(493mg,68%产率)。esms(m/z):656(m+1)。
[0194]
制备17
[0195]
(z)-2-(((苄氧基)羰基)氨基)-3-(2-溴-3-氟苯基)丙烯酸甲酯
[0196][0197]
将2-(苄氧羰基氨基)-2-二甲氧基磷酰基-乙酸甲酯(25.5g,76.9mmol)溶解于dcm(250ml)中,并将溶液冷却至0℃。加入1,8-二氮杂双环[5.4.0]十一碳-7-烯(12ml,79.9mmol),并将混合物在0℃搅拌30分钟。在0℃下经20分钟缓慢添加2-溴-3-氟-苯甲醛(13.0g,64mmol)于dcm(250ml)中的溶液。将混合物在0℃搅拌2小时。将混合物转移到分液漏斗中,依次用饱和nh4cl水溶液(200ml)和饱和nacl水溶液(200ml)洗涤。用na2so4干燥有机层,过滤,减压浓缩滤液,得到白色固体。将固体从热etoac中重结晶并通过过滤收集,得到呈白色固体的标题化合物(25g,80%产率)。esms(m/z):407(m+1)。
[0198]
制备18
[0199]
(r)-2-(((苄氧基)羰基)氨基)-3-(2-溴-3-氟苯基)丙酸甲酯
[0200]
bmd-e17046-003
[0201][0202]
在手套箱中,将(z)-2-(((苄氧基)羰基)氨基)-3-(2-溴-3-氟苯基)丙烯酸甲酯(26g,63.8mmol)添加至含有meoh(300ml)的高压釜容器中。加入[((r)-叔丁基甲基膦)(二叔丁基膦基)胺](1,5-环辛二烯)铑(i)四氟硼酸盐(430mg,0.8mmol),密封高压釜容器并从手套箱中取出。将混合物用h2气体冲洗15秒,并加压至100psi h2。将所得混合物在室温下搅拌15小时。将高压釜容器放空,并将混合物在减压下浓缩,得到呈白色固体的标题化合物,不经进一步纯化即适用(26.3g,98%产率)。esms(m/z):410(m+1)。
[0203]
为了获得分析纯的样品,将一部分粗产物(66mg)通过硅胶快速色谱法纯化,用15%etoac的己烷溶液洗脱,在所需色谱级分的溶剂蒸发之后,得到呈白色固体的产物。手性hplc分析(oj-h柱,0.2%异丙醇/meoh,1ml/分钟):主要对映异构体tr:2.981分钟,次要对映异构体tr:3.557分钟。测量》99%ee。
[0204]
制备19
[0205]
(r)-3-(2-溴-3-氟苯基)-2-((甲氧基羰基)氨基)丙酸甲酯
[0206][0207]
将(r)-2-(((苄基氧基)羰基)氨基)-3-(2-溴-3-氟苯基)丙酸甲酯(21.25g,51.8mmol)溶于dcm(110ml)中,并加入33% hbr于乙酸中的溶液(28.1ml,155mmol)。将所得混合物在室温下搅拌1小时。加入水(100ml)和dcm(100ml),将所得混合物剧烈搅拌15分钟。分离并弃去有机层。将dcm(200ml)和na2co3(35.1g,331.5mmol)加入到水层中。将氯甲酸甲酯(5.2ml)添加至混合物中,并将所得混合物在室温下搅拌过夜。将混合物用dcm萃取。将合并的有机萃取物用饱和nacl水溶液洗涤,用na2so4干燥,过滤,并将滤液减压浓缩,得到呈白色固体的标题化合物(13.6g,78%产率)。esms(m/z):333(m+1)。
[0208]
制备20
[0209]
(r)-5-溴-6-氟-1,2,3,4-四氢异喹啉-3-羧酸盐酸盐
[0210][0211]
将(r)-3-(2-溴-3-氟苯基)-2-((甲氧基羰基)氨基)丙酸甲酯(17g,50.9mmol)和
多聚甲醛(6.9g,76.4mmol)悬浮于乙酸(128ml)中。将溶液冷却至0℃并加入浓h2so4(43ml)。将所得混合物加热至35℃并搅拌过夜。将反应混合物冷却至室温并倒入水(200ml)中。将所得悬浮液用etoac萃取,并将合并的有机萃取物用饱和nacl洗涤,用na2so4干燥,过滤,并将滤液减压浓缩。将所得残余物溶解于浓hcl(231ml)中,并将所得混合物在配备有回流冷凝器的圆底烧瓶中加热至120℃过夜。将反应混合物冷却至室温,并在50℃减压浓缩。将得到的残余物在40℃真空干燥18小时,得到呈无色固体的标题化合物(15g,94%产率)。esms(m/z):273(m+1)。
[0212]
制备21
[0213]
(r)-(5-溴-6-氟-1,2,3,4-四氢异喹啉-3-基)甲醇
[0214][0215]
将(r)-5-溴-6-氟-1,2,3,4-四氢异喹啉-3-甲酸盐酸盐(9.95g,32.0mmol)悬浮于thf(80ml)中,并将混合物冷却至0℃。缓慢加入1m硼烷-四氢呋喃络合物的溶液(96ml,96mmol)。将混合物加热至60℃并搅拌过夜。将反应混合物冷却至0℃,加入meoh(20ml)和饱和nh4cl水溶液(40ml),将混合物在室温搅拌10分钟。添加5n hcl水溶液(60ml),并将所得混合物加热至45℃持续4小时。将混合物冷却至0℃,并加入21重量%浓nh4oh水溶液。将混合物温热至室温。所得混合物用3:1的chcl3:异丙醇萃取。将合并的有机萃取物用饱和nacl水溶液洗涤,经na2so4干燥,过滤,并在减压下浓缩滤液,得到呈米色固体状的标题化合物(9.5g,86%产率)。esms(m/z):259(m+1)。
[0216]
制备22
[0217]
(r)-5-溴-3-(((叔丁基二苯基甲硅烷基)氧基)甲基)-6-氟-1,2,3,4-四氢异喹啉
[0218][0219]
将(r)-(5-溴-6-氟-1,2,3,4-四氢异喹啉-3-基)甲醇(12.6g,48.3mmol)溶解于dcm(185ml)和dmf(146ml)的混合物中。加入咪唑(16.4g,241.5mmol)和4,4-二甲氨基吡啶(118mg,1mmol),然后加入叔丁基二苯基氯硅烷(13.8ml,53.1mmol)。将所得混合物在室温下搅拌1小时。加入meoh(100ml)并将混合物再搅拌1小时。加入饱和nh4cl水溶液(200ml),混合物用dcm萃取。用饱和氯化钠水溶液洗涤合并的有机萃取物,用na2so4干燥,过滤,减压浓缩滤液。所得残余物通过快速硅胶色谱来纯化,用2-50% etoac的己烷溶液梯度洗脱,在所需色谱级分的溶剂蒸发后得到呈油状的标题化合物(10.5g,45%产率)。esms(m/z):498m+1)。
[0220]
制备23
[0221]
(r)-5-溴-3-(((叔丁基二苯基甲硅烷基)氧基)甲基)-6-氟-3,4-二氢异喹啉
[0222][0223]
将(r)-5-溴-3-(((叔丁基二苯基甲硅烷基)氧基)甲基)-6-氟-1,2,3,4-四氢异喹啉(10.9g,21.9mmol)溶解于thf(200ml)中,并添加n-氯琥珀酰亚胺(4.4g,32.9mmol)。将所得混合物在室温下搅拌40分钟并减压浓缩。将所得残余物溶于0.75m koh于meoh(100ml)中的溶液中,并在室温搅拌30分钟。将反应混合物用水(50ml)稀释并用dcm萃取。用饱和nacl水溶液洗涤合并的有机萃取物,用na2so4干燥,过滤,浓缩滤液。所得残余物通过硅胶快速色谱来纯化,用5-50% etoac的己烷溶液梯度洗脱,在所需色谱级分的溶剂蒸发后得到呈无色油状的标题化合物(9.63g,88%产率)。esms(m/z):496(m+1)。
[0224]
制备24
[0225]
(1s,3r)-5-溴-3-(((叔丁基二苯基甲硅烷基)氧基)甲基)-6-氟-1-甲基-1,2,3,4-四氢异喹啉
[0226][0227]
在氮气下将(r)-5-溴-3-(((叔丁基二苯基甲硅烷基)氧基)甲基)-6-氟-3,4-二氢异喹啉(7.5g,15mmol)溶解于叔丁基甲基醚(190ml)中,并将溶液冷却至-15℃。在搅拌下滴加3m甲基氯化镁于thf中的溶液(20ml,60mmol),将反应混合物温热至室温并再搅拌2小时。将混合物冷却至0℃,依次加入meoh(4ml)和饱和nh4cl水溶液(4ml)。所得混合物用dcm萃取。用饱和氯化钠水溶液洗涤合并的有机萃取物,用na2so4干燥,过滤,减压浓缩滤液。将得到的残余物通过快速硅胶色谱纯化,用2-5%叔丁基甲基醚的dcm溶液梯度洗脱,在所需色谱级分的溶剂蒸发后得到呈黄色油状的标题化合物(1.7g,23%产率)。esms(m/z):512(m+1)。
[0228]
制备25
[0229]
1-[(1s,3r)-5-溴-3-[[叔丁基(二苯基)甲硅烷基]氧基甲基]-6-氟-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2-氯-6-氟-苯基)乙酮
[0230][0231]
将(1s,3r)-5-溴-3-(((叔丁基二苯基甲硅烷基)氧基)甲基)-6-氟-1-甲基-1,2,3,4-四氢异喹啉(5.2g,10.1mmol)溶解于dmf(101ml)中。依次加入2-(2-氯-6-氟-苯基)乙酸(2.5g,13.2mmol)、1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑[4,5-b]吡啶3-氧化物六氟磷酸盐(5g,13.2mmol)和dipea(8.8ml,50.6mmol),并将所得混合物在室温搅拌过夜。加入水(200ml),混合物用etoac萃取。用饱和氯化钠水溶液洗涤合并的有机萃取物,用na2so4干燥,过滤,减压浓缩滤液。将所得残余物通过快速硅胶色谱法来纯化,用0-25% etoac的己烷溶液梯度洗脱,在所需色谱级分的溶剂蒸发后得到呈白色泡沫状的标题化合物(6.3g,91%产率)。esms(m/z):682(m+1)。
[0232]
制备26
[0233]
1-[(1s,3r)-3-[[叔丁基(二苯基)甲硅烷基]氧基甲基]-6-氟-5-(3-羟基-3-甲基-丁-1-炔基)-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2-氯-6-氟-苯基)乙酮
[0234][0235]
将1-[(1s,3r)-5-溴-3-[[叔丁基(二苯基)甲硅烷基]氧基甲基]-6-氟-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2-氯-6-氟-苯基)乙酮(5.8g,8.5mmol)、三叔丁基膦(氯)(巴豆基)钯(ii)(675mg,1.7mmol)和2-噻吩羧酸亚铜盐(258mg,1.35mmol)加入到500ml圆底烧瓶中。将烧瓶抽空并用氮气回填。加入dmf(60ml)、dipea(6ml,42.5mmol)和2-甲基丁-3-炔-2-醇(4.1ml,42.2mmol)。将反应混合物加热至40℃并搅拌过夜。将混合物冷却至室温,用etoac稀释,分离各相。依次用水和饱和氯化钠水溶液洗涤有机层,用na2so4干燥,过滤,减压浓缩滤液。将得到的残余物通过快速硅胶色谱来纯化,用20-40% etoac的己烷溶液梯度洗脱,在所需色谱级分的溶剂蒸发后得到呈白色泡沫状的标题化合物(5.4g,93%产率)。esms(m/z):686(m+1)。
[0236]
制备27
[0237]
1-[(1s,3r)-3-[[叔丁基(二苯基)甲硅烷基]氧基甲基]-6-氟-5-(3-羟基-3-甲
基-丁基)-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2-氯-6-氟-苯基)乙酮
[0238][0239]
用氮气吹扫parr振荡器并加入5重量%硫化铂/碳(2.5g)。再次用氮气吹扫容器。加入etoac(50ml)。加入1-[(1s,3r)-3-[[叔丁基(二苯基)甲硅烷基]氧基甲基]-6-氟-5-(3-羟基-3-甲基-丁-1-炔基)-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2-氯-6-氟-苯基)乙酮(5.4g,7.9mmol)溶于etoac(50ml)中的溶液。用h2气体吹扫反应容器,并加压至60psi h2。将反应混合物在室温下振摇3小时。加入另外的悬浮在etoac(30ml)中的5重量%碳负载的硫化铂(1.21g),并将容器再增压至60psi h2。将反应容器再振摇8小时。将容器减压并用氮气吹扫。过滤所得悬浮液并在减压下浓缩滤液。将所得残余物通过快速硅胶色谱来纯化,用0-100% etoac的己烷溶液梯度洗脱(梯度0-100%),在所需色谱级分的溶剂蒸发后得到呈白色泡沫状的标题化合物(5.33g,7.72mmol)。ms(m/z):690.4(m+1)。
[0240]
制备28
[0241]
反式-2-((1s,3r)-3-(((叔丁基二苯基甲硅烷基)氧基)甲基)-2-(2-(2-氯-6-氟苯基)乙酰基)-6-氟-1-[0242]
甲基-1,2,3,4-四氢异喹啉-5-基)环丙烷-1-甲酸乙酯
[0243][0244]
将1-[(1s,3r)-5-溴-3-[[叔丁基(二苯基)甲硅烷基]氧基甲基]-6-氟-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2-氯-6-氟-苯基)乙酮(665mg,1mmol)、外消旋-[(反式-2-乙氧基羰基环丙基]-三氟硼酸钾(cas#1612792-88-7;参见pct/fr2013/053057;12月12日,2013)(257mg,1.2mmol)、二(1-金刚烷基)-正丁基-碘化鏻(50mg,0.1mmol)、[(二(1-金刚烷基)-丁基膦)-2-(2'-氨基-1,1'-联苯基)]甲磺酸钯(ii)(75mg,0.1mmol)和cs2co3(631mg,1.9mmol)悬浮于甲苯(10ml)和水(1ml)的混合物中。将混合物用氮气脱气5分钟,将小瓶密
封并在搅拌下加热至100℃过夜。将反应混合物冷却至室温并用etoac稀释。将所得混合物依次用水、饱和nahco3水溶液、水和饱和nacl水溶液洗涤,并分离各层。用mgso4干燥有机相并过滤,减压浓缩滤液。将所得残余物通过快速硅胶色谱来纯化,用0-100%乙基叔丁基醚的己烷溶液梯度洗脱,在所需色谱级分的溶剂蒸发后得到呈黄色固体的标题化合物的混合物(448mg,61%产率)。esms(m/z):716(m+1)。
[0245]
制备29
[0246]
1-[(1r,3r)-3-[[叔丁基(二苯基)甲硅烷基]氧基甲基]-6-氟-5-[(反式-2-(1-羟基-1-甲基-乙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2-氯-6-氟-苯基)乙酮
[0247][0248]
将反式-2-((1s,3r)-3-(((叔丁基二苯基甲硅烷基)氧基)甲基)-2-(2-(2-氯-6-氟苯基)乙酰基)-6-氟-1-甲基-1,2,3,4-四氢异喹啉-5-基)环丙烷-1-甲酸乙酯异构体1和反式-2-((1s,3r)-3-(((叔丁基二苯基甲硅烷基)氧基)甲基)-2-(2-(2-氯-6-氟苯基)乙酰基)-6-氟-1-甲基-1,2,3,4-四氢异喹啉-5-基)环丙烷-1-甲酸乙酯异构体2(440mg,0.6mmol)的混合物溶解于thf(4ml)中,并冷却至0℃。经10分钟缓慢添加1m甲基溴化镁于thf中的溶液(1.4ml,1.4mmol),并将所得反应混合物升温至室温且搅拌过夜。将反应混合物冷却至0℃,再加入1m甲基溴化镁于thf中的溶液(1.4ml,1.4mmol)。将反应混合物温热至室温并搅拌8小时。将混合物用meoh(130μl)淬灭,并在室温搅拌过夜。将反应混合物用etoac稀释,并将得到的溶液依次用饱和nahco3水溶液、水和饱和nacl水溶液洗涤。分离各层,有机相经mgso4干燥并过滤。减压浓缩滤液。将所得残余物通过快速硅胶色谱来纯化,用0-100%甲基叔丁基醚的己烷溶液梯度洗脱,在所需级分的溶剂蒸发后得到呈黄色固体的标题化合物的混合物(245mg,42%产率)。esms(m/z):702(m+1)。
[0249]
制备30
[0250]
1-[(1s,3r)-5-溴-3-[[叔丁基(二苯基)甲硅烷基]氧基甲基]-6-氟-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2,6-二氯苯基)乙酮
[0251][0252]
将(1s,3r)-5-溴-3-(((叔丁基二苯基甲硅烷基)氧基)甲基)-6-氟-1-甲基-1,2,3,4-四氢异喹啉(500mg,1mmol)、2-(2,6-二氯苯基)乙酸(260mg,1.3mmol)和1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑[4,5-b]吡啶鎓3-氧化物六氟磷酸盐(482mg,1.3mmol)悬浮于dmf(10ml)中。加入dipea(0.85ml,4.9ml),并在室温下搅拌混合物72小时。将反应混合物用etoac稀释,并依次用水和饱和nacl水溶液洗涤。分离各层,用na2so4干燥有机层,过滤,减压浓缩滤液。将得到的残余物通过快速硅胶色谱来纯化,用5-15% etoac的己烷溶液梯度洗脱,在所需级分的溶剂蒸发后得到呈白色泡沫状的标题化合物(680mg,0.97mmol)。esms(m/z):698(m+1)。
[0253]
制备31
[0254]
反式2-[(1s,3r)-3-[[叔丁基(二苯基)甲硅烷基]氧基甲基]-2-[2-(2,6-二氯苯基)乙酰基]-6-氟-1-甲基-3,4-二氢-1h-异喹啉-5-基]环丙烷羧酸乙酯
[0255][0256]
将1-[(1s,3r)-5-溴-3-[[叔丁基(二苯基)甲硅烷基]氧基甲基]-6-氟-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2,6-二氯苯基)乙酮(604mg,0.9mmol)、外消旋-[(反式-2-乙氧基羰基环丙基]-三氟硼酸钾(cas#1612792-88-7;参见pct/fr2013/053057;12月12日,2013)(228mg,1mmol)、二(1-金刚烷基)-正丁基-碘化鏻(44mg,0.1mmol)、[(二(1-金刚烷基)-丁基膦)-2-(2'-氨基-1,1'-联苯基)]甲磺酸钯(ii)(66mg,0.1mmol)和cs2co3(565mg,1.7mmol)悬浮于甲苯(10ml)和水(1ml)的混合物中。将所得混合物用氮气鼓泡5分钟,将反应密封,并将混合物在搅拌下加热至100℃过夜。将反应混合物冷却至室温,用etoac稀释,并依次用水、饱和nahco3水溶液、水和饱和nacl水溶液洗涤。分离各层,有机mgso4干燥有机相,过滤,减压浓缩滤液。将所得残余物通过快速硅胶色谱法来纯化,用0-100%甲基叔丁基醚的己烷溶液梯度洗脱,在所需色谱级分的溶剂蒸发后得到呈黄色固体的标题化合物,为反式非对映异构体的混合物(417mg,64%产率)。esms(m/z):732(m+1)。
[0257]
制备32
[0258]
1-[(1s,3r)-3-[[叔丁基(二苯基)甲硅烷基]氧基甲基]-6-氟-5-[反式-2-(1-羟基-1-甲基-乙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2,6-二氯苯基)乙酮
[0259][0260]
将反式2-[(1s,3r)-3-[[叔丁基(二苯基)甲硅烷基]氧基甲基]-2-[2-(2,6-二氯苯基)乙酰基]-6-氟-1-甲基-3,4-二氢-1h-异喹啉-5-基]环丙烷羧酸乙酯异构体1和反式2-[(1s,3r)-3-[[叔丁基(二苯基)甲硅烷基]氧基甲基]-2-[2-(2,6-二氯苯基)乙酰基]-6-氟-1-甲基-3,4-二氢-1h-异喹啉-5-基]环丙烷羧酸乙酯异构体2的混合物(405mg,0.6mmol)溶解于thf(3ml)中,并将混合物冷却至0℃。在10分钟内缓慢加入1m甲基溴化镁于thf中的溶液(2.8ml,2.8mmol),将反应混合物温热至室温并搅拌过夜。将反应混合物冷却至0℃并用meoh(220μl)淬灭。将反应混合物用etoac稀释,并依次用水和饱和nacl水溶液洗涤。分离各层,用mgso4干燥有机相,过滤,减压浓缩滤液。将所得残余物通过快速硅胶色谱法来纯化,用0-100%甲基叔丁基醚的己烷溶液梯度洗脱,在所需色谱级分的溶剂蒸发后得到呈黄色固体的标题化合物,为反式非对映异构体的混合物(336mg,55%产率)。esms(m/z):718(m+1)。
[0261]
实例1和2
[0262]
2-(2,6-二氯苯基)-1-[(1s,3r)-3-(羟基甲基)-5-[反式-2-(1-羟基-1-甲基-乙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙烯酮,异构体1
[0263]
以及
[0264]
2-(2,6-二氯苯基)-1-[(1s,3r)-3-(羟基甲基)-5-[反式-2-(1-羟基-1-甲基-乙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙烯酮,异构体2
[0265][0266]
将1m甲酸四丁基铵于thf(0.8ml)中的溶液加入到1-[(1s,3r)-3-[[叔丁基(二甲基)甲硅烷基]氧基甲基]-5-[(反式-2-(2-羟基-2-甲基-丙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2,6-二氯苯基)乙酮(170mg,0.3mmol)于thf(2.5ml)中的溶液中,并将得到的混合物搅拌30分钟。减压浓缩反应混合物。将所得残余物通过快速硅胶色谱来纯化,用0-65%etoac的己烷溶液梯度洗脱,进一步通过快速硅胶色谱纯化,用0-50% etoac
的dcm溶液梯度洗脱,在所需色谱级分的溶剂蒸发后得到标题化合物2-(2,6-二氯苯基)-1-[(1s,3r)-3-(羟基甲基)-5-[反式-2-(1-羟基-1-甲基-乙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙烯酮异构体2(20mg,14%产率)。将来自另外的快速色谱的混合级分通过反相色谱在18c硅胶上进一步纯化,使用5-95%含有碳酸氢铵的水的乙腈溶液的梯度,以在所需色谱级分的溶剂蒸发后得到标题化合物2-(2,6-二氯苯基)-1-[(1s,3r)-3-(羟基甲基)-5-[反式-2-(1-羟基-1-甲基-乙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙烯酮,异构体1(26mg,18%产率)。每个的esms(m/z):462(m+1)。1h nmr(400mhz,dmso-d6)异构体1:δ0.66-1.01(m,4h),1.08-1.27(m,6h),1.40-1.89(m,1h),2.00-2.08(m,1h),2.66-3.04(m,2h),3.20-3.32(m,2h),3.63-3.75(m,1h),4.09-4.31(m,3h),4.38-4.50(m,1h),4.98-5.25(m,2h),6.88-7.16(m,3h),7.33(t,1h),7.48(d,2h)。1h nmr(400mhz,dmso-d6)异构体2:δ0.66-1.01(m,4h),1.06-1.31(m,5h),1.40-1.89(m,2h),1.99-2.05(m,1h),2.67-3.00(m,2h),3.24-3.32(m,2h),3.66-3.77(m,1h),4.09-4.01(m,3h),4.40-4.49(m,1h),4.98-5.25(m,2h),6.92-7.15(m,3h),7.34(t,1h),7.48(d,2h)。
[0267]
实例3和4
[0268]
2-(2,6-二氯苯基)-1-[(1s,3r)-3-(羟基甲基)-5-[顺式-2-(1-羟基-1-甲基-乙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙烯酮,异构体1
[0269]
以及
[0270]
2-(2,6-二氯苯基)-1-[(1s,3r)-3-(羟基甲基)-5-[顺式-2-(1-羟基-1-甲基-乙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙烯酮,异构体2
[0271][0272]
基本上使用实例1和2中所述的方法,使用1-[(1s,3r)-3-[[叔丁基(二甲基)甲硅烷基]氧基甲基]-5-[顺式-2-(2-羟基-2-甲基-丙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2,6-二氯苯基)乙烯酮异构体1(100mg,173mmol),得到标题化合物2-(2,6-二氯苯基)-1-[(1s,3r)-3-(羟基甲基)-5-[顺式-2-(1-羟基-1-甲基-乙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙烯酮异构体1(61mg,76%产率)。esms(m/z):462(m+1)。1h nmr(400mhz,dmso-d6)异构体1:δ0.55(s,1h),0.65(s,2h),0.94-1.00(m,2h),1.07-1.22(m,7h),1.40-1.49(m,2h),1.98-2.07(m,1h),2.60-3.15(m,2h),3.20-3.29(m,1h),3.53-3.65(m,1h),4.09-4.34(m,2h),4.37-4.48(m,1h),4.98-5.34(m,2h),7.00-7.22(m,3h),7.30-7.37(m,1h),7.45-7.51(m,2h)。
[0273]
基本上使用实例1和2中所述的方法,使用1-[(1s,3r)-3-[[叔丁基(二甲基)甲硅烷基]氧基甲基]-5-[顺式-2-(2-羟基-2-甲基-丙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2,6-二氯苯基)乙烯酮异构体2(60mg,104mmol),得到标题化合物2-(2,6-二
氯苯基)-1-[(1s,3r)-3-(羟基甲基)-5-[顺式-2-(1-羟基-1-甲基-乙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙烯酮异构体2(42mg,87%产率)。每个的esms(m/z):462(m+1)。1h nmr(400mhz,dmso-d6)异构体1:δ0.81(s,3h),0.83-0.94(m,4h),1.14-1.26(m,5h),1.28-1.37(m,1h),1.50(d,1h),2.09-2.18(m,1h),2.66-3.05(m,2h),3.27-3.32(m,1h),3.56-3.67(m,1h),4.08-4.32(m,2h),4.39-4.48(m,1h),4.96-5.26(m,2h),7.00-7.16(m,3h),7.33(t,1h),7.48(d,2h)。
[0274]
实例5和6
[0275]
2-(2,6-二氯苯基)-1-[(1s,3r)-5-[4,4-二氟-3-羟基-3-甲基-丁基]-3-(羟基甲基)-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙烯酮,异构体1
[0276]
以及
[0277]
2-(2,6-二氯苯基)-1-[(1s,3r)-5-[4,4-二氟-3-羟基-3-甲基-丁基]-3-(羟基甲基)-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙烯酮,异构体2
[0278][0279]
将2-(2,6-二氯苯基)-1-[(1s,3r)-5-[(3r)-4,4-二氟-3-羟基-3-甲基-丁基]-3-(羟基甲基)-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙酮和2-(2,6-二氯苯基)-1-[(1s,3r)-5-[(3s)-4,4-二氟-3-羟基-3-甲基-丁基]-3-(羟基甲基)-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙酮(493mg,0.75mmol)的混合物溶解于thf(12ml)中。加入1m四丁基氟化铵于thf中的溶液(3ml,3mmol),将所得混合物在室温下搅拌1小时。将混合物用etoac稀释并用饱和nacl水溶液洗涤。分离有机层,用na2so4干燥,过滤,减压浓缩滤液。所得残余物通过硅胶快速色谱来纯化,使用0-75%etoac的己烷溶液梯度,在所需色谱级分蒸发后得到呈白色泡沫状的标题混合物(342mg,97%产率)。esms(m/z):470(m+1)。
[0280]
通过手性sfc(od-h柱,21
×
250mm)进一步纯化和分离两种非对映异构体,在80ml/分钟的流速和40℃的温度下用甲醇:co2(15:85)洗提,在所需色谱级分的溶剂蒸发后得到异构体1(156mg,46%产率)和异构体2(141mg,41%产率)。
[0281]
异构体1:esms(m/z):470(m+h)。分析性sfc tr:1.618分钟(od-h柱,4
×
150mm,15%甲醇/co2,5ml/分钟,225nm)。
[0282]
异构体2:esms(m/z):470(m+h)。分析性sfc tr:2.213分钟(od-h柱,4
×
150mm,15%甲醇/co2,5ml/分钟,225nm)。
[0283]
实例7
[0284]
2-(2-氯-6-氟-苯基)-1-[(1s,3r)-6-氟-3-(羟基甲基)-5-(3-羟基-3-甲基-丁基)-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙酮
[0285][0286]
将1-((1s,3r)-3-(((叔丁基二苯基甲硅烷基)氧基)甲基)-6-氟-5-(3-羟基-3-甲基丁基)-1-甲基-3,4-二氢异喹啉-2(1h)-基)-2-(2-氯-6-氟苯基)乙-1-酮(5.3g,7.7mmol)溶解于thf(129ml)中,并添加1m四丁基氟化铵于thf中的溶液(23ml,23mmol)。将混合物在室温下搅拌1小时。混合物用etoac稀释,用饱和nacl水溶液洗涤,分离各层。用na2so4干燥有机萃取物,过滤,减压浓缩滤液。将所得残余物通过快速硅胶色谱来纯化,用50-100% etoac的己烷溶液梯度洗脱,在所需色谱级分的溶剂蒸发后得到呈白色固体的标题化合物(2.75g,79%产率)。esms(m/z):452(m+1)。
[0287]
实例8和9
[0288]
2-(2-氯-6-氟-苯基)-1-[(1r,3r)-6-氟-3-(羟基甲基)-5-[反式-2-(1-羟基-1-甲基-乙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙烯酮,异构体1
[0289]
以及
[0290]
2-(2-氯-6-氟-苯基)-1-[(1r,3r)-6-氟-3-(羟基甲基)-5-[反式-2-(1-羟基-1-甲基-乙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙烯酮,异构体2
[0291][0292]
将1-[(1r,3r)-3-[[叔丁基(二苯基)甲硅烷基]氧基甲基]-6-氟-5-[反式-2-(1-羟基-1-甲基-乙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2-氯-6-氟-苯基)乙酮异构体1和1-[(1r,3r)-3-[[叔丁基(二苯基)甲硅烷基]氧基甲基]-6-氟-5-[反式-2-(1-羟基-1-甲基-乙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2-氯-6-氟-苯基)乙酮异构体2的混合物(1.1g,1.5mmol)溶解于thf(10ml)中,并将混合物冷却至0℃。滴加1m四丁基氟化铵于thf中的溶液(3ml,3mmol),搅拌反应混合物,同时温热至室温过夜。将反应混合物用etoac稀释,并依次用水和饱和nacl水溶液洗涤。分离所得层,用mgso4干燥有机层,过滤,减压浓缩滤液。将得到的残余物通过快速硅胶色谱法来纯化,用0-100%甲基叔丁基
醚的己烷溶液梯度洗脱,在所需色谱级分的溶剂蒸发后得到呈琥珀色油状的标题化合物的混合物(490mg,69%产率)。esms(m/z):464(m+1)。
[0293]
将两种非对映异构体进一步纯化并通过手性sfc分离(cellulose-2柱,21
×
250mm,20%异丙醇/co2,80ml/分钟,40℃),在所需色谱级分的溶剂蒸发后得到呈白色固体的2-(2-氯-6-氟-苯基)-1-[(1r,3r)-6-氟-3-(羟基甲基)-5-[反式-2-(1-羟基-1-甲基-乙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙烯酮,异构体1(136mg,28%产率;分析性hplc tr:2.769分钟)和2-(2-氯-6-氟-苯基)-1-[(1r,3r)-6-氟-3-(羟基甲基)-5-[反式-2-(1-羟基-1-甲基-乙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙烯酮,异构体2(121mg,25%产率;分析性hplc tr:3.368分钟)。
[0294]
异构体1:esms(m/z):464(m+h)。1h nmr(400mhz,cdcl3):δ0.94-1.17(m,2h),1.17-1.25(m,3h),1.25-1.44(m,4h),1.60(s,3h),1.80-1.90(m,1h),2.07-2.37(m,1h),2.91-3.23(m,3h),3.47-3.57(m,1h),3.92(t,j=16.1hz,1h),4.05(s,2h),4.42-4.66(m,1h),5.04(q,j=7.0hz,0.5h),5.21(q,j=7.0hz,0.5h)。6.83-7.09(m,3h),7.19-7.26(m,2h)。分析性scf tr2.769分钟(cellulose-2柱,4
×
150mm,20%异丙醇/co2,5ml/分钟,225nm)。
[0295]
异构体2:esms(m/z):464(m+h)。1h nmr(400mhz,cdcl3):δ0.71-0.81(m,1h),1.22(s,3h),1.24(s,3h),1.33-1.44(m,4h),1.48-1.59(m,1h),1.59-1.69(m,2h),1.69-1.81(m,1h),2.87-3.08(m,1h),3.08-3.31(m,1h),3.47-3.57(m,1h),3.70-3.95(m,1h),3.97-4.11(m,2h),4.43-4.60(m,1h),5.01-5.26(m,1h),6.84-7.08(m,3h),7.20-7.26(m,2h)。分析性sfc t
r 3.368分钟(cellulose-2柱,4
×
150mm,20%异丙醇/co2,5ml/分钟,225nm)。
[0296]
实例10和11
[0297]
2-(2,6-二氯苯基)-1-[(1s,3r)-6-氟-3-(羟基甲基)-5-[反式-(1-羟基-1-甲基-乙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙烯酮,异构体1
[0298]
以及
[0299]
2-(2,6-二氯苯基)-1-[(1s,3r)-6-氟-3-(羟基甲基)-5-[反式-(1-羟基-1-甲基-乙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙烯酮,异构体2
[0300][0301]
将1-[(1s,3r)-3-[[叔丁基(二苯基)甲硅烷基]氧基甲基]-6-氟-5-[反式-2-(1-羟基-1-甲基-乙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2,6-二氯苯基)乙酮异构体1和1-[(1s,3r)-3-[[叔丁基(二苯基)甲硅烷基]氧基甲基]-6-氟-5-[反式-2-(1-羟基-1-甲基-乙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]-2-(2,6-二氯苯基)乙酮异构体2的混合物(333mg,0.5mmol)溶解于thf(4.6ml)中,并将混合物冷却至0℃。滴加1m四丁
基氟化铵溶液于thf(0.50ml,0.50mmol)中的溶液,并将所得反应混合物升温至室温且搅拌过夜。将反应混合物用etoac稀释,并依次用水和饱和nacl水溶液洗涤。分离各层,用mgso4干燥有机相,过滤,减压浓缩滤液。将得到的残余物通过快速硅胶色谱法来纯化,用0-100%甲基叔丁基醚的己烷溶液梯度洗脱,在所需色谱级分的溶剂蒸发后得到呈琥珀色油状的标题化合物的混合物(211mg,72%收率)。esms(m/z):480(m+1)。
[0302]
将两种非对映异构体进一步纯化并通过手性sfc分离(cellulose-2柱,21
×
250mm,20%ipa/co2,80ml/分钟,40℃),在所需色谱级分的溶剂蒸发后得到呈白色固体的标题化合物2-(2,6-二氯苯基)-1-[(1s,3r)-6-氟-3-(羟基甲基)-5-[反式-(1-羟基-1-甲基-乙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙烯酮,异构体1(41mg,21%产率;分析性hplc tr:3.095分钟)和2-(2,6-二氯苯基)-1-[(1s,3r)-6-氟-3-(羟基甲基)-5-[反式-(1-羟基-1-甲基-乙基)环丙基]-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙烯酮,异构体2(47mg,24%产率;分析性hplc tr:3.793分钟)。
[0303]
异构体1:esms(m/z):480(m+h)。1h nmr(400mhz,cdcl3):δ0.92-1.07(m,2h),1.20-1.27(m,5h),1.60-1.66(d,j=6.6hz,1h),1.80-1.90(m,1h)。2.93-3.23(m,2h),3.47-3.60(m,1h),3.81-4.00(m,1h),4.23(s,2h),4.44-4.70(m,1h),5.06(q,j=6.7hz,0.5h),5.23(q,j=6.7hz,0.5h),6.84-7.04(m,2h),7.16-7.23(m,1h),7.36(d,j=8.1hz,2h)。分析性sfc t
r 3.095分钟(cellulose-2柱,4.6
×
150mm,20%ipa/co2,5ml/分钟,225nm)。
[0304]
异构体2:esms(m/z):480(m+h)。1h nmr(400mhz,cdcl3):δ0.70-0.83(m,1h),1.09-1.18(m,1h),1.23(d,j=6.1hz,1h),1.32(s,3h),1.35-1.46(m,4h),1.64(d,j=6.7hz,2h),1.69-1.82(m,1h),2.90-3.10(m,1h),3.16-3.37(m,1h),3.50-3.61(m,1h),3.70-4.00(m,1h),4.23(s,2h),4.48-4.63(m,1h),5.09(q,j=6.7hz,0.6h),5.24(q,j=6.7hz,0.4h)。6.85-7.05(m,2h),7.15-7.23(m,1h),7.36(d,j=8.0hz,2h)。分析性sfc t
r 3.793分钟(cellulose-2柱,4.6
×
150mm,20%ipa/co2,5ml/分钟,225nm)。
[0305]
人d1受体pam测定
[0306]
本发明化合物的pam活性可以基本上如svensson等人,an allosteric potentiator of the dopamine d1 receptor increases locomotor activity in human d1 knock-in mices without casusing stereotypy or tachyphylaxis,j.pharmacol.exp.ther.(2017)360:117-128中所描述的来测量。
[0307]
更具体地,使用pbabe-bleo逆转录病毒载体、通过基因转导产生稳定表达人d1受体(登录号nm_000794)的hek293细胞,并用zeocin
tm
(invivogen)选择。在约80%汇合时,使用tryple
tm
express(gibco)收获细胞,悬浮在fbs加8%dmso中,并储存在液氮中。在测定当天,将细胞解冻且再悬浮于stim缓冲液(补充有0.1%bsa,20mm hepes,500μm ibmx和100μm抗坏血酸的汉克斯平衡盐溶液)中。
[0308]
使用声学分配(labcyte)将测试化合物用dmso连续稀释(1:2)到测定板(proxiplate-384plus,perkinelmer)中以提供20个浓度用于全响应曲线。将测试化合物(80nl)加入到5μl含有2000个细胞的stim缓冲液和5μl于stim缓冲液中的2x浓度多巴胺溶液中,其将产生ec
20
水平响应(储备溶液中24nm,或最终12nm),并且孔中的最终dmso浓度为
0.8%。将板在室温下温育60分钟的总反应时间。
[0309]
根据制造商的说明,使用检测(cisbio)定量camp产生。通常,将含有抗-camp穴状化合物(5μl)和d2-缀合物(来自试剂盒)(5μl)的裂解缓冲液加入孔中,将板再温育60至90分钟,并使用envision
tm
板读数器(perkinelmer)检测时间分辨荧光。使用camp标准曲线将荧光数据转化为camp浓度,并使用4参数非线性逻辑斯谛方程(genedata screener,版本13.0.5-标准)进行分析。对于增效剂模式浓度-响应曲线,结果表示为在单独多巴胺的ec
20
浓度下的响应(标准化为0%)与对多巴胺的最大响应(定义为对5μm多巴胺的响应,最终浓度,归一化为100%)之间的窗口百分比。
[0310]
基于对照激动剂(多巴胺)的最大和最小响应计算绝对ec
50
值。%增强(%最高/%top)由浓度响应曲线的拟合顶部确定。某些实例化合物的绝对ec
50
和%最高示于下表1中:
[0311]
表1:
[0312][0313]
表1中上述实例化合物的绝对ec
50
值说明响应多巴胺的人d1受体信号传导增强,并
说明权利要求1的化合物作为人多巴胺d1受体的正变构调节剂的活性。本发明的实例化合物1-6和8-11代表手性化合物,并且如本文实施例中所述,已经作为单独的立体异构体制备和测试。参见以上实例1-6、8-11和表1。单个立体异构体的组合数据(实例1-6、8-11的绝对ec
50
范围为5.6nm至86.7nm)证明每个单个立体异构体代表本发明的d1正变构调节剂实施例。本文提供的实例的单个立体异构体的绝对立体化学的表征和测定在本领域普通技术人员的技能范围内,并且用于此类测定的方法在药物化学文献中是熟知的(参见例如chiral analysis(second edition)advances in spectroscopy,chromatography and emerging methods,2018)。例如,绝对构型通常通过nmr基于cda的使用来确定:涉及手性助剂和对映体底物之间的共价结合的非对映体衍生物采用优选的构象,其可以基于由结合到手性鉴别剂中的芳环引起的差异屏蔽来预测。
[0314]
人d1受体敲入小鼠的产生
[0315]
可以通过标准技术产生其中鼠多巴胺1(d1)受体被其人类对应物替代的转基因小鼠(一般参见svensson等人,j.pharmacol.exp.ther.(2017)360:117
–
128)。例如,将小鼠基因组片段从rp23细菌人工染色体文库中亚克隆并再克隆到pgk-neo靶向载体中。用外显子2中的人d1受体开放阅读框替换小鼠开放阅读框。外显子2上游的neo选择标记侧翼为frt位点,用于随后的去除。外显子2侧翼有loxp选择位点,允许选择通过与表达cre核酸酶基因的小鼠杂交产生d1敲除小鼠。
[0316]
c57bl/6n胚胎干细胞系b6-3在含有20%fbs和2
×
106单位/l白血病抑制因子的高葡萄糖dmem中的小鼠胚胎成纤维细胞的有丝分裂灭活饲养层上生长。将1000万个胚胎干细胞加30微克线性化载体dna电穿孔并进行g418选择(200μg/ml)。分离克隆并通过southern印迹进行分析。
[0317]
将含有预期大小插入片段的克隆插入胚泡中,并通过pcr对所得小鼠进行基因分型。将雄性嵌合体与含有flp核酸酶基因的雌性嵌合体杂交以消除选择标记。通过pcr鉴定含有无选择标记的人d1受体的后代。将雄性杂合体与雌性c57bl/6小鼠交配。将含有人d1受体的雄性和雌性后代交配,并通过pcr鉴定纯合子。发现纯合子的行为和繁殖是正常的,并且所述群体在随后的世代中保持纯合子状态。
[0318]
基础(习惯性)运动活性
[0319]
本发明化合物的体内功效可被证明是通过使用小鼠运动活性的d1受体来作用。使用自动系统跟踪小鼠的运动以测量运动活性。在尺寸为45
×
25
×
20cm的透明塑料鞋盒笼中进行小鼠运动活动行为的监测,其中1cm深的木片用于吸收性垫料,并用通风过滤的塑料笼顶部覆盖。将笼子置于矩形框架中,其含有8
×
4构型的12个光电管光束的网格(kinder scientific,poway,ca),该网格位于距笼子地板2.5厘米处,用于检测身体运动(步行)并由计算机记录。
[0320]
将雄性人d1受体敲入小鼠置于室中并使其适应室60分钟。在习惯期间,小鼠表现出如预期的随时间的运动减少。在施用本发明化合物后,发现动物运动以剂量依赖性方式增加。
[0321]
将小鼠随机分配到各治疗组。在剂量反应研究中,将每只小鼠分别置于一个运动活性盒中60分钟适应期。然后使用在20%羟丙基-β-环糊精载体中的测试化合物并使用10ml/kg剂量体积口服给予小鼠。给药后,将小鼠放回到lma盒中,在60分钟的测量期内,每
10分钟间隔记录每只小鼠的总行走次数。使用单向anova进行统计分析,然后使用dunnett比较检验进行事后分析。
[0322]
基本上如上所述测定实例7的化合物,发现其以剂量依赖性方式增加基础运动(下表2)。
[0323]
表2:
[0324][0325]
表2中所示的实例7的基础运动活性数据说明,本发明的化合物、特别是实例7,在适应环境的动物的运动活化中是有效的。该活性被认为是经由变构增强的、d1受体中枢活化的结果(参见例如svensson等人,j.pharmacol.exp.ther.(2017)360:117
–
128)。表2中提供的实例7的数据说明了本发明化合物增强内源性多巴胺介导的反应的药理学有利的体内功效。表2中提供的实例7的数据进一步说明了实例7和具有式i的化合物的药理学有利的口服生物利用度。
[0326]
血浆和脑水平:
[0327]
将实例7的2-(2-氯-6-氟-苯基)-1-[(1s,3r)-6-氟-3-(羟基甲基)-5-(3-羟基-3-甲基-丁基)-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙烯酮以3mg/kg至60mg/kg在进食条件下口服给予雄性小鼠,并在给药后1小时测定血浆和脑浓度。如先前所述体外测定化合物的未结合分数(zamek-gliszczynski mj等人,validation of 96-well equilibrium dialysis with non-radiolabeled drug for definitive measurement of protein binding and application to clinical development of highly-bounddrugs.j.pharm.sci.,(2011)100:2498-2507)。如先前所述测定未结合脑浓度(cu,脑)与未结合血浆浓度(cu,血浆)的比率(kpuu)(raub tj等人,brain exposure of two selective dual cdk4 and cdk6 inhibitors and the antitumor activity of cdk4 and cdk6 inhibition in combination with temozolomide in an intracranial glioblastoma xenograft,drug metab.dispos.(2015)43:1360-71)。下表3中给出的实例7的数据是每个剂量下3只动物的平均值。“con.”指浓度。
[0328]
表3:
[0329][0330]
本发明的化合物,例如实例7,2-(2-氯-6-氟-苯基)-1-[(1s,3r)-6-氟-3-(羟甲基)-5-(3-羟基-3-甲基-丁基)-1-甲基-3,4-二氢-1h-异喹啉-2-基]乙烯酮,显示药理学性质的有利组合,例如响应于多巴胺的人d1受体信号传导增强、高口服体内可用性、中枢神经系统可用性和在习惯于环境的动物的运动活化中的体内功效。例如,实例7证明了人d1受体信号传导响应于多巴胺的增强(10.2
±
1.40nm(n=15)),并且当以10、30和60mg/kg po口服施用时,在习惯于环境的人d1受体敲入小鼠的运动活化中具有显著的体内功效,说明了该化合物的有利的口服生物利用度。此外,当在宽剂量范围内对正常大鼠体内给药时,实例7通常具有良好的耐受性,并且在该体内实验中显示出有利的毒性缺乏。因此,实例7证明了有利的药理学性质的有利组合,支持可能用作口服施用的治疗剂用于多巴胺d1受体增效,以及用于治疗帕金森病、精神分裂症、adhd和/或阿尔茨海默病。
[0331]
通过cyp3a4(fm
cyp3a4
)代谢清除的化合物的分数的测定
[0332]
药物通过细胞色素p450(cyp)的氧化代谢的总清除率分数是潜在的受害药物-药物相互作用介导的副作用的重要指示。(一般参见ogu,chris c和maxa,jan l,drug interactions due to cytochrome p450,bumc proceedings 2000;13:421-423)。通过cyp氧化途径、特别是仅通过单一cyp(例如cyp3a4)的给定药物的总清除率的分数越大,在用于治疗时发现药物对不期望的受害药物-药物相互作用敏感的可能性越大。为了测定fm
cyp3a4
(通过cyp3a4代谢的分数),首先在人肝细胞中测定p450介导的氧化的分数(fm
cyp
)。然后使用重组cyp表型分析(rcp)测定cyp3a4介导的氧化(fm
cyp3a4_rcp
)相对于其它p450的相对贡献。使用以下等式计算fm
cyp3a4
:
[0333]
fm
cyp3a4
=fm
cyp x fm
cyp3a4_rcp
[0334]
人肝细胞中细胞色素p450(cyp)介导的氧化代谢(fm
cyp
)分数的测定
[0335]
在存在和不存在泛p450抑制剂1-氨基苯并三唑(abt)的情况下,在使用人肝细胞的测定中,由药物的清除率确定p450在化合物的总体代谢中的相对贡献。基本上如mcginnity df等人(2004)evaluation of fresh and cryopreserved hepatocytes as in vitro drug metabolism tools for the prediction of metabolic clearance,drug metab dispos,32:1247-1253中所述来测定冷藏保存的人肝细胞中的固有清除率。通常,测定温育物含有0.3μm测试化合物和106个细胞/ml肝细胞,进行或不进行使用abt(1mm)的0.5小时预温育。在15、30、60和90分钟温育后通过lc/ms测量母体化合物损失。固有清除率(μl/分钟/106个细胞)使用以下等式针对
±
abt计算。cyp的代谢分数是abt的抑制百分比。
[0336]
cl
int
=-k
dep
×
温育体积/106细胞,
[0337]
其中k
dep
,底物耗尽常数(min-1
),是使用线性回归从y轴上对数转换的剩余%对x轴上的时间(min-1
)确定的斜率。
[0338]
使用重组cyp表型分析(fm
cyp3a4_rcp
)测定cyp3a4介导的代谢的分数
first-pass drug metabolism,curr drug metab 8:676-684中发现的以下等式来确定:
[0353]fg
=q
gut
/(q
gut
+fu
gut x cl
int cyp3a,gut
)
[0354]qgut
=q
villi x cl
perm
/(q
villi
+cl
perm
)
[0355]
cl
perm
=p
app
×
肠表面积
[0356]
cl
int,cyp3a,gut
=cl
int,cyp3a,liver
/fu
mic
×
肠中的微粒体蛋白(mg)
×
0.4(内部计算)
[0357]
其中q
gut
是取决于绒毛状血流和化合物的渗透性的混合流动项。
[0358]
fu
gut
是肠细胞中未结合药物的分数
[0359]qvilli
是绒毛状血流
[0360]
cl
perm
是定义通过肠细胞的渗透性的清除术语
[0361]
p
app
是在内部mdckii细胞系中测量的被动渗透性
[0362]
fu
mic
是人肝微粒体中未结合药物的分数
[0363]
人肠参数来自yang等人,2007和gertz m等人(2010)prediction of human intestinal first-pass metabolism of 25cyp3a substrates from in vitro clearance and permeability data,drug metab dispos 38:1147-1158。
[0364]
用于评估通过cyp3a4抑制的受害者ddi的机械静态模型
[0365]
试验化合物在伊曲康唑(itraconazole)存在下的药物auc暴露量的潜在变化可通过使用基本上如han b等人(2013)prediction of cyp3a mediated drug-drug interactions:estimation of gut wall and hepatic contributions,ascpt annual meeting,indianapolis,in所述的机械静态模型测定:
[0366]
auc
po,inh
/auc
po
=[1/(ax fm+(1-fm))]x[1/(x x(1-fg)+fg)]
[0367]
a=1/(1+[i]h/ki)
[0368]
x=1/(1+[i]
gut
/ki)
[0369]
其中fm是肝cyp3a4代谢的分数
[0370]fg
是肠细胞中逃避肠代谢的分数
[0371]
ki是伊曲康唑从酶上的解离常数
[0372]
[i]h是伊曲康唑在肝脏中的浓度
[0373]
[i]
gut
是肠中伊曲康唑的浓度
[0374]
伊曲康唑作为cyp3a4抑制剂的参数(a=0.10且x=0.10)来自olkkola kt等人(1994)midazolam should be avoided in patients receiving the systemic antimycotics ketoconazole or itraconazole,clin pharmacol ther 55:481-485。
[0375]
代表干预化合物的实例7化合物基本上如上所述进行测试,发现其主要通过肝细胞中的非氧化过程代谢,而不是通过p450的氧化代谢(由cyp代谢的分数为32.3%)。在rcp测定中没有检查该化合物,但是假定100%cyp3a4代谢的最坏情况的参变量用于预测在伊曲康唑存在下的auc比。预测的auc比小于2。因此,认为本发明的化合物具有最小的通过任何cyp、包括cyp3a4的受害药物-药物相互作用风险(参见表4)。
[0376]
表4.实例7化合物的fm
cyp3a4
、fg和预测的auc比
[0377]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1