ALC1抑制剂和与PARPi的协同作用的制作方法
背景技术:
1、为了识别单链断裂和双链断裂(ssb/dsb),核聚adp核糖聚合酶(parp)是dna损伤反应(ddr)的早期关键因子。使用代谢物nad+,parp-1和parp-2将聚adp-核糖(par)链添加到染色质组分以及属于ddr的因子中,并且parp-3经由单adp核糖基化靶向染色质组分。通过识别发生特定改变的dna损伤诱导的结构,parp被募集到dna损害处,这打开了它们的par化活性,进而调节它们的活性以及其他ddr和染色质蛋白的活性,从而促进ddr(raychaudhuri和nussenzweig,2017)。
2、这种催化活性可以被nad+类似物抑制,并且已经变得特别令人感兴趣并且在临床上可用于遗传确定的癌症中。值得注意的是,通过在brca1或brca2缺陷的情况下靶向合成致死,所谓的parp抑制剂(parpi)用于治疗同源修复(hr)缺陷型癌症和其他癌症。这被认为是通过降低parp活性和/或通过将parp-1/2/3生物化学地“捕获”在染色质上而发生的(murai等人,2012,2014)。虽然“捕获”在分子上仍然定义不清,但所述术语定义了parp-1/2/3酶在受损染色质上的增强的募集、缔合和/或保留,通常通过用parp抑制剂(parpi)处理parp-1酶或者parp-1/2/3酶在其初始募集后释放的减少(这导致延长的保留)来诱导。这在生物化学上本身表现为parp酶与受损基因组区域/基因座的增强的稳态缔合/保留/结合(“捕获”)。
3、由于parpi的临床出现,parp-1已经成为越来越多的癌症的强大靶标,包括与免疫肿瘤学疗法组合(如当前的lynparza/keytruda试验),举许多例子中的一个。此外,一线parpi疗法和在种系brca-1/2突变之外的情况下的应用正在变得可能。
4、重要的是,临床parpi化合物都通过阻断底物nad+的结合而在活性位点的催化中心处结合基本上相同的位置,从而阻止聚(adp-核糖)合成,这主要是由于它们与烟酰胺(nad+核苷酸的一部分)的结构相似。
5、然而,parpi在临床上在肿瘤杀伤和患者结局方面展现出大不相同的临床功效。这些parpi作用的一个根本差异是它们促进parp-1/-2在染色质上的高度不同水平的捕获。目前普遍认为,与临床上不太有用的parpi相比,最强大且在临床上最有效的parpi将parp-1更强烈地捕获在dna断裂的位点处。在parp捕获后,dna损害变得更具细胞毒性,尤其是在于dna链断裂修复中具有遗传或表观遗传缺陷的突变肿瘤细胞(如功能hr缺陷型brca-1/2突变肿瘤细胞)中。此外,目前尚不清楚parp-1酶相比于parp-2酶相比于parp-3酶所做出的相对或优势贡献是什么,因为所有酶都参与感测dna链断裂并且募集到dna损伤位点处,并且parp-1和parp-2两者都促进染色质因子的par化,并且所有现有的临床parpi分子几乎都无法区分这两种相关的par聚合酶parp-1和parp-2。进而,认为parp捕获会导致癌细胞中的dna复制应激、基因组不稳定性和细胞死亡(lord和ashworth,2012)。例如,认为parp1的增强的捕获会导致杀伤癌细胞(尤其是具有缺陷型dna修复途径的癌症)的能力增加(zandarashvili等人,2020)。图1示出了经由parpi进行的parp捕获的细胞毒性机制。
6、因此,“parp捕获”被描述为parp-1或parp-2或parp-3与活细胞中的染色质的(增强的)缔合。使用parpi,可以破坏有助于parp-1与dna结合的变构机制(zandarashvili等人,2020),其中基于体外parp-1、parpi和dna相互作用,一些parpi有助于保留机制,而其他parpi促进促释放机制(zandarashvili等人,2020)。parpi对parp捕获的不同机制示于图3中。用他拉唑帕尼(talazoparib)处理的u2os细胞显示出增强的gfp标记的parp2在诱导的dna损害处的保留,而用维利帕尼处理的细胞显示出总体较少的parp2向dna损害处的募集。
7、parp-1或parp-2对于将ssb修复蛋白充分募集到损伤位点处是必要的。经由par化,染色质重塑酶或组蛋白修饰酶在dna损伤位点处被激活,这导致染色质压实的变化(luijsterburg等人,2016;mehrotra等人,2011;sellou等人,2016;smeenk等人,2013;timinszky等人,2009)。
8、parp-1酶和/或parp-2酶的激活导致许多蛋白质(尤其是特定的染色质重塑酶,尤其包括含宏结构域的核小体重塑蛋白(remodeler)alc1(chd1l))的募集(ahel等人,2009;gottschalk等人,2009;lehmann等人,2017;singh等人,2017)。宏结构域通常结合adp-核糖、寡adp-核糖和聚adp-核糖(karras等人,2005),因此含有宏结构域的蛋白质做出反应并募集到基因组上的parp激活位点处,包括在dna损伤期间以及与癌症相关时。重要的是,par或寡adp-核糖与alc1的宏结构域的结合稳健地打开了染色质重塑活性(ahel等人,2009;gottschalk等人,2009;lehmann等人,2017;singh等人,2017),这揭示了alc1作为变构调节的染色质重塑酶,是同类中的第一种,也是极少数催化活性直接受par调节的酶之一。此外,alc1是经验证的癌基因,并且在brca1/2缺陷型卵巢癌和乳腺癌样品中通常与parp1一起进行基因扩增(参见图2)。alc1抑制剂(如抑制alc1的atp酶功能和或核小体重塑功能的小分子)可以增强parpi的作用,导致增强的癌细胞杀伤和/或降低脱靶效应,从而减轻非癌细胞的细胞毒性。改变alc1的表达水平(在这种情况下,通过基于crispr的敲除)会影响癌细胞对parp抑制剂的敏感性的事实也为改变alc1的活性水平可以克服parp抑制剂耐药性提供了机会,因为去除alc1将parpi奥拉帕尼稳健地增强到可能足以规避parpi耐药性的水平,诸如(但不限于)brca缺陷状态的逆转(例如,通过内部缺失或通过表观遗传brca1/2基因沉默的丧失)。由于癌细胞对parp抑制剂的敏感性普遍被认为(也)源于parpi将parp1捕获在染色质上(并且可能还捕获parp2酶-这在本领域中尚未正式确立)的能力,我们假设小分子alc1抑制剂可能通过对parp1和/或parp2捕获的直接或间接影响而介导parpi致敏作用,绕过parpi耐药性和/或促进癌细胞杀伤。具体地,我们假设用小分子抑制alc1会增强parp1/2在dna损伤位点处的捕获,从而间接促进dna损伤和癌细胞杀伤。
9、最近,已经报道了首批靶向alc1(chd1l)的小分子抑制剂(abbott等人,2020)。所述出版物揭示了alc1抑制剂由于其致癌功能在驱动恶性结直肠癌(crc)方面(特别是通过影响crc中wnt/tcf驱动的上皮细胞向间充质细胞转化(emt))的作用。用一种特定的alc1抑制剂进行的分析进一步显示了显著dna损伤的证据,如通过dna损伤标记物γh2ax所测量的。这使得作者假设alc1可能增强结直肠癌标准护理dna损伤性化学疗法(如依托泊苷,其与dna和拓扑异构酶ii形成三元复合物,阻止dna链在dna复制后重新连接,从而导致dna链断裂和细胞死亡)的功效。重要的是,在此出版物中没有提出关于alc1抑制剂对parp-1/2/4捕获的影响的启示和实验证据。
10、为了分析引起不同捕获效应的机制,假设染色质重塑蛋白alc1(chd1l)可调节parp保留。含有par结合宏结构域的alc1调节蛋白质向dna损害处的募集。(ahel等人,2009;gottschalk等人,2009;lehmann等人,2017;singh等人,2017)。如图5和图6所示,在u2os细胞中经由alci抑制alc1导致parp-2在dna损害处的特异性保留。
11、由于alc1在几种肿瘤中被上调并且是肝细胞癌中经验证的癌基因(cheng等人,2013;li等人,2019;su等人,2014),经由导致parp-2捕获的小分子抑制剂抑制alc1可以驱动dna损伤反应的稳健变化。
12、诸位发明人假设经由小分子操纵alc1活性可能足以绕过低水平或高水平的parpi耐药性。因此,可以利用alc1和parp-2来完善肿瘤学中的parp靶向疗法。
13、此假设得到了本发明的一部分,即经由小分子抑制剂进行的alc1操纵通过parp-1、parp-2和/或parp-3捕获以及尚未描述的染色质/dna损伤相关parp家族成员的捕获来影响对dna损伤的反应。这些化合物(旨在抑制atp依赖性染色质重塑蛋白alc1(chd1l)的酶活性)将增强dna损伤的积累,从而在体外介导对brca缺陷的合成致死,因为基于临床前和临床证据,parp抑制已经被充分证明具有brca-1/2肿瘤抑制功能丧失的合成致死性,抑制parpi耐药机制,开辟alc1抑制作为具有完整hr途径的癌症的治疗方法,和/或允许经由促进parp-1/2/3捕获通过破坏alc1的癌基因功能(如hcc)来靶向alc1扩增的肿瘤。此外,由于alc1基因是在诱导的dna损伤处进行parp-染色质重排的关键介质(sellou等人,2016),靶向alc1活性的小分子可能在不影响parp-1/2/3在细胞核内或细胞核外的其他作用或不影响非dna损伤诱导的parp酶的情况下影响parp-1/2/3对染色质重排的核dna损伤相关功能,这可能得到脱靶效应降低和/或副作用减少的“第二代parpi”。
14、parp-1和parp-2以及parp-3在dna损伤位点处富集的动力学可以使用如本领域针对parp-1建立的活细胞成像来可视化(ahel等人,2009;gottschalk等人,2009)。野生型parp-1和parp-2迅速募集到由激光微束辐照诱导的dna损害处。经由在永生化细胞系中瞬时转染gfp标记的parp-1和parp-2,我们测量了alc1i对它们在dna损伤位点处的募集和保留的影响。
15、诸位发明人成功地鉴定到alc1的atp酶结构域内的变构结合口袋,并且使用此信息为alc1活性的抑制剂建模。鉴于alc1与各种增殖性疾病(特别是那些brca1/2缺陷型疾病)的上述相关性。因此,本发明提供了一类新型的化合物来治疗或改善肿瘤疾病,特别是特征在于alc1例如由于表达增加而活性增加的肿瘤疾病。
16、此外,诸位发明人确定,通过使用parpi和alc1抑制剂(优选本发明的变构alc1抑制剂)的组合,可以出乎意料地增强parpi的效果。因此,本发明尤其提供了(i)对parpi敏感的肿瘤的有效疗法,(ii)介导parpi致敏作用,(iii)绕过parpi耐药性,(iv)允许减少所施用的parpi的量,和/或(v)通过对parp-1、parp-2和/或parp-3捕获的直接或间接影响而促进癌细胞杀伤。
技术实现思路
1、在第一方面,本发明涉及一种克罗莫结构域-解旋酶-dna-结合蛋白1样蛋白(alc1)的变构抑制剂,其中所述抑制剂与由跨越seq id no:1的氨基酸残基101至219的氨基酸延伸段形成的变构结合口袋特异性结合。
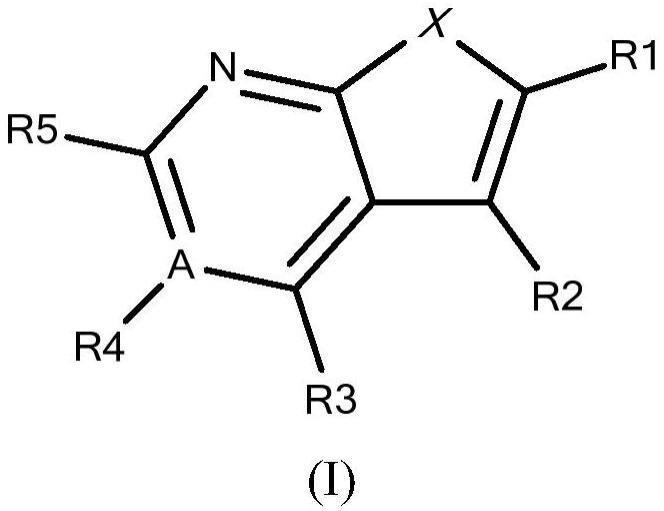
2、在进一步方面,本发明涉及一种式(i)的化合物:
3、
4、及其异构体、盐、溶剂化物、化学保护形式和前药,其中:
5、x是n或s;
6、a是c或n;
7、r1是-co-or6、-co-r7或-co-nr6ra,优选地r1是-co-or6;
8、r2是-r7、-nhr8、-o-r7、-c-o-r7、br、-c3-8-环烷基(优选环丙基)或-c4-8-环烯基(优选环己烯基);
9、或者
10、r1和r2一起形成任选地优选被1、2或3个独立地选自以下的取代基取代的5、6或7元碳环或杂环:-oh、-no2、-cn、-br、-cl、-f、-i、-o-c1-3-烷基、=o、[-o-ch2-ch2]-nh-ch(oh)-o-tbu;
11、r3是h、=o、-oh、-o-r7、-r7或-(ch2)m-l,其中m为0、1或2,并且l是任选地优选被1、2或3个独立地选自以下的取代基取代的5、6或7元碳环或杂环:-oh、-no2、-cn、-br、-cl、-f、-i、-o-c1-3-烷基、-羟基c1-3-烷基和=o;
12、r4是h或-c1-3-烷基,优选h;
13、r5是-(ch2)m-l或-(ch2)m-(ch=ch)-l,其中m为0、1或2,优选0或1,并且l是任选地优选被1、2、3或4个独立地选自以下的取代基取代的5、6或7元碳环或杂环、金刚烷基、c1-4-烷基或-n(ch3)2:-oh、-no2、-cn、-co-or6、-br、-cl、-f、-i、-r9、-o-r9、=o和[-o-ch2-ch2]q-nh-生物素,其中q为1、2、3或4,或者两个相邻的取代基形成5、6或7元碳环或杂环;
14、或者
15、r4和r5一起形成任选地优选被1、2或3个独立地选自以下的取代基取代的5、6或7元碳环:-oh、-no2、-cn、-br、-cl、-f、-i、-o-r9、-r9、=ch-ra和-ch2-ra,优选地r4和r5一起形成c5-7-环烷基;
16、r6是h、任选经取代的-c1-6-烷基、-c2-6-烯基、-c2-6-炔基,优选地r6是h;
17、r7是任选地优选被1、2或3个独立地选自以下的取代基取代的-c1-3-烷基、-c2-3-烯基、-c2-3-炔基:-br、-cl、-f、-i、-ch3、-och3或-sch3;
18、r8是h或c1-6-烷基,优选h,
19、r9是任选地被1、2或3个选自以下的取代基取代的-c1-6-烷基、-c2-6-烯基、-c2-6-炔基、-c1-6-烷基-芳基或c1-6-烷基-杂芳基(优选异噁唑、噻唑、四唑、1,2,4-噻二唑、1,2,3-噻二唑、1,2,5-噻二唑、吡啶、1,2,4-噁二唑、吡嗪或吡唑):-br、-cl、-f、-i、-no2、-cn、-conh2、-conh-c1-3-烷基(优选-conh-ch3)、-nh-co-c1-3-烷基(优选-nh-co-ch3)、-c1-6-烷基(优选-ch3、乙基、丙基、叔丁基或戊基)、-c1-3-卤代烷基(优选-cf3或-chf2)、-o-chf2、-o-cf3、碳环(优选环丙基、环己基或苯基)、-o-碳环(优选苯氧基)、杂环(优选吡唑基)、-co-杂环(优选-co-(1-吡咯烷基))、-so2-ch3、-so2-n(ch3)2、-o-c1-4-烷基(优选-och3)、-o-c1-3-烷基-o-c1-3-烷基(优选-o-ch2-o-ch3)、-sch3,或者当r9是-c1-6-烷基-芳基时,则所述芳基部分上的两个相邻取代基能够形成任选经取代的5、6或7元碳环或杂环;
20、ra是h、任选地优选被1、2或3个独立地选自以下的取代基取代的碳环或杂环:-oh、-no2、-cn、-br、-cl、-f、-i、-r9、-o-r7、-o-(ch2)o-r9、-so2nh2和=o,其中o为0或1。
21、在又另一方面,本发明涉及一种式(i)的化合物:
22、
23、及其异构体、盐、溶剂化物、化学保护形式和前药,其中:
24、x是n或s;
25、a是c或n;
26、r1是-co-or6、-co-r7或-co-nr6ra,优选地r1是-co-or6;
27、r2是-r7、-nhr8、-c-o-r7;
28、或者
29、r1和r2一起形成任选地优选被1、2或3个独立地选自以下的取代基取代的5、6或7元碳环或杂环:-oh、-no2、-cn、-br、-cl、-f、-i、-o-c1-3-烷基和=o;
30、r3是h、=o、-oh、-o-r7、-r7或-(ch2)m-l,其中m为0、1或2,并且l是任选地优选被1、2或3个独立地选自以下的取代基取代的5、6或7元碳环或杂环:-oh、-no2、-cn、-br、-cl、-f、-i、-o-c1-3-烷基、-羟基c1-3-烷基和=o;
31、r4是h或-c1-3-烷基,优选h;
32、r5是-(ch2)m-l或-(ch2)m-(ch=ch)-l,其中m为0、1或2,优选0或1,并且l是任选地优选被1、2、3或4个独立地选自以下的取代基取代的5、6或7元碳环或杂环或金刚烷基:-oh、-no2、-cn、-co-or6、-br、-cl、-f、-i、-r9、-o-r9和=o,或者两个相邻的取代基形成5、6或7元碳环或杂环;
33、或者
34、r4和r5一起形成任选地优选被1、2或3个独立地选自以下的取代基取代的5、6或7元碳环:-oh、-no2、-cn、-br、-cl、-f、-i、-o-r9、-r9、=ch-ra,优选地r4和r5一起形成c5-7-环烷基;
35、r6是h、任选经取代的-c1-6-烷基、-c2-6-烯基、-c2-6-炔基,优选地r6是h;
36、r7是任选地优选被1、2或3个独立地选自以下的取代基取代的-c1-3-烷基、-c2-3-烯基、-c2-3-炔基:-br、-cl、-f、-i、-ch3、-och3或-sch3;
37、r8是h或c1-6-烷基,优选h,
38、r9是任选地被1、2或3个选自以下的取代基取代的-c1-6-烷基、-c2-6-烯基、-c2-6-炔基或-c1-6-烷基-芳基:-br、-cl、-f、-i、-ch3、-och3或-sch3
39、ra是h、任选地优选被1、2或3个独立地选自以下的取代基取代的碳环或杂环:-oh、-no2、-cn、-br、-cl、-f、-i、-r9、-o-r7、-o-(ch2)o-r9、-so2nh2和=o,其中o为0或1。
40、在第二方面,本发明涉及一种双官能化合物,所述双官能化合物包含本发明的第一或进一步方面的alc1的变构抑制剂以及将e3泛素连接酶募集到alc1的化合物(e3募集剂(recruiter)),其中所述alc1的变构抑制剂和所述e3募集剂任选地通过接头共价连接。
41、在第三方面,本发明涉及一种药物组合物,所述药物组合物包含所述alc1的变构抑制剂和药学上可接受的赋形剂。
42、在第四方面,本发明涉及一种用于在治疗或改善患者的增殖性疾病中使用的alc1抑制剂(alc1i),其中所述方法包括施用所述alc1i以及任选地施用聚(adp-核糖)聚合酶抑制剂(parpi)。
43、在第五方面,本发明涉及一种用于在治疗或改善患者的增殖性疾病中使用的parpi,其中所述方法包括施用所述parpi以及施用所述alc1i。
44、在第六方面,本发明涉及一种试剂盒(kit of parts),所述试剂盒包含分开包装的parpi和alc1i或含有parpi和alc1i的组合物,优选地具有治疗或改善增殖性疾病的使用说明书。
- 还没有人留言评论。精彩留言会获得点赞!